
Myocardial Oedema as a Consequence of Viral Infection and Persistence—A Narrative Review with Focus on COVID-19 and Post COVID Sequelae
 , , ,
, , ,
Abstract
:1. Introduction
2. Physiological Background of Myocardial Fluid Filtration
2.1. Starling Forces and Microvascular Fluid Filtration
2.2. The Glycocalyx as Key Regulator of the Endothelial Barrier
3. Pathophysiology
3.1. Glycocalyx Disintegration
3.2. Intercellular Junctions and Key Signalling Processes
3.3. Inflammation and Myocardial Oedema
3.4. Detection of Myocardial Oedema and Myocarditis
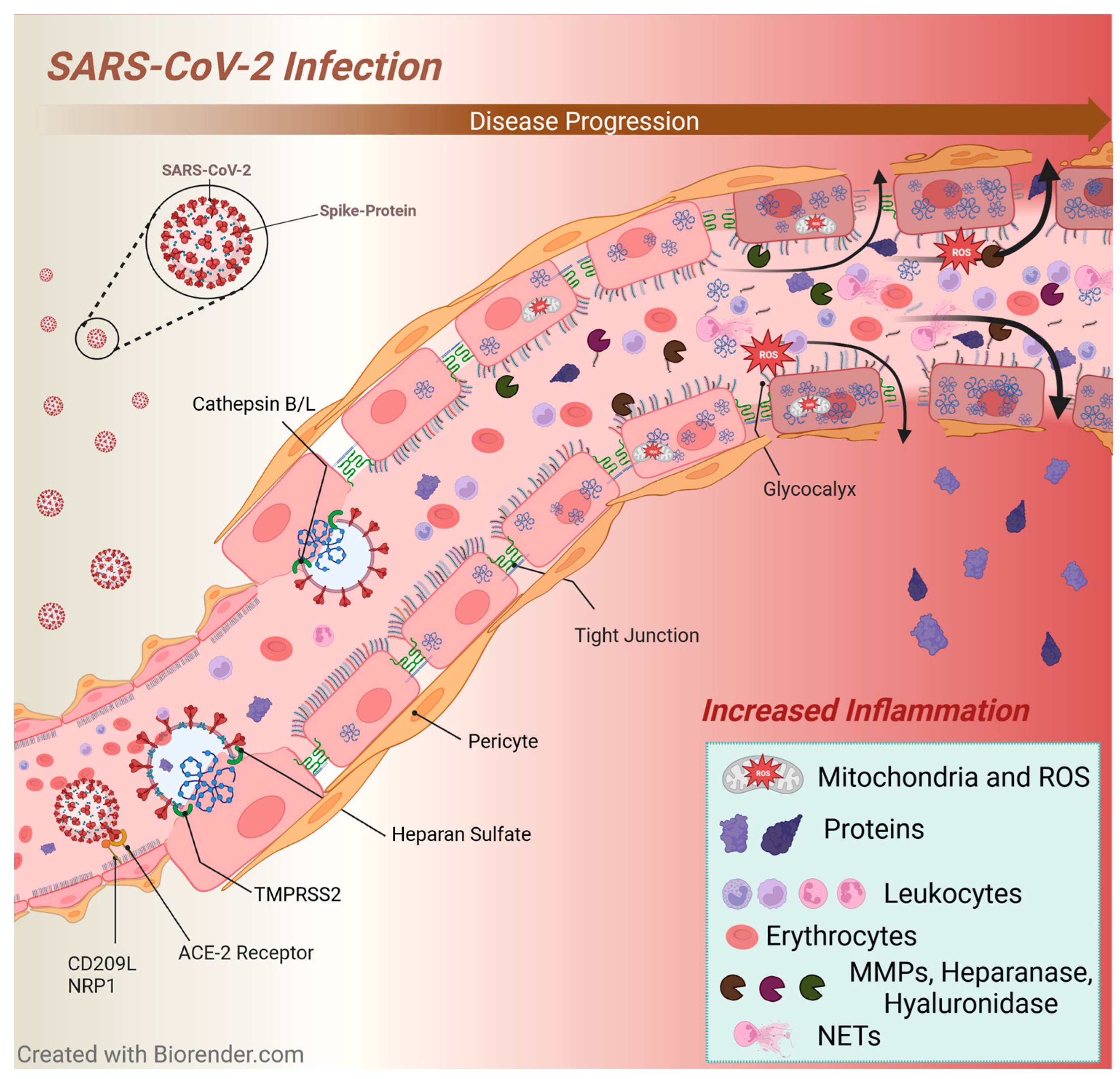
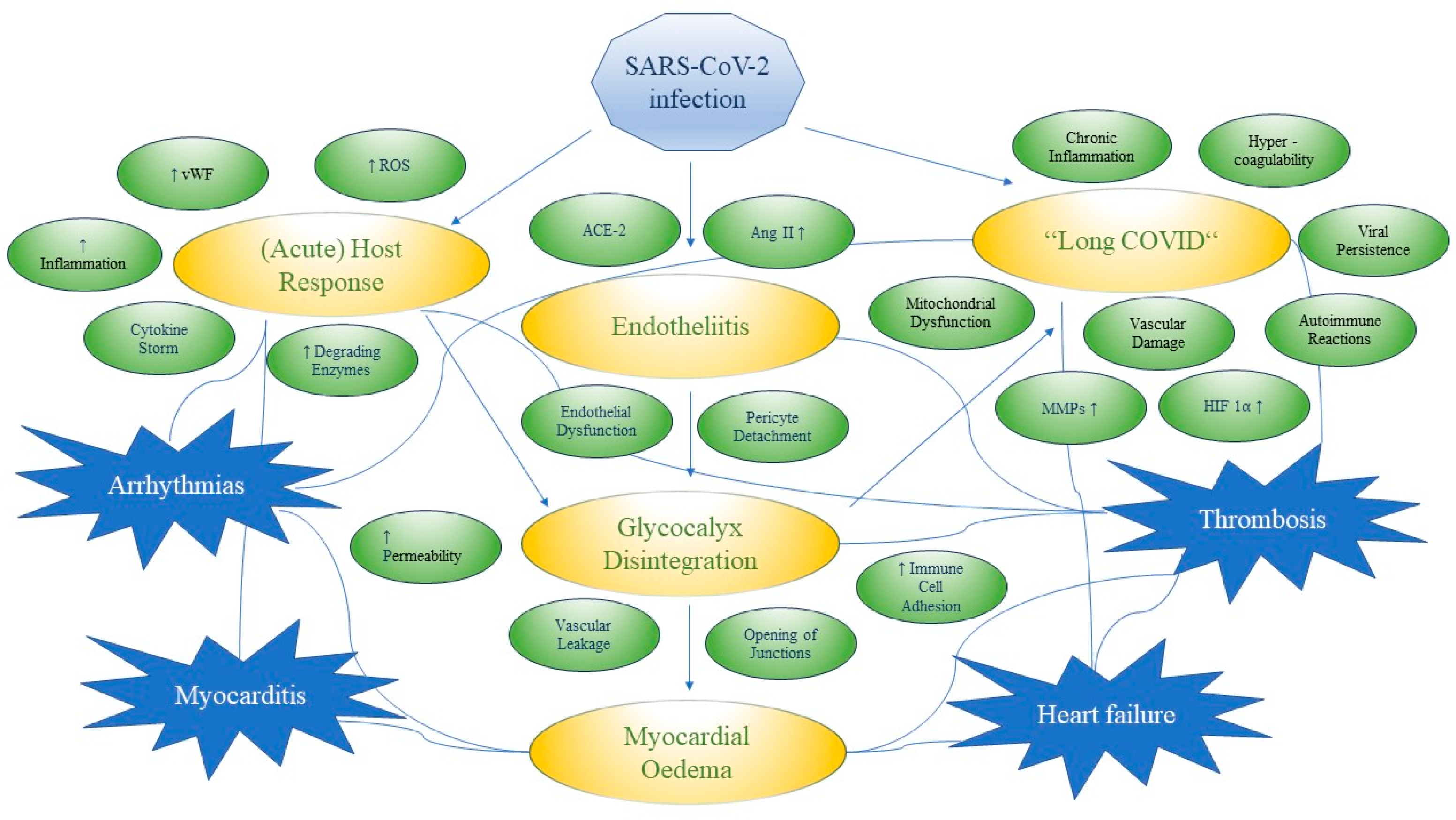
3.5. SARS-CoV-2 Infection

3.5.1. Myocarditis in SARS-CoV-2 Infection
3.5.2. Arrhythmias in SARS-CoV-2 Infection
3.5.3. Glycocalyx Changes and Inflammation in SARS-CoV-2 Infection
3.5.4. Immunothrombosis in SARS-CoV-2 Infection
3.5.5. Possible Long-Term Effects of SARS-CoV-2 Infection

3.6. Cardiotrophic Viruses and Myocarditis

{kind=link}
{kind=link}
| Virus | Glycocalyx Components | Literature Reference |
|---|---|---|
| Parvovirus B19 | heparan sulphate, sialic acid | [405] |
| Human herpesvirus 1, 6 | heparan sulphate, syndecan-1 | [406,407] |
| Epstein–Barr virus | glycoproteins, hyaluronan synthesis | [408,409] |
| Human cytomegalovirus | heparan sulphate | [410,411] |
| Enteroviruses | heparan sulphate, P-selectin glycoprotein ligand-1, sialylated glycan | [412,413,414] |
| Adenovirus | heparan sulphate, sialic acid | [415,416,417] |
| Hepatitis C virus | heparan sulphate, syndecan-1 | [418,419] |
| SARS-CoV-2 | heparan sulphate proteoglycans | [241,420] |
| Influenza virus | sialic acids, hyaluronan synthesis | [415,421] |
4. Clinical Implications and Treatment Options
4.1. Myocardial Oedema and Myocarditis
4.2. Endothelial Damage and Glycocalyx Disintegration
4.3. Thrombosis
5. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM10 (a disintegrin and metalloproteinase 10) |
| AF (atrial fibrillation) |
| AJ (adherens junctions) |
| ANP (A-type natriuretic peptide) |
| Ang-1 (angiopoietin-1) |
| Ang-2 (angiopoietin-2) |
| AngII (angiotensin II) |
| cAMP (cyclic adenosine monophosphate) |
| CD39 (ecto-ADPase) |
| CD209L (cluster of differentiation 209) |
| CMR (cardiac magnetic resonance imaging) |
| CNP (C-type natriuretic peptide) |
| CVD (cardiovascular disease) |
| DAMPs (damage-associated molecular patterns) |
| EMB (endomyocardial biopsy) |
| Epac1 (guanine nucleotide exchange factor 3) |
| ERK1/2 (extracellular signal-regulated kinase 1/2) |
| ESL (endothelial surface layer) |
| ET (extracellular trap) |
| FDG (fluorodeoxyglucose) |
| G-CSF (granulocyte colony-stimulating factor) |
| GDM (guideline directed medical) |
| GEFs (guanine nucleotide exchange factors) |
| HFrEF (heart failure with reduced ejection fraction) |
| HFpEF (heart failure with preserved ejection fraction) |
| HIF (hypoxia-inducible factor) |
| HO-1 (heme oxygenase 1) |
| ICAM (intercellular adhesion molecule) |
| IFN (interferon) |
| IL (interleukin) |
| LDL (low-density lipoprotein) |
| LGE (late gadolinium enhancement) |
| LPS (lipopolysaccharide) |
| LVEF (left ventricular ejection fraction) |
| mECM (myocardial extracellular matrix) |
| MCS (mechanical circulatory support) |
| MMPs (matrix metalloproteinases) |
| MO (myocardial oedema) |
| NET (neutrophil extracellular trap) |
| NO (nitric oxide) |
| NRF2 (nuclear factor erythroid-2-related factor 2) |
| NRP1 (neuropilin 1) |
| NT-proBNP (N-terminal prohormone of BNP) |
| OAS-RNase L (oligoadenylate-ribonuclease L) |
| PAMP (pathogen-associated molecular pattern) |
| PET (positron emission tomography) |
| PKA (protein kinase A) |
| PKR (protein kinase R) |
| POTS (postural orthostatic tachycardia syndrome) |
| PRRs (pattern recognition receptors) |
| RAAS (renin-angiotensin-aldosterone system) |
| ROS (reactive oxygen species) |
| S (surface filtration area) |
| S1P (sphingosine-1-phosphate) |
| SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) |
| SGLT-2i (sodium-glucose cotransporter 2 inhibitors) |
| SPARC (Secreted Protein Acidic and Rich in Cysteine) |
| SOD-1 (superoxide dismutase 1) |
| Tiam1 (T-lymphoma invasion and metastasis inducing protein 1) |
| TF (tissue factor) |
| TIMPs (tissue inhibitors of metalloproteinases) |
| TLR (Toll-like receptor) |
| TNF-α (tumor necrosis factor-α) |
| TMPRSS2 (transmembrane protease serine 2) |
| TJ (tight junctions) |
| Trio (Trio Rho guanine nucleotide exchange factor) |
| Vav2 (guanine nucleotide exchange factor 2) |
| VA-ECMO (veno-arterial extra-corporal membrane oxygenation) |
| VE-cadherin (vascular endothelial-cadherin) |
| VEGF (vascular endothelial growth factor) |
| vWF (von Willebrand factor) |
| ZO (zonula occludens) |
| α-klotho (alpha-klotho) |
| Δ P (difference between capillary and interstitial hydrostatic pressure) |
| Δ Π (difference between osmotic pressure across the membrane derived from plasma proteins) |
| σ (protein reflection coefficient) |
| ΠC (intracapillary colloid osmotic pressure) |
| ΠG (subglycocalyx colloid osmotic pressure) |
| PC (intracapillary hydrostatic pressure) |
| PI (interstitial hydrostatic pressure) |
References
- Vasques-Nóvoa, F.; Angélico-Gonçalves, A.; Alvarenga, J.M.G.; Nobrega, J.; Cerqueira, R.J.; Mancio, J.; Leite-Moreira, A.F.; Roncon-Albuquerque, R. Myocardial oedema: Pathophysiological basis and implications for the failing heart. ESC Heart Fail. 2022, 9, 958–976. [Google Scholar] [CrossRef] [PubMed]
- Mehlhorn, U.; Geissler, H.J.; Laine, G.A.; Allen, S.J. Myocardial fluid balance. Eur. J. Cardio-Thorac. Surg. 2001, 20, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Laine, G.A.; Allen, S.J. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ. Res. 1991, 68, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.V.; Laine, G.A.; Stewart, R.H.; Cox, C.S.; Quick, C.M.; Allen, S.J.; Fischer, U.M. Mechanics of the left ventricular myocardial interstitium: Effects of acute and chronic myocardial edema. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2428–H2434. [Google Scholar] [CrossRef]
- Rubboli, A.; Sobotka, P.A.; Euler, D.E. Effect of acute edema on left ventricular function and coronary vascular resistance in the isolated rat heart. Am. J. Physiol.-Heart Circ. Physiol. 1994, 267, H1054–H1061. [Google Scholar] [CrossRef]
- Miyamoto, M.; McClure, D.E.; Schertel, E.R.; Andrews, P.J.; Jones, G.A.; Pratt, J.W.; Ross, P.; Myerowitz, P.D. Effects of hypoproteinemia-induced myocardial edema on left ventricular function. Am. J. Physiol. 1998, 274, H937–H944. [Google Scholar] [CrossRef] [PubMed]
- Karolle, B.L.; Carlson, R.E.; Aisen, A.M.; Buda, A.J. Transmural distribution of myocardial edema by NMR relaxometry following myocardial ischemia and reperfusion. Am. Heart J. 1991, 122, 655–664. [Google Scholar] [CrossRef]
- Röttgen, R.; Christiani, R.; Freyhardt, P.; Gutberlet, M.; Schultheiss, H.P.; Hamm, B.; Kühl, U. Magnetic resonance imaging findings in acute myocarditis and correlation with immunohistological parameters. Eur. Radiol. 2011, 21, 1259–1266. [Google Scholar] [CrossRef]
- Friedrich, M.G.; Strohm, O.; Schulz-Menger, J.; Marciniak, H.; Luft, F.C.; Dietz, R. Contrast media-enhanced magnetic resonance imaging visualizes myocardial changes in the course of viral myocarditis. Circulation 1998, 97, 1802–1809. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Andres-Villarreal, M.; Ruiz-Meana, M.; Inserte, J.; Barba, I. Myocardial edema: A translational view. J. Mol. Cell. Cardiol. 2012, 52, 931–939. [Google Scholar] [CrossRef]
- Duncker, D.J.; Bache, R.J. Regulation of coronary blood flow during exercise. Physiol. Rev. 2008, 88, 1009–1086. [Google Scholar] [CrossRef] [PubMed]
- Bassenge, E.; Heusch, G. Endothelial and neuro-humoral control of coronary blood flow in health and disease. Rev. Physiol. Biochem. Pharmacol. 1990, 116, 77–165. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, W.H.; Goresky, C.A. Transcapillary exchange in the working left ventricle of the dog. Circ. Res. 1971, 29, 181–207. [Google Scholar] [CrossRef]
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Schafer, A.E.; Blaxall, B.C. Extracellular matrix-mediated cellular communication in the heart. J. Mol. Cell. Cardiol. 2016, 91, 228. [Google Scholar] [CrossRef]
- Al-Kofahi, M.; Omura, S.; Tsunoda, I.; Sato, F.; Becker, F.; Gavins, F.N.E.; Woolard, M.D.; Pattillo, C.; Zawieja, D.; Muthuchamy, M.; et al. IL-1β reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed. Pharmacother. 2018, 107, 1591–1600. [Google Scholar] [CrossRef]
- Abassi, Z.; Khoury, E.E.; Karram, T.; Aronson, D. Edema formation in congestive heart failure and the underlying mechanisms. Front. Cardiovasc. Med. 2022, 9, 933215. [Google Scholar] [CrossRef]
- Davis, K.L.; Laine, G.A.; Geissler, H.J.; Mehlhorn, U.; Brennan, M.; Allen, S.J. Effects of myocardial edema on the development of myocardial interstitial fibrosis. Microcirculation 2000, 7, 269–280. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.J.; Schalkwijk, C.G.; Bronzwaer, J.G.F.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Van Heerebeek, L.; Borbély, A.; Niessen, H.W.M.; Bronzwaer, J.G.F.; Van Der Velden, J.; Stienen, G.J.M.; Linke, W.A.; Laarman, G.J.; Paulus, W.J. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation 2006, 113, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Patterson, E.K.; Slessarev, M.; Gill, S.E.; Martin, C.; Daley, M.; Miller, M.R.; Patel, M.A.; dos Santos, C.C.; Bosma, K.J.; et al. Endothelial Injury and Glycocalyx Degradation in Critically Ill Coronavirus Disease 2019 Patients: Implications for Microvascular Platelet Aggregation. Crit. Care Explor. 2020, 2, e0194. [Google Scholar] [CrossRef] [PubMed]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Paz, L.; Capodanno, D.; Montalescot, G.; Angiolillo, D.J. Coronavirus Disease 2019-Associated Thrombosis and Coagulopathy: Review of the Pathophysiological Characteristics and Implications for Antithrombotic Management. J. Am. Heart Assoc. 2021, 10, e019650. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Starling, E.H. On the Absorption of Fluids from the Connective Tissue Spaces. J. Physiol. 1896, 19, 312–326. [Google Scholar] [CrossRef]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef]
- Staverman, A.J. The theory of measurement of osmotic pressure. Recl. Des Trav. Chim. Des Pays-Bas 1951, 70, 344–352. [Google Scholar] [CrossRef]
- Michel, C.C. Fluid movements through capillary walls. In Handbook of Physiology. The Cardiovascular System Microcirculation, Section 2; American Physiology Society: Bethesda, MD, USA, 1984; Volume 4, pp. 375–409. [Google Scholar]
- Chilian, W.M.; Eastham, C.L.; Layne, S.M.; Marcus, M.L. Small vessel phenomena in the coronary microcirculation: Phasic intramyocardial perfusion and coronary microvascular dynamics. Prog. Cardiovasc. Dis. 1988, 31, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Bassingthwaighte, J.B. A concurrent flow model for extraction during transcapillary passage. Circ. Res. 1974, 35, 483–503. [Google Scholar] [CrossRef] [PubMed]
- Chilian, W.M. Microvascular pressures and resistances in the left ventricular subepicardium and subendocardium. Circ. Res. 1991, 69, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Koyama, T.; Kikuchi, Y.; Kakiuchi, Y.; Nagashima, C. An analysis of water movement between myocardial tissue and capillary blood during reactive hyperemia. Jpn. J. Physiol. 1979, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Laine, G.A.; Granger, H.J. Microvascular, interstitial, and lymphatic interactions in normal heart. Am. J. Physiol. 1985, 249, H834–H842. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Marzilli, M.; Sabbah, H.N.; Lee, T. Systolic and diastolic pressure gradients within the left ventricular wall. Am. J. Physiol. 1980, 238, H625–H630. [Google Scholar] [CrossRef]
- Pilati, C.F. Macromolecular transport in canine coronary microvasculature. Am. J. Physiol. 1990, 258, H748–H753. [Google Scholar] [CrossRef]
- Mehlhorn, U.; Davis, K.L.; Laine, G.A.; Geissler, H.J.; Allen, S.J. Myocardial fluid balance in acute hypertension. Microcirculation 1996, 3, 371–378. [Google Scholar] [CrossRef]
- Mehlhorn, U.; Allen, S.J.; Davis, K.L.; Geissler, H.J.; Warters, R.D.; Rainer de Vivie, E. Increasing the colloid osmotic pressure of cardiopulmonary bypass prime and normothermic blood cardioplegia minimizes myocardial oedema and prevents cardiac dysfunction. Cardiovasc. Surg. 1998, 6, 274–281. [Google Scholar] [CrossRef]
- Navar, P.D.; Navar, L.G. Relationship between colloid osmotic pressure and plasma protein concentration in the dog. Am. J. Physiol. 1977, 233, H295–H298. [Google Scholar] [CrossRef]
- Curry, F.E.; Michel, C.C. The Colloid Osmotic Pressure Across the Glycocalyx: Role of Interstitial Fluid Sub-Compartments in Trans-Vascular Fluid Exchange in Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 729873. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, P.S.; Levick, J.R. Chronic peripheral oedema: The critical role of the lymphatic system. Clin. Med. 2004, 4, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.; Egbrink, M.G.A.O. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Vink, H.; Duling, B.R. Capillary endothelial surface layer selectively reduces plasma solute distribution volume. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H285–H289. [Google Scholar] [CrossRef]
- Ori, A.; Wilkinson, M.C.; Fernig, D.G. A Systems Biology Approach for the Investigation of the Heparin/Heparan Sulfate Interactome. J. Biol. Chem. 2011, 286, 19892. [Google Scholar] [CrossRef]
- Wiig, H.; Swartz, M.A. Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef] [PubMed]
- Wearn, J.T. The extent of the capillary bed of the heart. J. Exp. Med. 1928, 47, 273–290. [Google Scholar] [CrossRef]
- Laine, G.A. Microvascular changes in the heart during chronic arterial hypertension. Circ. Res. 1988, 62, 953–960. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Bertrand, P.B.; Willems, E.; Gielen, E.; Mullens, W.; Giri, S.; Tang, W.H.W.; Raman, S.V.; Verhaert, D. Global myocardial oedema in advanced decompensated heart failure. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 787–794. [Google Scholar] [CrossRef]
- Takegawa, R.; Kabata, D.; Shimizu, K.; Hisano, S.; Ogura, H.; Shintani, A.; Shimazu, T. Serum albumin as a risk factor for death in patients with prolonged sepsis: An observational study. J. Crit. Care 2019, 51, 139–144. [Google Scholar] [CrossRef]
- Boyd, J.H.; Forbes, J.; Nakada, T.A.; Walley, K.R.; Russell, J.A. Fluid resuscitation in septic shock: A positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit. Care Med. 2011, 39, 259–265. [Google Scholar] [CrossRef]
- Wiig, H.; Rubin, K.; Reed, R.K. New and active role of the interstitium in control of interstitial fluid pressure: Potential therapeutic consequences. Acta Anaesthesiol. Scand. 2003, 47, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M.; Chew, M.; Nneji, G.; Neal, C.; Barry, J.; Michel, C. Quasi-periodic substructure in the microvessel endothelial glycocalyx: A possible explanation for molecular filtering? J. Struct. Biol. 2001, 136, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Rostgaard, J.; Qvortrup, K. Electron microscopic demonstrations of filamentous molecular sieve plugs in capillary fenestrae. Microvasc. Res. 1997, 53, 1–13. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflug. Arch. 2000, 440, 653–666. [Google Scholar] [CrossRef]
- Lipowsky, H.H. Microvascular rheology and hemodynamics. Microcirculation 2005, 12, 5–15. [Google Scholar] [CrossRef]
- Vink, H.; Constantinescu, A.A.; Spaan, J.A.E. Oxidized lipoproteins degrade the endothelial surface layer: Implications for platelet-endothelial cell adhesion. Circulation 2000, 101, 1500–1502. [Google Scholar] [CrossRef]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef]
- Busch, B.; Ljungman, C.; Heldin, C.M.; Waskson, E.; Öbrink, B. Surface properties of cultured endothelial cells. Haemostasis 1979, 8, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Damiano, E.R. The effect of the endothelial-cell glycocalyx on the motion of red blood cells through capillaries. Microvasc. Res. 1998, 55, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Adamson, R.H.; Huxley, V.H.; Curry, F.E. Single capillary permeability to proteins having similar size but different charge. Am. J. Physiol. 1988, 254, H304–H312. [Google Scholar] [CrossRef]
- Henry, C.B.S.; Duling, B.R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am. J. Physiol. 1999, 277, H508–H514. [Google Scholar] [CrossRef] [PubMed]
- Adamson, R.H. Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J. Physiol. 1990, 428, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.R.; Blumenstock, F.A.; Cooper, J.A.; Malik, A.B. Role of albumin arginyl sites in albumin-induced reduction of endothelial hydraulic conductivity. J. Cell. Physiol. 1989, 141, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, E.E.; Hamelin, M. Interaction of serum proteins with lung endothelial glycocalyx: Its effect on endothelial permeability. Am. J. Physiol. 1984, 247, H206–H217. [Google Scholar] [CrossRef]
- Turner, M.R.; Clough, G.; Michel, C.C. The effects of cationised ferritin and native ferritin upon the filtration coefficient of single frog capillaries. Evidence that proteins in the endothelial cell coat influence permeability. Microvasc. Res. 1983, 25, 205–222. [Google Scholar] [CrossRef]
- Levick, J.R.; Michel, C.C. The effect of bovine albumin on the permeability of frog mesenteric capillaries. Q. J. Exp. Physiol. Cogn. Med. Sci. 1973, 58, 87–97. [Google Scholar] [CrossRef]
- Lopez-Quintero, S.V.; Amaya, R.; Pahakis, M.; Tarbell, J.M. The endothelial glycocalyx mediates shear-induced changes in hydraulic conductivity. Am. J. Physiol. Circ. Physiol. 2009, 296, H1451–H1456. [Google Scholar] [CrossRef]
- Jacob, M.; Rehm, M.; Loetsch, M.; Paul, J.O.; Bruegger, D.; Welsch, U.; Conzen, P.; Becker, B.F. The endothelial glycocalyx prefers albumin for evoking shear stress-induced, nitric oxide-mediated coronary dilatation. J. Vasc. Res. 2007, 44, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Bolli, R.; Qiu, Y.; Tang, X.L.; Murphree, S.S.; French, B.A. Gene therapy with extracellular superoxide dismutase attenuates myocardial stunning in conscious rabbits. Circulation 1998, 98, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Kobayashi, M.; Kimura, S.; Nishinaga, M.; Takeuchi, K.; Ozawa, T.; Shimada, K.; Kobayashi, M.; Kimura, S.; Nishinaga, M.; et al. Anticoagulant heparin-like glycosaminoglycans on endothelial cell surface. Jpn. Circ. J. 1991, 55, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Tovar, A.M.F.; De Mattos, D.A.; Stelling, M.P.; Sarcinelli-Luz, B.S.L.; Nazareth, R.A.; Mourão, P.A.S. Dermatan sulfate is the predominant antithrombotic glycosaminoglycan in vessel walls: Implications for a possible physiological function of heparin cofactor II. Biochim. Biophys. Acta 2005, 1740, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.K.; Cepinskas, G.; Fraser, D.D. Endothelial Glycocalyx Degradation in Critical Illness and Injury. Front. Med. 2022, 9, 898592. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, B.M.; Vink, H.; Spaan, J.A.E. The Endothelial Glycocalyx Protects Against Myocardial Edema. Circ. Res. 2003, 92, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Bruegger, D.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology 2006, 104, 1223–1231. [Google Scholar] [CrossRef]
- Malik, A.B.; Siflinger-Birnboim, A. Vascular Endothelial Barrier Function and Its Regulation. In Biological Barriers to Protein Delivery; Springer: Boston, MA, USA, 1993; pp. 231–267. [Google Scholar] [CrossRef]
- Mann, G.E. Alterations of myocardial capillary permeability by albumin in the isolated, perfused rabbit heart. J. Physiol. 1981, 319, 311–323. [Google Scholar] [CrossRef]
- Wiesinger, A.; Peters, W.; Chappell, D.; Kentrup, D.; Reuter, S.; Pavenstädt, H.; Oberleithner, H.; Kümpers, P. Nanomechanics of the Endothelial Glycocalyx in Experimental Sepsis. PLoS ONE 2013, 8, e80905. [Google Scholar] [CrossRef]
- Rienks, M.; Carai, P.; van Teeffelen, J.; Eskens, B.; Verhesen, W.; Hemmeryckx, B.; Johnson, D.M.; van Leeuwen, R.; Jones, E.A.; Heymans, S.; et al. SPARC preserves endothelial glycocalyx integrity, and protects against adverse cardiac inflammation and injury during viral myocarditis. Matrix Biol. 2018, 74, 21–34. [Google Scholar] [CrossRef]
- Epstein, F.H.; Parrillo, J.E. Pathogenetic Mechanisms of Septic Shock. N. Engl. J. Med. 1993, 328, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.H.C.; Alonso, S.; Ng, L.F.P.; Thein, T.L.; Pang, V.J.X.; Leo, Y.S.; Lye, D.C.B.; Yeo, T.W. Increased Serum Hyaluronic Acid and Heparan Sulfate in Dengue Fever: Association with Plasma Leakage and Disease Severity. Sci. Rep. 2017, 7, srep46191. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [PubMed]
- Bansch, P.; Nelson, A.; Ohlsson, T.; Bentzer, P. Effect of charge on microvascular permeability in early experimental sepsis in the rat. Microvasc. Res. 2011, 82, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.B.S.; Duling, B.R. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2815–H2823. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Tedder, T.F. Leukocyte interactions with vascular endothelium. New insights into selectin-mediated attachment and rolling. J. Immunol. 1995, 155, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.W.; Stone, P.; Shasby, D.M. Cationic neutrophil proteins increase transendothelial albumin movement. J. Appl. Physiol. 1987, 62, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Hoover, R.L.; Briggs, R.T.; Karnovsky, M.J. The adhesive interaction between polymorphonuclear leukocytes and endothelial cells in vitro. Cell 1978, 14, 423–428. [Google Scholar] [CrossRef]
- Morita, Y.; Clemens, M.G.; Miller, L.S.; Rangan, U.; Kondo, S.; Miyasaka, M.; Yoshikawa, T.; Bulkley, G.B. Reactive oxidants mediate TNF-alpha-induced leukocyte adhesion to rat mesenteric venular endothelium. Am. J. Physiol. 1995, 269, H1833–H1842. [Google Scholar] [CrossRef]
- Carden, D.L.; Smith, J.K.; Korthuis, R.J. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ. Res. 1990, 66, 1436–1444. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222, e12860. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.J.; Donnelly, J.L. Hypoxia induced disruption of the cardiac endothelial glycocalyx: Implications for capillary permeability. Cardiovasc. Res. 1993, 27, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A.E. Elevated capillary tube hematocrit reflects degradation of endothelial cell glycocalyx by oxidized LDL. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1051–H1057. [Google Scholar] [CrossRef] [PubMed]
- Gardner, G.; Banka, C.L.; Roberts, K.A.; Mullick, A.E.; Rutledge, J.C. Modified LDL–Mediated Increases in Endothelial Layer Permeability Are Attenuated with 17β-Estradiol. Arter. Thromb. Vasc. Biol. 1999, 19, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, S.; Penn, M.S.; Saidel, G.M.; Chisolm, G.M. Exogenous Oxidized Low-Density Lipoprotein Injures and Alters the Barrier Function of Endothelium in Rats In Vivo. Circ. Res. 1997, 80, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lehr, H.A.; Frei, B.; Olofsson, A.M.; Carew, T.E.; Arfors, K.E. Protection From Oxidized LDL–Induced Leukocyte Adhesion to Microvascular and Macrovascular Endothelium In Vivo by Vitamin C but Not by Vitamin E. Circulation 1995, 91, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Harris, N.R.; Granger, D.N. Oxidized low-density lipoproteins and microvascular responses to ischemia-reperfusion. Am. J. Physiol. Circ. Physiol. 1996, 271, H2508–H2514. [Google Scholar] [CrossRef]
- Lehr, H.A.; Becker, M.; Marklund, S.L.; Hübner, C.; Arfors, K.E.; Kohlschütter, A.; Messmer, K. Superoxide-dependent stimulation of leukocyte adhesion by oxidatively modified LDL in vivo. Arter. Thromb. A J. Vasc. Biol. 1992, 12, 824–829. [Google Scholar] [CrossRef]
- Beręsewicz, A.; Czarnowska, E.; Mączewski, M. Ischemic preconditioning and superoxide dismutase protect against endothelial dysfunction and endothelium glycocalyx disruption in the postischemic guinea-pig hearts. Myocard. Ischemia Reper. 1998, 87–97. [Google Scholar] [CrossRef]
- Mulivor, A.W.; Lipowsky, H.H. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1672–H1680. [Google Scholar] [CrossRef]
- Kurzelewski, M.; Czarnowska, E.; Beresewicz, A. Superoxide- and nitric oxide-derived species mediate endothelial dysfunction, endothelial glycocalyx disruption, and enhanced neutrophil adhesion in the post-ischemic guinea-pig heart. J. Physiol. Pharmacol. 2005, 56, 163–178. [Google Scholar]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Poledniczek, M.; Neumayer, C.; Kopp, C.W.; Schlager, O.; Gremmel, T.; Jozkowicz, A.; Gschwandtner, M.E.; Koppensteiner, R.; Wadowski, P.P. Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches. Biomedicines 2023, 11, 2284. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Niccoli, G.; De Maria, G.; Brugaletta, S.; Montone, R.A.; Vergallo, R.; Benenati, S.; Magniani, G.; D’Amario, D.; Porto, I.; et al. Coronary microvascular obstruction and dysfunction in patients with acute myocardial infarction. Nat. Rev. Cardiol. 2023, 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Pedersen, S.H.; Jensen, J.S.; Mogelvang, R.; Johansson, P.I. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit. Care 2013, 17, R32. [Google Scholar] [CrossRef]
- Algoet, M.; Janssens, S.; Himmelreich, U.; Gsell, W.; Pusovnik, M.; Van den Eynde, J.; Oosterlinck, W. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med. 2023, 33, 357–366. [Google Scholar] [CrossRef]
- Pellegrini, D.; Kawakami, R.; Guagliumi, G.; Sakamoto, A.; Kawai, K.; Gianatti, A.; Nasr, A.; Kutys, R.; Guo, L.; Cornelissen, A.; et al. Microthrombi as a Major Cause of Cardiac Injury in COVID-19: A Pathologic Study. Circulation 2021, 143, 1031–1042. [Google Scholar] [CrossRef]
- Reffelmann, T.; Kloner, R.A. The no-reflow phenomenon: A basic mechanism of myocardial ischemia and reperfusion. Basic Res. Cardiol. 2006, 101, 359–372. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Bekkers, S.C.A.M.; Yazdani, S.K.; Virmani, R.; Waltenberger, J. Microvascular Obstruction: Underlying Pathophysiology and Clinical Diagnosis. J. Am. Coll. Cardiol. 2010, 55, 1649–1660. [Google Scholar] [CrossRef]
- Crea, F.; Montone, R.A.; Rinaldi, R. Pathophysiology of Coronary Microvascular Dysfunction. Circ. J. 2022, 86, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Panzer, B.; Kopp, C.W.; Neumayer, C.; Koppensteiner, R.; Jozkowicz, A.; Poledniczek, M.; Gremmel, T.; Jilma, B.; Wadowski, P.P. Toll-like Receptors as Pro-Thrombotic Drivers in Viral Infections: A Narrative Review. Cells 2023, 12, 1865. [Google Scholar] [CrossRef] [PubMed]
- Guagliumi, G.; Sonzogni, A.; Pescetelli, I.; Pellegrini, D.; Finn, A.V. Microthrombi and ST-Segment-Elevation Myocardial Infarction in COVID-19. Circulation 2020, 142, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Montone, R.A.; Iannaccone, G.; Meucci, M.C.; Gurgoglione, F.; Niccoli, G. Myocardial and Microvascular Injury Due to Coronavirus Disease 2019. Eur. Cardiol. 2020, 15, e52. [Google Scholar] [CrossRef]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Polverini, P.J. The pathophysiology of angiogenesis. Crit. Rev. Oral Biol. Med. 1995, 6, 230–247. [Google Scholar] [CrossRef]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. Biomed Res. Int. 2015, 2015, 549412. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Fingleton, B.; Rothenberg, M.L.; Matrisian, L.M. Matrix metalloproteinases: Biologic activity and clinical implications. J. Clin. Oncol. 2000, 18, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, Y.; Miller, A.; Shapiro, S.; Lahat, N. Hypoxia of endothelial cells leads to MMP-2-dependent survival and death. Am. J. Physiol. Cell Physiol. 2005, 289, C1321–C1331. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Singh, P.; Shami, A.; Kluza, E.; Pan, M.; Djordjevic, D.; Michaelsen, N.B.; Kennbäck, C.; van der Wel, N.N.; Orho-Melander, M.; et al. Spatial Transcriptional Mapping Reveals Site-Specific Pathways Underlying Human Atherosclerotic Plaque Rupture. J. Am. Coll. Cardiol. 2023, 81, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Ando, S.I.; Imaizumi, T.; Harada, S.; Hirooka, Y.; Takeshita, A. Atrial natriuretic peptide increases human capillary filtration and venous distensibility. J. Hypertens. 1992, 10, 451–457. [Google Scholar] [CrossRef]
- Huxley, V.H.; Tucker, V.L.; Verburg, K.M.; Freeman, R.H. Increased capillary hydraulic conductivity induced by atrial natriuretic peptide. Circ. Res. 1987, 60, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Bruegger, D.; Jacob, M.; Rehm, M.; Loetsch, M.; Welsch, U.; Conzen, P.; Becker, B.F. Atrial natriuretic peptide induces shedding of endothelial glycocalyx in coronary vascular bed of guinea pig hearts. Am. J. Physiol. Circ. Physiol. 2005, 289, H1993–H1999. [Google Scholar] [CrossRef]
- Jacob, M.; Saller, T.; Chappell, D.; Rehm, M.; Welsch, U.; Becker, B.F. Physiological levels of A-, B- and C-type natriuretic peptide shed the endothelial glycocalyx and enhance vascular permeability. Basic Res. Cardiol. 2013, 108, 347. [Google Scholar] [CrossRef]
- Oberleithner, H.; Peters, W.; Kusche-Vihrog, K.; Korte, S.; Schillers, H.; Kliche, K.; Oberleithner, K. Salt overload damages the glycocalyx sodium barrier of vascular endothelium. Pflug. Arch. 2011, 462, 519–528. [Google Scholar] [CrossRef]
- Selcuk, M.; Keskin, M.; Cinar, T.; Gunay, N.; Dogan, S.; Cicek, V.; Kilic, S.; Asal, S.; Yavuz, S.; Keser, N.; et al. Prognostic significance of N-Terminal Pro-BNP in patients with COVID-19 pneumonia without previous history of heart failure. Eur. Heart J. 2021, 42 (Suppl. S1), ehab724.0866. [Google Scholar] [CrossRef]
- Huelsmann, M.; Neuhold, S.; Resl, M.; Strunk, G.; Brath, H.; Francesconi, C.; Adlbrecht, C.; Prager, R.; Luger, A.; Pacher, R.; et al. PONTIAC (NT-proBNP Selected PreventiOn of cardiac eveNts in a populaTion of dIabetic patients without A history of Cardiac disease): A Prospective Randomized Controlled Trial. J. Am. Coll. Cardiol. 2013, 62, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Torisawa, T.; Oiwa, K.; Tsukita, S. AMPK-dependent phosphorylation of cingulin reversibly regulates its binding to actin filaments and microtubules. Sci. Rep. 2018, 8, 15550. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M. Molecular basis of the core structure of tight junctions. Cold Spring Harb. Perspect. Biol. 2010, 2, a002907. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Paschoud, S.; Pulimeno, P.; Foglia, A.; Citi, S. The cytoplasmic plaque of tight junctions: A scaffolding and signalling center. Biochim. Biophys. Acta 2008, 1778, 601–613. [Google Scholar] [CrossRef]
- Schossleitner, K.; Rauscher, S.; Gröger, M.; Friedl, H.P.; Finsterwalder, R.; Habertheuer, A.; Sibilia, M.; Brostjan, C.; Födinger, D.; Citi, S.; et al. Evidence That Cingulin Regulates Endothelial Barrier Function In Vitro and In Vivo. Arter. Thromb. Vasc. Biol. 2016, 36, 647–654. [Google Scholar] [CrossRef]
- Dejana, E.; Vestweber, D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 2013, 116, 119–144. [Google Scholar] [CrossRef]
- Bravi, L.; Dejana, E.; Lampugnani, M.G. VE-cadherin at a glance. Cell Tissue Res. 2014, 355, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J. Endothelial permeability and VE-cadherin: A wacky comradeship. Cell Adhes. Migr. 2014, 8, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Waschke, J.; Curry, F.E.; Adamson, R.H.; Drenckhahn, D. Regulation of actin dynamics is critical for endothelial barrier functions. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1296–H1305. [Google Scholar] [CrossRef]
- Stevens, T.; Garcia, J.G.N.; Shasby, D.M.; Bhattacharya, J.; Malik, A.B. Mechanisms regulating endothelial cell barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L419–L422. [Google Scholar] [CrossRef] [PubMed]
- Prasain, N.; Stevens, T. The actin cytoskeleton in endothelial cell phenotypes. Microvasc. Res. 2009, 77, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G.N. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Shasby, D.M.; Shasby, S.S.; Sullivan, J.M.; Peach, M.J. Role of endothelial cell cytoskeleton in control of endothelial permeability. Circ. Res. 1982, 51, 657–661. [Google Scholar] [CrossRef]
- García-Ponce, A.; Citalán-Madrid, A.F.; Velázquez-Avila, M.; Vargas-Robles, H.; Schnoor, M. The role of actin-binding proteins in the control of endothelial barrier integrity. Thromb. Haemost. 2015, 113, 20–36. [Google Scholar] [CrossRef]
- Wang, L.; Dudek, S.M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc. Res. 2009, 77, 39–45. [Google Scholar] [CrossRef]
- Xu, M.; Waters, C.L.; Hu, C.; Wysolmerski, R.B.; Vincent, P.A.; Minnear, F.L. Sphingosine 1-phosphate rapidly increases endothelial barrier function independently of VE-cadherin but requires cell spreading and Rho kinase. Am. J. Physiol. Cell Physiol. 2007, 293, C1309–C1318. [Google Scholar] [CrossRef]
- Wilkerson, B.A.; Argraves, K.M. The role of sphingosine-1-phosphate in endothelial barrier function. Biochim. Biophys. Acta 2014, 1841, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Ghosh, C.C.; Mukherjee, A.; Parikh, S.M. Angiopoietin-1 requires IQ domain GTPase-activating protein 1 to activate Rac1 and promote endothelial barrier defense. Arter. Thromb. Vasc. Biol. 2011, 31, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Dierkes, M.; Küppers, V.; Vockel, M.; Tomm, J.; Zeuschner, D.; Rossaint, J.; Zarbock, A.; Koh, G.Y.; Peters, K.; et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 2015, 212, 2267–2287. [Google Scholar] [CrossRef]
- Zeng, Y.; Adamson, R.H.; Curry, F.R.E.; Tarbell, J.M. Sphingosine-1-phosphate protects endothelial glycocalyx by inhibiting syndecan-1 shedding. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H363–H372. [Google Scholar] [CrossRef]
- Schlegel, N.; Waschke, J. cAMP with other signaling cues converges on Rac1 to stabilize the endothelial barrier- a signaling pathway compromised in inflammation. Cell Tissue Res. 2014, 355, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Lampugnani, M.G.; Zanetti, A.; Breviario, F.; Balconi, G.; Orsenigo, F.; Corada, M.; Spagnuolo, R.; Betson, M.; Braga, V.; Dejana, E. VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol. Biol. Cell 2002, 13, 1175–1189. [Google Scholar] [CrossRef]
- Kopperud, R.K.; Rygh, C.B.; Karlsen, T.V.; Krakstad, C.; Kleppe, R.; Hoivik, E.A.; Bakke, M.; Tenstad, O.; Selheim, F.; Lidén; et al. Increased microvascular permeability in mice lacking Epac1 (Rapgef3). Acta Physiol. 2017, 219, 441–452. [Google Scholar] [CrossRef]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef]
- Michel, C.C.; Curry, F.E. Microvascular permeability. Physiol. Rev. 1999, 79, 703–761. [Google Scholar] [CrossRef]
- Curry, F.R.E.; Adamson, R.H. Tonic regulation of vascular permeability. Acta Physiol. 2013, 207, 628–649. [Google Scholar] [CrossRef]
- Schlegel, N.; Baumer, Y.; Drenckhahn, D.; Waschke, J. Lipopolysaccharide-induced endothelial barrier breakdown is cyclic adenosine monophosphate dependent in vivo and in vitro. Crit. Care Med. 2009, 37, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, N.; Waschke, J. Impaired cAMP and Rac 1 signaling contribute to TNF-alpha-induced endothelial barrier breakdown in microvascular endothelium. Microcirculation 2009, 16, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Spindler, V.; Werthmann, R.C.; Bünemann, M.; Waschke, J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J. Cell. Physiol. 2009, 220, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Tanislav, C.; Troidl, C.; Schulz, R.; Hamm, C.; Gündüz, D. cAMP controls the restoration of endothelial barrier function after thrombin-induced hyperpermeability via Rac1 activation. Physiol. Rep. 2014, 2, e12175. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Orsenigo, F. Endothelial adherens junctions at a glance. J. Cell Sci. 2013, 126 Pt. 12, 2545–2549. [Google Scholar] [CrossRef]
- Küppers, V.; Vockel, M.; Nottebaum, A.F.; Vestweber, D. Phosphatases and kinases as regulators of the endothelial barrier function. Cell Tissue Res. 2014, 355, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Schlegel, N.; Waschke, J. Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 2010, 87, 243–253. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vasc. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1208. [Google Scholar] [CrossRef]
- Garatti, L.; Wu, M.A.; Ammirati, E.; Sacco, A. Systemic leak capillary syndrome with myocardial involvement and cardiogenic shock: A case report. Eur. Heart J.-Case Reports 2022, 6, ytac262. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, S.; Wertheim, H.; Simmons, C.P.; Screaton, G.; Wills, B. Cardiovascular manifestations of the emerging dengue pandemic. Nat. Rev. Cardiol. 2014, 11, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Gomes de Sousa, F.T.; Tjang, L.V.; Pahmeier, F.; Zhu, C.; Ruan, R.; Blanc, S.F.; Patel, T.S.; Worthington, C.M.; Glasner, D.R.; et al. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-β signaling. Nat. Commun. 2022, 13, 7630. [Google Scholar] [CrossRef] [PubMed]
- Dongaonkar, R.M.; Stewart, R.H.; Quick, C.M.; Uray, K.L.; Cox, C.S.; Laine, G.A. Award article: Microcirculatory Society Award for Excellence in Lymphatic Research: Time course of myocardial interstitial edema resolution and associated left ventricular dysfunction. Microcirculation 2012, 19, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.W.; Schertel, E.R.; Schaefer, S.L.; Esham, K.E.; McClure, D.E.; Heck, C.F.; Myerowitz, P.D. Acute transient coronary sinus hypertension impairs left ventricular function and induces myocardial edema. Am. J. Physiol. 1996, 271, H834–H841. [Google Scholar] [CrossRef] [PubMed]
- Dongaonkar, R.M.; Stewart, R.H.; Geissler, H.J.; Laine, G.A. Myocardial microvascular permeability, interstitial oedema, and compromised cardiac function. Cardiovasc. Res. 2010, 87, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Taherzadeh, M.; Das, A.K.; Warren, J.B. Nifedipine increases microvascular permeability via a direct local effect on postcapillary venules. Am. J. Physiol. 1998, 275, H1388–H1394. [Google Scholar] [CrossRef]
- Monmeneu, J.V.; Bodí, V.; Sanchis, J.; López-Lereu, M.P.; Mainar, L.; Núñez, J.; Chaustre, F.; Rumiz, E.; Chorro, F.J.; Llácer, Á. Cardiac magnetic resonance evaluation of edema after ST-elevation acute myocardial infarction. Rev. Esp. Cardiol. 2009, 62, 858–866. [Google Scholar] [CrossRef]
- Fehrmann, A.; Treutlein, M.; Rudolph, T.; Rudolph, V.; Weiss, K.; Giese, D.; Bunck, A.C.; Maintz, D.; Baeßler, B. Myocardial T1 and T2 mapping in severe aortic stenosis: Potential novel insights into the pathophysiology of myocardial remodelling. Eur. J. Radiol. 2018, 107, 76–83. [Google Scholar] [CrossRef]
- Alabed, S.; Saunders, L.; Garg, P.; Shahin, Y.; Alandejani, F.; Rolf, A.; Puntmann, V.O.; Nagel, E.; Wild, J.M.; Kiely, D.G.; et al. Myocardial T1-mapping and extracellular volume in pulmonary arterial hypertension: A systematic review and meta-analysis. Magn. Reson. Imaging 2021, 79, 66–75. [Google Scholar] [CrossRef]
- Spieker, M.; Haberkorn, S.; Gastl, M.; Behm, P.; Katsianos, S.; Horn, P.; Jacoby, C.; Schnackenburg, B.; Reinecke, P.; Kelm, M.; et al. Abnormal T2 mapping cardiovascular magnetic resonance correlates with adverse clinical outcome in patients with suspected acute myocarditis. J. Cardiovasc. Magn. Reson. 2017, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Gräni, C.; Eichhorn, C.; Bière, L.; Murthy, V.L.; Agarwal, V.; Kaneko, K.; Cuddy, S.; Aghayev, A.; Steigner, M.; Blankstein, R.; et al. Prognostic Value of Cardiac Magnetic Resonance Tissue Characterization in Risk Stratifying Patients with Suspected Myocarditis. J. Am. Coll. Cardiol. 2017, 70, 1964. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, L.W.; Tillisch, J.H. Maintenance of cardiac output with normal filling pressures in patients with dilated heart failure. Circulation 1986, 74, 1303–1308. [Google Scholar] [CrossRef]
- Laine, G.A.; Allen, S.J.; Katz, J.; Gabel, J.C.; Drake, R.E. Effect of systemic venous pressure elevation on lymph flow and lung edema formation. J. Appl. Physiol. 1986, 61, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.D.; Ramanathan, K.B.; McGee, J.E.; Newman, K.P.; Weber, K.T. Oxidative stress and cardiomyocyte necrosis with elevated serum troponins: Pathophysiologic mechanisms. Am. J. Med. Sci. 2011, 342, 129–134. [Google Scholar] [CrossRef]
- Colombo, P.C.; Onat, D.; Harxhi, A.; Demmer, R.T.; Hayashi, Y.; Jelic, S.; Lejemtel, T.H.; Bucciarelli, L.; Kebschull, M.; Papapanou, P.; et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur. Heart J. 2014, 35, 448–454. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Libby, P. The Heart in COVID-19: Primary Target or Secondary Bystander? JACC Basic Transl. Sci. 2020, 5, 537–542. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arter. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.F.; Islam, N.; Alyonycheva, T.N.; Safier, L.B.; Hajjar, K.A.; Posnett, D.N.; Schoenborn, M.A.; Schooley, K.A.; et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J. Clin. Investig. 1997, 99, 1351–1360. [Google Scholar] [CrossRef]
- Roy, C.; Tabiasco, J.; Caillon, A.; Delneste, Y.; Merot, J.; Favre, J.; Guihot, A.L.; Martin, L.; Nascimento, D.C.; Ryffel, B.; et al. Loss of vascular expression of nucleoside triphosphate diphosphohydrolase-1/CD39 in hypertension. Purinergic Signal. 2018, 14, 73–82. [Google Scholar] [CrossRef]
- Robson, S.C.; Kaczmarek, E.; Siegel, J.B.; Candinas, D.; Koziak, K.; Millan, M.; Hancock, W.W.; Bach, F.H. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J. Exp. Med. 1997, 185, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.H.; Kuo, M.L.; Chen, C.A.; Chou, C.H.; Lai, K.B.; Lee, C.N.; Hsieh, C.Y. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 2003, 22, 1517–1527. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, T.; Iguchi, K.; Kajiwara, S.; Kato, M.; Kanda, H.; Hirokawa, Y.; Arima, K.; Mizokami, A.; Sugimura, Y. Interleukin-6 induces VEGF secretion from prostate cancer cells in a manner independent of androgen receptor activation. Prostate 2018, 78, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Soga, N.; Namba, N.; McAllister, S.; Cornelius, L.; Teitelbaum, S.L.; Dowdy, S.F.; Kawamura, J.; Hruska, K.A. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp. Cell Res. 2001, 269, 73–87. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Busse, R.; Fleming, I. Vascular endothelium and blood flow. Handb. Exp. Pharmacol. 2006, 176, 43–78. [Google Scholar]
- Moncada, S.; Higgs, E.A. Nitric oxide and the vascular endothelium. Handb. Exp. Pharmacol. 2006, 176, 213–254. [Google Scholar]
- Mcdonald, D.M.; Thurston, G.; Baluk, P. Endothelial Gaps as Sites for Plasma Leakage in Inflammation. Microcirculation 1999, 6, 7–22. [Google Scholar] [CrossRef]
- Siao, C.J.; Lorentz, C.U.; Kermani, P.; Marinic, T.; Carter, J.; McGrath, K.; Padow, V.A.; Mark, W.; Falcone, D.J.; Leona, C.G.; et al. ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J. Exp. Med. 2012, 209, 2291–2305. [Google Scholar] [CrossRef]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, C.; Chen, Z.; Bankaitis, V.; Tzima, E.; Sheibani, N.; Burridge, K. Pericytes Regulate Vascular Basement Membrane Remodeling and Govern Neutrophil Extravasation during Inflammation. PLoS ONE 2012, 7, e45499. [Google Scholar] [CrossRef]
- Díaz-Flores, L.; Gutiérrez, R.; Madrid, J.F.; Varela, H.; Valladares, F.; Acosta, E.; Martín-Vasallo, P.; Díaz-Flores, J. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol. Histopathol. 2009, 24, 909–969. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, C.; Hällgren, R.; Elvin, A.; Gerdin, B.; Tufveson, G. Hyaluronidase ameliorates rejection-induced edema. Transpl. Int. 1999, 12, 235–243. [Google Scholar] [CrossRef]
- Jackson, D.G. Leucocyte Trafficking via the Lymphatic Vasculature- Mechanisms and Consequences. Front. Immunol. 2019, 10, 471. [Google Scholar] [CrossRef]
- Schwager, S.; Detmar, M. Inflammation and Lymphatic Function. Front. Immunol. 2019, 10, 308. [Google Scholar] [CrossRef]
- Pfister, F.; Feng, Y.; Vom Hagen, F.; Hoffmann, S.; Molema, G.; Hillebrands, J.L.; Shani, M.; Deutsch, U.; Hammes, H.P. Pericyte MigrationA Novel Mechanism of Pericyte Loss in Experimental Diabetic Retinopathy. Diabetes 2008, 57, 2495–2502. [Google Scholar] [CrossRef]
- Curry, F.R.E. Atrial natriuretic peptide: An essential physiological regulator of transvascular fluid, protein transport, and plasma volume. J. Clin. Investig. 2005, 115, 1458–1461. [Google Scholar] [CrossRef]
- Chappell, D.; Bruegger, D.; Potzel, J.; Jacob, M.; Brettner, F.; Vogeser, M.; Conzen, P.; Becker, B.F.; Rehm, M. Hypervolemia increases release of atrial natriuretic peptide and shedding of the endothelial glycocalyx. Crit. Care 2014, 18, 538. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.M.; Schulz-Menger, J.; Holmvang, G.; Kramer, C.M.; Carbone, I.; Sechtem, U.; Kindermann, I.; Gutberlet, M.; Cooper, L.T.; Liu, P.; et al. Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J. Am. Coll. Cardiol. 2018, 72, 3158–3176. [Google Scholar] [CrossRef] [PubMed]
- Iles, L.M.; Ellims, A.H.; Llewellyn, H.; Hare, J.L.; Kaye, D.M.; McLean, C.A.; Taylor, A.J. Histological validation of cardiac magnetic resonance analysis of regional and diffuse interstitial myocardial fibrosis. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 14–22. [Google Scholar] [CrossRef]
- Palmisano, A.; Benedetti, G.; Faletti, R.; Rancoita, P.M.V.; Gatti, M.; Peretto, G.; Sala, S.; Boccia, E.; Francone, M.; Galea, N.; et al. Early T1 Myocardial MRI Mapping: Value in Detecting Myocardial Hyperemia in Acute Myocarditis. Radiology 2020, 295, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Beijnink, C.W.H.; van der Hoeven, N.W.; Konijnenberg, L.S.F.; Kim, R.J.; Bekkers, S.C.A.M.; Kloner, R.A.; Everaars, H.; El Messaoudi, S.; van Rossum, A.C.; van Royen, N.; et al. Cardiac MRI to Visualize Myocardial Damage after ST-Segment Elevation Myocardial Infarction: A Review of Its Histologic Validation. Radiology 2021, 301, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Masci, P.G.; Pavon, A.G.; Muller, O.; Iglesias, J.F.; Vincenti, G.; Monney, P.; Harbaoui, B.; Eeckhout, E.; Schwitter, J. Relationship between CMR-derived parameters of ischemia/reperfusion injury and the timing of CMR after reperfused ST-segment elevation myocardial infarction. J. Cardiovasc. Magn. Reson. 2018, 20, 50. [Google Scholar] [CrossRef]
- Templin, C.; Manka, R.; Cammann, V.L.; Szawan, K.A.; Gotschy, A.; Karolyi, M.; Winnik, S.; Moch, H.; Meyer, P.; Moreo, A.; et al. Takotsubo Syndrome in Coronavirus Disease 2019. Am. J. Cardiol. 2021, 138, 118. [Google Scholar] [CrossRef]
- Tsao, C.W.; Strom, J.B.; Chang, J.D.; Manning, W.J. COVID-19 Associated Stress (Takotsubo) Cardiomyopathy. Circ. Cardiovasc. Imaging 2020, 13, e011222. [Google Scholar] [CrossRef]
- Plácido, R.; Cunha Lopes, B.; Almeida, A.G.; Rochitte, C.E. The role of cardiovascular magnetic resonance in takotsubo syndrome. J. Cardiovasc. Magn. Reson. 2016, 18, 68. [Google Scholar] [CrossRef]
- Gutberlet, M.; Spors, B.; Thoma, T.; Bertram, H.; Denecke, T.; Felix, R.; Noutsias, M.; Schultheiss, H.-P.; Kühl, U. Suspected chronic myocarditis at cardiac MR: Diagnostic accuracy and association with immunohistologically detected inflammation and viral persistence. Radiology 2008, 246, 401–409. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, B.T.; Yuan, W.F.; Zhao, C.X. Frontiers of COVID-19-related myocarditis as assessed by cardiovascular magnetic resonance. World J. Clin. Cases 2022, 10, 6784. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.P.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 2636–2648. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Piechota-Polańczyk, A.; Andreas, M.; Kopp, C.W. Cardiovascular Disease Management in the Context of Global Crisis. Int. J. Environ. Res. Public Health 2022, 20, 689. [Google Scholar] [CrossRef] [PubMed]
- Siripanthong, B.; Asatryan, B.; Hanff, T.C.; Chatha, S.R.; Khanji, M.Y.; Ricci, F.; Muser, D.; Ferrari, V.A.; Nazarian, S.; Santangeli, P.; et al. The Pathogenesis and Long-Term Consequences of COVID-19 Cardiac Injury. JACC Basic Transl. Sci. 2022, 7, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Z. Bench-to-bedside: Innovation of small molecule anti-SARS-CoV-2 drugs in China. Eur. J. Med. Chem. 2023, 257, 115503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Li, Y.H.; Wang, L.L.; Liu, H.Q.; Lu, S.Y.; Liu, Y.; Li, K.; Liu, B.; Li, S.Y.; Shao, F.M.; et al. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 414. [Google Scholar] [CrossRef]
- Xie, Y.; Yin, W.; Zhang, Y.; Shang, W.; Wang, Z.; Luan, X.; Tian, G.; Aisa, H.A.; Xu, Y.; Xiao, G.; et al. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res. 2021, 31, 1212–1214. [Google Scholar] [CrossRef]
- Cheema, H.A.; Rehman, A.U.; Elrashedy, A.A.; Mohsin, A.; Shahid, A.; Ehsan, M.; Ayyan, M.; Ismail, H.; Almas, T. Antiandrogens for the treatment of COVID-19 patients: A meta-analysis of randomized controlled trials. J. Med. Virol. 2023, 95, e28740. [Google Scholar] [CrossRef]
- Shang, W.; Dai, W.; Yao, C.; Xu, L.; Tao, X.; Su, H.; Li, J.; Xie, X.; Xu, Y.; Hu, M.; et al. In vitro and in vivo evaluation of the main protease inhibitor FB2001 against SARS-CoV-2. Antiviral Res. 2022, 208, 105450. [Google Scholar] [CrossRef]
- Hou, N.; Shuai, L.; Zhang, L.; Xie, X.; Tang, K.; Zhu, Y.; Yu, Y.; Zhang, W.; Tan, Q.; Zhong, G.; et al. Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLpro. ACS Cent. Sci. 2022, 9, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.C.; Mao, Y.W.; Hu, J.W.; Hsieh, H.Y.; Shih, T.M.; Lu, L.P.; Chang, W.H.; Huang, C.H.; Lin, C.H.; Lin, C.H.; et al. Development of Ultrapure and Potent Tannic Acids asa Pan-coronal Antiviral Therapeutic. ACS Pharmacol. Transl. Sci. 2022, 5, 400. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Watashi, K.; Saso, W.; Shionoya, K.; Iwanami, S.; Hirokawa, T.; Shirai, T.; Kanaya, S.; Ito, Y.; Kim, K.S.; et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience 2021, 24, 102367. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; AlOmar, S.Y.; Almuqri, E.A.; Rudayni, H.A.; Kumar, V. Genomics-guided identification of potential modulators of SARS-CoV-2 entry proteases, TMPRSS2 and Cathepsins B/L. PLoS ONE 2021, 16, e0256141. [Google Scholar] [CrossRef]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Central Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Bermejo-Jambrina, M.; Eder, J.; Kaptein, T.M.; van Hamme, J.L.; Helgers, L.C.; Vlaming, K.E.; Brouwer, P.J.M.; van Nuenen, A.C.; Spaargaren, M.; de Bree, G.J.; et al. Infection and transmission of SARS-CoV-2 depend on heparan sulfate proteoglycans. EMBO J. 2021, 40, e106765. [Google Scholar] [CrossRef]
- Dai, J.; Wang, Y.; Wang, H.; Gao, Z.; Wang, Y.; Fang, M.; Shi, S.; Zhang, P.; Wang, H.; Su, Y.; et al. Toll-Like Receptor Signaling in Severe Acute Respiratory Syndrome Coronavirus 2-Induced Innate Immune Responses and the Potential Application Value of Toll-Like Receptor Immunomodulators in Patients with Coronavirus Disease 2019. Front. Microbiol. 2022, 13, 948770. [Google Scholar] [CrossRef]
- Capone, C.; Faraco, G.; Park, L.; Cao, X.; Davisson, R.L.; Iadecola, C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Kobayashi, M.; Li, Q.; Yamanishi, S.; Katsumura, K.; Minami, M.; Wu, D.M.; Puro, D.G. Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J. Physiol. 2004, 561, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Tilton, R.G.; Kilo, C.; Williamson, J.R.; Murch, D.W. Differences in pericyte contractile function in rat cardiac and skeletal muscle microvasculatures. Microvasc. Res. 1979, 18, 336–352. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-Coronavirus Activating Proteases TMPRSS2 and HAT Are Expressed at Multiple Sites in Human Respiratory and Gastrointestinal Tracts. PLoS ONE 2012, 7, e35876. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, A.; Kawakami, R.; Kawai, K.; Gianatti, A.; Pellegrini, D.; Kutys, R.; Guo, L.; Mori, M.; Cornelissen, A.; Sato, Y.; et al. ACE2 (Angiotensin-Converting Enzyme 2) and TMPRSS2 (Transmembrane Serine Protease 2) Expression and Localization of SARS-CoV-2 Infection in the Human Heart. Arter. Thromb. Vasc. Biol. 2021, 41, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Gkogkou, E.; Barnasas, G.; Vougas, K.; Trougakos, I.P. Expression profiling meta-analysis of ACE2 and TMPRSS2, the putative anti-inflammatory receptor and priming protease of SARS-CoV-2 in human cells, and identification of putative modulators. Redox Biol. 2020, 36, 101615. [Google Scholar] [CrossRef]
- Batah, S.S.; Fabro, A.T. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir. Med. 2021, 176, 106239. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Tavazzi, G.; Pellegrini, C.; Maurelli, M.; Belliato, M.; Sciutti, F.; Bottazzi, A.; Sepe, P.A.; Resasco, T.; Camporotondo, R.; Bruno, R.; et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur. J. Heart Fail. 2020, 22, 911–915. [Google Scholar] [CrossRef]
- Nakamura, Y.; Katano, H.; Nakajima, N.; Sato, Y.; Suzuki, T.; Sekizuka, T.; Kuroda, M.; Izutani, Y.; Morimoto, S.; Maruyama, J.; et al. SARS-CoV-2 is localized in cardiomyocytes: A postmortem biopsy case. Int. J. Infect. Dis. 2021, 111, 43. [Google Scholar] [CrossRef]
- Wong, S.H.; Lui, R.N.S.; Sung, J.J.Y. Covid-19 and the digestive system. J. Gastroenterol. Hepatol. 2020, 35, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Nijst, P.; Kiefer, K.; Tang, W.H.W. Endothelial Glycocalyx as Biomarker for Cardiovascular Diseases: Mechanistic and Clinical Implications. Curr. Heart Fail. Rep. 2017, 14, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Hülsmann, M.; Schörgenhofer, C.; Lang, I.M.; Wurm, R.; Gremmel, T.; Koppensteiner, R.; Steinlechner, B.; Schwameis, M.; Jilma, B. Sublingual functional capillary rarefaction in chronic heart failure. Eur. J. Clin. Investig. 2018, 48, e12869. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Steinlechner, B.; Zimpfer, D.; Schlöglhofer, T.; Schima, H.; Hülsmann, M.; Lang, I.M.; Gremmel, T.; Koppensteiner, R.; Zehetmayer, S.; et al. Functional capillary impairment in patients with ventricular assist devices. Sci. Rep. 2019, 9, 5909. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Schörgenhofer, C.; Rieder, T.; Ertl, S.; Pultar, J.; Serles, W.; Sycha, T.; Mayer, F.; Koppensteiner, R.; Gremmel, T.; et al. Microvascular rarefaction in patients with cerebrovascular events. Microvasc. Res. 2022, 140, 104300. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, W.; Liang, X.; Shi, L.; Zhang, P.; Wang, Y.; Wang, Y.; Yang, H. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health 2021, 21, 1533. [Google Scholar] [CrossRef]
- Gerotziafas, G.T.; Catalano, M.; Colgan, M.P.; Pecsvarady, Z.; Wautrecht, J.C.; Fazeli, B.; Olinic, D.M.; Farkas, K.; Elalamy, I.; Falanga, A.; et al. Guidance for the Management of Patients with Vascular Disease or Cardiovascular Risk Factors and COVID-19: Position Paper from VAS-European Independent Foundation in Angiology/Vascular Medicine. Thromb. Haemost. 2020, 120, 1597–1628. [Google Scholar] [CrossRef]
- Ronco, C.; Reis, T.; Husain-Syed, F. Management of acute kidney injury in patients with COVID-19. Lancet Respir. Med. 2020, 8, 738–742. [Google Scholar] [CrossRef]
- Ullah, W.; Saeed, R.; Sarwar, U.; Patel, R.; Fischman, D.L. COVID-19 Complicated by Acute Pulmonary Embolism and Right-Sided Heart Failure. JACC Case Rep. 2020, 2, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and viral sepsis: Observations and hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, P.; Khanji, M.Y.; Cooper Jr, L.T.; Chahal, C.A.A. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm. 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Burashnikov, A. Overview of Basic Mechanisms of Cardiac Arrhythmia. Card. Electrophysiol. Clin. 2011, 3, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Avolio, E.; Madeddu, P. Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vasc. Pharmacol. 2016, 86, 53–63. [Google Scholar] [CrossRef]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 413767. [Google Scholar] [CrossRef]
- Bontekoe, J.; Lee, J.; Bansal, V.; Syed, M.; Hoppensteadt, D.; Maia, P.; Walborn, A.; Liles, J.; Brailovsky, E.; Fareed, J. Biomarker Profiling in Stage 5 Chronic Kidney Disease Identifies the Relationship between Angiopoietin-2 and Atrial Fibrillation. Clin. Appl. Thromb. 2018, 24, 269S–276S. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Ng, C.Y.; Zhang, Z.; Liu, T.; Li, G. Association of Plasma Transforming Growth Factor-β1 Levels and the Risk of Atrial Fibrillation: A Meta-Analysis. PLoS ONE 2016, 11, e0155275. [Google Scholar] [CrossRef]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef]
- Chang, S.-H.; Yeh, Y.-H.; Lee, J.-L.; Hsu, Y.-J.; Kuo, C.-T.; Chen, W.-J. Transforming growth factor-β-mediated CD44/STAT3 signaling contributes to the development of atrial fibrosis and fibrillation. Basic Res. Cardiol. 2017, 112, 58. [Google Scholar] [CrossRef]
- Qiao, G.; Xia, D.; Cheng, Z.; Zhang, G. miR-132 in atrial fibrillation directly targets connective tissue growth factor. Mol. Med. Rep. 2017, 16, 4143–4150. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.L.; Chintalgattu, V. Pericytes in the Heart. Adv. Exp. Med. Biol. 2019, 1122, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yi, X.; Ma, L.; Zhou, Y. Hepatocyte growth factor and basic fibroblast growth factor regulate atrial fibrosis in patients with atrial fibrillation and rheumatic heart disease via the mitogen-activated protein kinase signaling pathway. Exp. Ther. Med. 2013, 6, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Freestone, B.; Chong, A.Y.; Lim, H.S.; Blann, A.; Lip, G.Y.H. Angiogenic factors in atrial fibrillation: A possible role in thrombogenesis? Ann. Med. 2005, 37, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Doubt, T.J.; Hogan, P.M. Effects of hydrostatic pressure on conduction and excitability in rabbit atria. J. Appl. Physiol. 1978, 45, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xue, Y.M.; Guo, H.M.; Deng, C.Y.; Peng, D.W.; Yang, H.; Wei, W.; Liu, Y.; Liu, F.Z.; Wang, Z.Y.; et al. High hydrostatic pressure induces atrial electrical remodeling through upregulation of inflammatory cytokines. Life Sci. 2020, 242, 117209. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, C.Y.; Xue, Y.M.; Yang, H.; Wei, W.; Liu, F.Z.; Guo, H.M.; Liu, Y.; Wang, Z.Y.; Zhang, M.Z.; et al. High hydrostatic pressure induces atrial electrical remodeling through angiotensin upregulation mediating FAK/Src pathway activation. J. Mol. Cell. Cardiol. 2020, 140, 10–21. [Google Scholar] [CrossRef]
- Granger, D.N.; Rodrigues, S.F.; Yildirim, A.; Senchenkova, E.Y. Microvascular Responses to Cardiovascular Risk Factors. Microcirculation 2010, 17, 192–205. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Jilma, B.; Kopp, C.W.; Ertl, S.; Gremmel, T.; Koppensteiner, R. Glycocalyx as Possible Limiting Factor in COVID-19. Front. Immunol. 2021, 12, 607306. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634. [Google Scholar] [CrossRef]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-γ-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Šoltés, L.; Mendichi, R.; Kogan, G.; Schiller, J.; Stankovská, M.; Arnhold, J. Degradative action of reactive oxygen species on hyaluronan. Biomacromolecules 2006, 7, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Maciej-Hulme, M.L. New Insights Into Human Hyaluronidase 4/Chondroitin Sulphate Hydrolase. Front. Cell Dev. Biol. 2021, 9, 767924. [Google Scholar] [CrossRef]
- Matzner, Y.; Bar-Ner, M.; Yahalom, J.; Ishai-Michaeli, R.; Fuks, Z.; Vlodavsky, I. Degradation of heparan sulfate in the subendothelial extracellular matrix by a readily released heparanase from human neutrophils. Possible role in invasion through basement membranes. J. Clin. Investig. 1985, 76, 1306. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D. The Weibel-Palade body: The storage granule for von Willebrand factor and P-selectin. Thromb. Haemost. 1993, 70, 105–110. [Google Scholar] [CrossRef]
- Gupta, R.M.; Libby, P.; Barton, M. Linking regulation of nitric oxide to endothelin-1: The Yin and Yang of vascular tone in the atherosclerotic plaque. Atherosclerosis 2020, 292, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.; Havervall, S.; Rosell, A.; Aguilera, K.; Parv, K.; Von Meijenfeldt, F.A.; Lisman, T.; Mackman, N.; Thålin, C.; Phillipson, M. Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients with COVID-19. Arter. Thromb. Vasc. Biol. 2021, 41, 988–994. [Google Scholar] [CrossRef]
- Hidalgo, A. A NET-thrombosis axis in COVID-19. Blood 2020, 136, 1118–1119. [Google Scholar] [CrossRef]
- Gluckman, T.J.; Bhave, N.M.; Allen, L.A.; Chung, E.H.; Spatz, E.S.; Ammirati, E.; Baggish, A.L.; Bozkurt, B.; Cornwell, W.K.; Harmon, K.G.; et al. 2022 ACC Expert Consensus Decision Pathway on Cardiovascular Sequelae of COVID-19 in Adults: Myocarditis and Other Myocardial Involvement, Post-Acute Sequelae of SARS-CoV-2 Infection, and Return to Play: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2022, 79, 1717. [Google Scholar] [CrossRef]
- Hanneman, K.; Houbois, C.; Schoffel, A.; Gustafson, D.; Iwanochko, R.M.; Wintersperger, B.J.; Chan, R.; Fish, J.E.; Howe, K.L.; Thavendiranathan, P. Combined Cardiac Fluorodeoxyglucose-Positron Emission Tomography/Magnetic Resonance Imaging Assessment of Myocardial Injury in Patients Who Recently Recovered From COVID-19. JAMA Cardiol. 2022, 7, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Barmada, A.; Klein, J.; Ramaswamy, A.; Brodsky, N.N.; Jaycox, J.R.; Sheikha, H.; Jones, K.M.; Habet, V.; Campbell, M.; Sumida, T.S.; et al. Cytokinopathy with aberrant cytotoxic lymphocytes and profibrotic myeloid response in SARS-CoV-2 mRNA vaccine–associated myocarditis. Sci. Immunol. 2023, 8, eadh3455. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, T.K.; Kompaniyets, L.; Lavery, A.M.; Hsu, J.; Ko, J.Y.; Yusuf, H.; Romano, S.D.; Gundlapalli, A.V.; Oster, M.E.; Harris, A.M. Association Between COVID-19 and Myocarditis Using Hospital-Based Administrative Data-United States, March 2020-January 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1228–1232. [Google Scholar] [CrossRef]
- Alvarez-Garcia, J.; Jaladanki, S.; Rivas-Lasarte, M.; Cagliostro, M.; Gupta, A.; Joshi, A.; Ting, P.; Mitter, S.S.; Bagiella, E.; Mancini, D.; et al. New Heart Failure Diagnoses Among Patients Hospitalized for COVID-19. J. Am. Coll. Cardiol. 2021, 77, 2260–2262. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Yang, Z.; Tang, D.; Huang, L.; Xia, L. Tissue Characterization by Mapping and Strain Cardiac MRI to Evaluate Myocardial Inflammation in Fulminant Myocarditis. J. Magn. Reson. Imaging 2020, 52, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Hinojar, R.; Varma, N.; Child, N.; Goodman, B.; Jabbour, A.; Yu, C.Y.; Gebker, R.; Doltra, A.; Kelle, S.; Khan, S.; et al. T1 Mapping in Discrimination of Hypertrophic Phenotypes: Hypertensive Heart Disease and Hypertrophic Cardiomyopathy: Findings From the International T1 Multicenter Cardiovascular Magnetic Resonance Study. Circ. Cardiovasc. Imaging 2015, 8, e003285. [Google Scholar] [CrossRef]
- Wang, H.; Li, R.; Zhou, Z.; Jiang, H.; Yan, Z.; Tao, X.; Li, H.; Xu, L. Cardiac involvement in COVID-19 patients: Mid-term follow up by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2021, 23, 14. [Google Scholar] [CrossRef]
- Rinaldi, R.; Basile, M.; Salzillo, C.; Grieco, D.L.; Caffè, A.; Masciocchi, C.; Lilli, L.; Damiani, A.; La Vecchia, G.; Iannaccone, G.; et al. Myocardial Injury Portends a Higher Risk of Mortality and Long-Term Cardiovascular Sequelae after Hospital Discharge in COVID-19 Survivors. J. Clin. Med. 2022, 11, 5964. [Google Scholar] [CrossRef]
- Liu, T.; Wu, D.; Yan, W.; Wang, X.; Zhang, X.; Ma, K.; Chen, H.; Zeng, Z.; Qin, Y.; Wang, H.; et al. Twelve-Month Systemic Consequences of Coronavirus Disease 2019 (COVID-19) in Patients Discharged From Hospital: A Prospective Cohort Study in Wuhan, China. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2022, 74, 1953. [Google Scholar] [CrossRef]
- Seecheran, R.; Narayansingh, R.; Giddings, S.; Rampaul, M.; Furlonge, K.; Abdool, K.; Bhagwandass, N.; Seecheran, N.A. Atrial Arrhythmias in a Patient Presenting with Coronavirus Disease-2019 (COVID-19) Infection. J. Investig. Med. High Impact Case Rep. 2020, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef] [PubMed]
- Inciardi, R.M.; Adamo, M.; Lupi, L.; Cani, D.S.; Di Pasquale, M.; Tomasoni, D.; Italia, L.; Zaccone, G.; Tedino, C.; Fabbricatore, D.; et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur. Heart J. 2020, 41, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Colon, C.M.; Barrios, J.G.; Chiles, J.W.; McElwee, S.K.; Russell, D.W.; Maddox, W.R.; Kay, G.N. Atrial Arrhythmias in COVID-19 Patients. JACC Clin. Electrophysiol. 2020, 6, 1189–1190. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A Myocardial Nox2 Containing NAD(P)H Oxidase Contributes to Oxidative Stress in Human Atrial Fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Liu, Z.; Zhu, Y.; Lu, J.; Ge, C.; Zhang, C.; Li, N.; Jin, N.; Li, Y.; Tian, M.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2022, 12, 780768. [Google Scholar] [CrossRef]
- Muszyński, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation—Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Wang, K.; Viveiros, A.; Kellner, M.J.; Penninger, J.M. Angiotensin-converting enzyme 2—At the heart of the COVID-19 pandemic. Cell 2023, 186, 906–922. [Google Scholar] [CrossRef]
- Jakovac, H.; Ferenčić, A.; Stemberger, C.; Mohar Vitezić, B.; Cuculić, D. Detection of SARS-CoV-2 Antigens in the AV-Node of a Cardiac Conduction System-A Case Report. Trop. Med. Infect. Dis. 2022, 7, 43. [Google Scholar] [CrossRef]
- Ferreira, A.J.; Moraes, P.L.; Foureaux, G.; Andrade, A.B.; Santos, R.A.S.; Almeida, A.P. The angiotensin-(1-7)/Mas receptor axis is expressed in sinoatrial node cells of rats. J. Histochem. Cytochem. 2011, 59, 761–768. [Google Scholar] [CrossRef]
- Nádasy, G.L.; Balla, A.; Cojocaru, E.; Cojocaru, C.; Vlad, C.-E.; Eva, L. Role of the Renin-Angiotensin System in Long COVID’s Cardiovascular Injuries. Biomed 2023, 11, 2004. [Google Scholar] [CrossRef]
- Khazaal, S.; Harb, J.; Rima, M.; Annweiler, C.; Wu, Y.; Cao, Z.; Khattar, Z.A.; Legros, C.; Kovacic, H.; Fajloun, Z.; et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules 2022, 27, 2903. [Google Scholar] [CrossRef]
- Carpenter, R.M.; Young, M.K.; Petri, W.A.O.; Lyons, G.R.; Gilchrist, C.; Carey, R.M.; Petri, W.A. Repressed Ang 1-7 in COVID-19 Is Inversely Associated with Inflammation and Coagulation. mSphere 2022, 7, e0022022. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Kiat, H.; McLachlan, C.S. Atrial fibrillation in COVID-19: A review of possible mechanisms. FASEB J. 2020, 34, 11347–11354. [Google Scholar] [CrossRef] [PubMed]
- Disertori, M.; Rigoni, M.; Pace, N.; Casolo, G.; Masè, M.; Gonzini, L.; Lucci, D.; Nollo, G.; Ravelli, F. Myocardial Fibrosis Assessment by LGE Is a Powerful Predictor of Ventricular Tachyarrhythmias in Ischemic and Nonischemic LV Dysfunction: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 9, 1046–1055. [Google Scholar] [CrossRef]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J.; Iba, T.; Levy, J.H. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020, 7, e438–e440. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Panzer, B.; Józkowicz, A.; Kopp, C.W.; Gremmel, T.; Panzer, S.; Koppensteiner, R. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. Int. J. Mol. Sci. 2023, 24, 2492. [Google Scholar] [CrossRef]
- Mehta, P.; Porter, J.C.; Manson, J.J.; Isaacs, J.D.; Openshaw, P.J.M.; McInnes, I.B.; Summers, C.; Chambers, R.C. Therapeutic blockade of granulocyte macrophage colony-stimulating factor in COVID-19-associated hyperinflammation: Challenges and opportunities. Lancet Respir. Med. 2020, 8, 822–830. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Cohen, P.L. Imperfect storm: Is interleukin-33 the Achilles heel of COVID-19? Lancet Rheumatol. 2020, 2, e779–e790. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, J.; Liu, C.; Su, L.; Zhang, D.; Fan, J.; Yang, Y.; Xiao, M.; Xie, J.; Xu, Y.; et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol. Med. 2020, 26, 97. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.S.; Shu, T.; Kang, L.; Wu, D.; Zhou, X.; Liao, B.W.; Sun, X.L.; Zhou, X.; Wang, Y.Y. Temporal profiling of plasma cytokines, chemokines and growth factors from mild, severe and fatal COVID-19 patients. Signal Transduct. Target. Ther. 2020, 5, 100. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Benest, A.V.; Kruse, K.; Savant, S.; Thomas, M.; Laib, A.M.; Loos, E.K.; Fiedler, U.; Augustin, H.G. Angiopoietin-2 is critical for cytokine-induced vascular leakage. PLoS ONE 2013, 8, e70459. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Sivakumar, P.; Gupta, S.; Sarkar, S.; Sen, S. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol. Cell. Biochem. 2008, 307, 159–167. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Van Almen, G.C.; Van Aelst, L.N.L.; Van Cleemput, J.; Droogné, W.; Jin, Y.; Van De Werf, F.; Carmeliet, P.; Vanhaecke, J.; Papageorgiou, A.P.; et al. Matricellular proteins and matrix metalloproteinases mark the inflammatory and fibrotic response in human cardiac allograft rejection. Eur. Heart J. 2013, 34, 1930–1941. [Google Scholar] [CrossRef]
- Vanhoutte, D.; Schellings, M.W.M.; Götte, M.; Swinnen, M.; Herias, V.; Wild, M.K.; Vestweber, D.; Chorianopoulos, E.; Cortés, V.; Rigotti, A.; et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation 2007, 115, 475–482. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Chen, Y.; Ma, J.; Yang, Y.; Aodeng, S.; Cui, Q.; Wen, K.; Xiao, M.; Xie, J.; et al. Syndecan-1, an indicator of endothelial glycocalyx degradation, predicts outcome of patients admitted to an ICU with COVID-19. Mol. Med. 2021, 27, 151. [Google Scholar] [CrossRef] [PubMed]
- Rovas, A.; Osiaevi, I.; Buscher, K.; Sackarnd, J.; Tepasse, P.R.; Fobker, M.; Kühn, J.; Braune, S.; Göbel, U.; Thölking, G.; et al. Microvascular dysfunction in COVID-19: The MYSTIC study. Angiogenesis 2021, 24, 145. [Google Scholar] [CrossRef] [PubMed]
- Goonewardena, S.N.; Grushko, O.G.; Wells, J.; Herty, L.; Rosenson, R.S.; Haus, J.M.; Hummel, S.L. Immune-Mediated Glycocalyx Remodeling in Hospitalized COVID-19 Patients. Cardiovasc. Drugs Ther. 2023, 37, 307. [Google Scholar] [CrossRef] [PubMed]
- Queisser, K.A.; Mellema, R.A.; Middleton, E.A.; Portier, I.; Manne, B.K.; Denorme, F.; Beswick, E.J.; Rondina, M.T.; Campbell, R.A.; Petrey, A.C. COVID-19 generates hyaluronan fragments that directly induce endothelial barrier dysfunction. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Buijsers, B.; Yanginlar, C.; de Nooijer, A.; Grondman, I.; Maciej-Hulme, M.L.; Jonkman, I.; Janssen, N.A.F.; Rother, N.; de Graaf, M.; Pickkers, P.; et al. Increased Plasma Heparanase Activity in COVID-19 Patients. Front. Immunol. 2020, 11, 575047. [Google Scholar] [CrossRef] [PubMed]
- Roshdy, A.; Zaher, S.; Fayed, H.; Coghlan, J.G. COVID-19 and the Heart: A Systematic Review of Cardiac Autopsies. Front. Cardiovasc. Med. 2021, 7, 626975. [Google Scholar] [CrossRef]
- Godoy, L.C.; Goligher, E.C.; Lawler, P.R.; Slutsky, A.S.; Zarychanski, R. Anticipating and managing coagulopathy and thrombotic manifestations of severe COVID-19. CMAJ 2020, 192, E1156–E1161. [Google Scholar] [CrossRef]
- Denorme, F.; Vanhoorelbeke, K.; De Meyer, S.F. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Front. Immunol. 2019, 10, 495067. [Google Scholar] [CrossRef]
- Kalagara, T.; Moutsis, T.; Yang, Y.; Pappelbaum, K.I.; Farken, A.; Cladder-Micus, L.; Vidal-Y-Sy, S.; John, A.; Bauer, A.T.; Moerschbacher, B.M.; et al. The endothelial glycocalyx anchors von Willebrand factor fibers to the vascular endothelium. Blood Adv. 2018, 2, 2347–2357. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Aiyegbusi, O.L.; Hughes, S.E.; Turner, G.; Rivera, S.C.; McMullan, C.; Chandan, J.S.; Haroon, S.; Price, G.; Davies, E.H.; Nirantharakumar, K.; et al. Symptoms, complications and management of long COVID: A review. J. R. Soc. Med. 2021, 114, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Martin, S.; Shchendrygina, A.; Hoffmann, J.; Ka, M.M.; Giokoglu, E.; Vanchin, B.; Holm, N.; Karyou, A.; Laux, G.S.; et al. Long-term cardiac pathology in individuals with mild initial COVID-19 illness. Nat. Med. 2022, 28, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, L. SARS CoV-2 related microvascular damage and symptoms during and after COVID-19: Consequences of capillary transit-time changes, tissue hypoxia and inflammation. Physiol. Rep. 2021, 9, e14726. [Google Scholar] [CrossRef]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G.; Cappuccinelli, P.; Fitzgerald, G.; Bacci, M.L.; et al. Long Covid: Where we stand and challenges ahead. Cell Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef]
- Nunn, A.V.W.; Guy, G.W.; Brysch, W.; Bell, J.D. Understanding Long COVID; Mitochondrial Health and Adaptation—Old Pathways, New Problems. Biomedicines 2022, 10, 3113. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Leone, O.; Pieroni, M.; Rapezzi, C.; Olivotto, I. The spectrum of myocarditis: From pathology to the clinics. Virchows Arch. 2019, 475, 279–301. [Google Scholar] [CrossRef]
- Andréoletti, L.; Lévêque, N.; Boulagnon, C.; Brasselet, C.; Fornes, P. Viral causes of human myocarditis. Arch. Cardiovasc. Dis. 2009, 102, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Kühl, U.; Pauschinger, M.; Seeberg, B.; Lassner, D.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Viral Persistence in the Myocardium Is Associated with Progressive Cardiac Dysfunction. Circulation 2005, 112, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef] [PubMed]
- Jeserich, M.; Brunner, E.; Kandolf, R.; Olschewski, M.; Kimmel, S.; Friedrich, M.G.; Föll, D.; Bode, C.; Geibel, A. Diagnosis of viral myocarditis by cardiac magnetic resonance and viral genome detection in peripheral blood. Int. J. Cardiovasc. Imaging 2013, 29, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.-P.; Baumeier, C.; Aleshcheva, G.; Bock, C.-T.; Escher, F. Viral Myocarditis—From Pathophysiology to Treatment. J. Clin. Med. 2021, 10, 5240. [Google Scholar] [CrossRef]
- Verdonschot, J.; Hazebroek, M.; Merken, J.; Debing, Y.; Dennert, R.; Brunner-La Rocca, H.P.; Heymans, S. Relevance of cardiac parvovirus B19 in myocarditis and dilated cardiomyopathy: Review of the literature. Eur. J. Heart Fail. 2016, 18, 1430–1441. [Google Scholar] [CrossRef]
- Escher, F.; Kühl, U.; Gross, U.; Westermann, D.; Poller, W.; Tschöpe, C.; Lassner, D.; Schultheiss, H.-P. Aggravation of left ventricular dysfunction in patients with biopsy-proven cardiac human herpesvirus A and B infection. J. Clin. Virol. 2015, 63, 1–5. [Google Scholar] [CrossRef]
- Bonavita, C.M.; Cardin, R.D. Don’t Go Breaking My Heart: MCMV as a Model for HCMV-Associated Cardiovascular Diseases. Pathog 2021, 10, 619. [Google Scholar] [CrossRef]
- Magno Palmeira, M.; Umemura Ribeiro, H.Y.; Garcia Lira, Y.; Machado Jucá Neto, F.O.; Da Silva Rodrigues, I.A.; Fernandes Da Paz, L.N.; Nascimento Pinheiro, M.D.C. Heart failure due to cytomegalovirus myocarditis in immunocompetent young adults: A case report. BMC Res. Notes 2016, 9, 391. [Google Scholar] [CrossRef]
- Roubille, C.; Brunel, A.S.; Gahide, G.; Kovacsik, H.V.; Le Quellec, A. Cytomegalovirus (CMV) and acute myocarditis in an immunocompetent patient. Intern. Med. 2010, 49, 131–133. [Google Scholar] [CrossRef]
- Watanabe, M.; Panetta, G.L.; Piccirillo, F.; Spoto, S.; Myers, J.; Serino, F.M.; Costantino, S.; Di Sciascio, G. Acute Epstein-Barr related myocarditis: An unusual but life-threatening disease in an immunocompetent patient. J. Cardiol. Cases 2019, 21, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.P.; Piper, C.; Sowade, O.; Waagstein, F.; Kapp, J.F.; Wegscheider, K.; Groetzbach, G.; Pauschinger, M.; Escher, F.; Arbustini, E.; et al. Betaferon in chronic viral cardiomyopathy (BICC) trial: Effects of interferon-β treatment in patients with chronic viral cardiomyopathy. Clin. Res. Cardiol. 2016, 105, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Pauschinger, M.; Bowles, N.E.; Fuentes-Garcia, F.J.; Pham, V.; Kühl, U.; Schwimmbeck, P.L.; Schultheiss, H.P.; Towbin, J.A. Detection of adenoviral genome in the myocardium of adult patients with idiopathic left ventricular dysfunction. Circulation 1999, 99, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, S.Z.; Tabassum, R.; Hamed, H.; Rehman, S.U.; Qadri, I. Hepatitis C Virus-Associated Extrahepatic Manifestations in Lung and Heart and Antiviral Therapy-Related Cardiopulmonary Toxicity. Viral Immunol. 2017, 30, 633–641. [Google Scholar] [CrossRef]
- Matsumori, A.; Matoba, Y.; Sasayama, S. Dilated cardiomyopathy associated with hepatitis C virus infection. Circulation 1995, 92, 2519–2525. [Google Scholar] [CrossRef]
- Matsumori, A.; Shimada, T.; Chapman, N.M.; Tracy, S.M.; Mason, J.W. Myocarditis and heart failure associated with hepatitis C virus infection. J. Card. Fail. 2006, 12, 293–298. [Google Scholar] [CrossRef]
- Mathez, G.; Cagno, V. Viruses Like Sugars: How to Assess Glycan Involvement in Viral Attachment. Microorganisms 2021, 9, 1238. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef]
- Pickles, R.J.; Fahrner, J.A.; Petrella, J.M.; Boucher, R.C.; Bergelson, J.M. Retargeting the coxsackievirus and adenovirus receptor to the apical surface of polarized epithelial cells reveals the glycocalyx as a barrier to adenovirus-mediated gene transfer. J. Virol. 2000, 74, 6050–6057. [Google Scholar] [CrossRef]
- Targosz-Korecka, M.; Kubisiak, A.; Kloska, D.; Kopacz, A.; Grochot-Przeczek, A.; Szymonski, M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci. Rep. 2021, 11, 12157. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Liu, L.; Wang, Y. Viral proteins recognized by different TLRs. J. Med. Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Katare, P.B.; Nizami, H.L.; Paramesha, B.; Dinda, A.K.; Banerjee, S.K. Activation of toll like receptor 4 (TLR4) promotes cardiomyocyte apoptosis through SIRT2 dependent p53 deacetylation. Sci. Rep. 2020, 10, 19232. [Google Scholar] [CrossRef] [PubMed]
- Garmaroudi, F.S.; Marchant, D.; Hendry, R.; Luo, H.; Yang, D.; Ye, X.; Shi, J.; McManus, B.M. Coxsackievirus B3 replication and pathogenesis. Future Microbiol. 2015, 10, 629–653. [Google Scholar] [CrossRef]
- Hsu, T.C.; Wu, W.J.; Chen, M.C.; Tsay, G.J. Human parvovirus B19 non-structural protein (NS1) induces apoptosis through mitochondria cell death pathway in COS-7 cells. Scand. J. Infect. Dis. 2004, 36, 570–577. [Google Scholar] [CrossRef]
- Bachelier, K.; Biehl, S.; Schwarz, V.; Kindermann, I.; Kandolf, R.; Sauter, M.; Ukena, C.; Yilmaz, A.; Sliwa, K.; Bock, C.-T.; et al. Parvovirus B19-induced vascular damage in the heart is associated with elevated circulating endothelial microparticles. PLoS ONE 2017, 12, e0176311. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, K.B.; Schwimmbeck, P.L.; Kühl, U.; Seeberg, B.; Schultheiss, H.P. Endothelium-dependent flow-mediated vasodilation of systemic arteries is impaired in patients with myocardial virus persistence. Circulation 2004, 110, 2938–2945. [Google Scholar] [CrossRef]
- Belkin, M.N.; Uriel, N. Heart health in the age of highly active antiretroviral therapy: A review of HIV cardiomyopathy. Curr. Opin. Cardiol. 2018, 33, 317–324. [Google Scholar] [CrossRef]
- Matsumori, A.; Yamada, T.; Suzuki, H.; Matoba, Y.; Sasayama, S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Heart 1994, 72, 561–566. [Google Scholar] [CrossRef]
- Kawai, C. From Myocarditis to Cardiomyopathy: Mechanisms of Inflammation and Cell Death. Circulation 1999, 99, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Neu, C.; Thiele, Y.; Horr, F.; Beckers, C.; Frank, N.; Marx, G.; Martin, L.; Kraemer, S.; Zechendorf, E. DAMPs Released from Proinflammatory Macrophages Induce Inflammation in Cardiomyocytes via Activation of TLR4 and TNFR. Int. J. Mol. Sci. 2022, 23, 15522. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series. J. Am. Coll. Cardiol. 2018, 72, 2213. [Google Scholar] [CrossRef] [PubMed]
- Magnani, J.W.; Suk Danik, H.J.; Dec, G.W.; DiSalvo, T.G. Survival in biopsy-proven myocarditis: A long-term retrospective analysis of the histopathologic, clinical, and hemodynamic predictors. Am. Heart J. 2006, 151, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.M.; McNamara, D. Myocarditis. N. Engl. J. Med. 2000, 343, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, C.; Ocampo, C.; Saura, M.; Leppo, M.; Wei, X.Q.; Quick, R.; Moncada, S.; Liew, F.Y.; Lowenstein, C.J. The role of inducible nitric oxide synthase in the host response to Coxsackievirus myocarditis. Proc. Natl. Acad. Sci. USA 1998, 95, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Kühl, U.; Lassner, D.; Pauschinger, M.; Gross, U.M.; Seeberg, B.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Prevalence of erythrovirus genotypes in the myocardium of patients with dilated cardiomyopathy. J. Med. Virol. 2008, 80, 1243–1251. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef]
- Kühl, U.; Lassner, D.; Von Schlippenbach, J.; Poller, W.; Schultheiss, H.P. Interferon-Beta improves survival in enterovirus-associated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1295–1296. [Google Scholar] [CrossRef]
- Pauschinger, M.; Doerner, A.; Kuehl, U.; Schwimmbeck, P.L.; Poller, W.; Kandolf, R.; Schultheiss, H.P. Enteroviral RNA replication in the myocardium of patients with left ventricular dysfunction and clinically suspected myocarditis. Circulation 1999, 99, 889–895. [Google Scholar] [CrossRef]
- Pietsch, H.; Escher, F.; Aleshcheva, G.; Lassner, D.; Bock, C.T.; Schultheiss, H.P. Detection of parvovirus mRNAs as markers for viral activity in endomyocardial biopsy-based diagnosis of patients with unexplained heart failure. Sci. Rep. 2020, 10, 22354. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, U.; Lassner, D.; Dorner, A.; Rohde, M.; Escher, F.; Seeberg, B.; Hertel, E.; Tschope, C.; Skurk, C.; Gross, U.M.; et al. A distinct subgroup of cardiomyopathy patients characterized by transcriptionally active cardiotropic erythrovirus and altered cardiac gene expression. Basic Res. Cardiol. 2013, 108, 372. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.L.; Frisancho-Kiss, S.; Rose, N.R. Viruses as adjuvants for autoimmunity: Evidence from Coxsackievirus-induced myocarditis. Rev. Med. Virol. 2005, 15, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol. 2008, 3, 127–155. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A. Viral Myocarditis and Dilated Cardiomyopathy: Etiology and Pathogenesis. Curr. Pharm. Des. 2016, 22, 408–426. [Google Scholar] [CrossRef]
- Rose, N.R. Viral myocarditis. Curr. Opin. Rheumatol. 2016, 28, 383–389. [Google Scholar] [CrossRef]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar] [CrossRef]
- Cunningham, M.W. T cell mimicry in inflammatory heart disease. Mol. Immunol. 2004, 40, 1121–1127. [Google Scholar] [CrossRef]
- Elamm, C.; Fairweather, D.L.; Cooper, L.T. Republished: Pathogenesis and diagnosis of myocarditis. Postgrad. Med. J. 2012, 88, 539–544. [Google Scholar] [CrossRef]
- Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. [Google Scholar] [CrossRef]
- Heymans, S.; Eriksson, U.; Lehtonen, J.; Cooper, L.T. The Quest for New Approaches in Myocarditis and Inflammatory Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 2348–2364. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; Von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Halder, S.; Agbandje-Mckenna, M. Parvovirus glycan interactions. Curr. Opin. Virol. 2014, 7, 108–118. [Google Scholar] [CrossRef]
- Conti, C.; Cirone, M.; Sgro, R.; Altieri, F.; Zompetta, C.; Faggioni, A. Early interactions of human herpesvirus 6 with lymphoid cells: Role of membrane protein components and glycosaminoglycans in virus binding. J. Med. Virol. 2000, 62, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Hadigal, S.; Koganti, R.; Yadavalli, T.; Agelidis, A.; Suryawanshi, R.; Shukla, D. Heparanase-Regulated Syndecan-1 Shedding Facilitates Herpes Simplex Virus 1 Egress. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmeier, K.M. Hyaluronan production in synoviocytes as a consequence of viral infections: HAS1 activation by Epstein-Barr virus and synthetic double- and single-stranded viral RNA analogs. J. Biol. Chem. 2008, 283, 16781–16789. [Google Scholar] [CrossRef]
- Mitra, D.; Hasan, M.H.; Bates, J.T.; Bierdeman, M.A.; Ederer, D.R.; Parmar, R.C.; Fassero, L.A.; Liang, Q.; Qiu, H.; Tiwari, V.; et al. The degree of polymerization and sulfation patterns in heparan sulfate are critical determinants of cytomegalovirus entry into host cells. PLoS Pathog. 2021, 17, e1009803. [Google Scholar] [CrossRef]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef]
- McLeish, N.J.; Williams, Ç.H.; Kaloudas, D.; Roivainen, M.M.; Stanway, G. Symmetry-related clustering of positive charges is a common mechanism for heparan sulfate binding in enteroviruses. J. Virol. 2012, 86, 11163–11170. [Google Scholar] [CrossRef]
- Tan, C.W.; Poh, C.L.; Sam, I.-C.; Chan, Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J. Virol. 2013, 87, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Koike, S. Cellular receptors for enterovirus A71. J. Biomed. Sci. 2020, 27, 23. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martínez, I.E.; Ramos-Martínez, E.; Segura-Velázquez, R.Á.; Saavedra-Montañez, M.; Cervantes-Torres, J.B.; Cerbón, M.; Papy-Garcia, D.; Zenteno, E.; Sánchez-Betancourt, J.I. Heparan Sulfate and Sialic Acid in Viral Attachment: Two Sides of the Same Coin? Int. J. Mol. Sci. 2022, 23, 9842. [Google Scholar] [CrossRef] [PubMed]
- Schröer, K.; Alshawabkeh, M.; Schellhorn, S.; Bronder, K.; Zhang, W.; Ehrhardt, A. Influence of Heparan Sulfate Proteoglycans and Factor X on species D Human Adenovirus Uptake and Transduction. Viruses 2022, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Fender, P.; Schoehn, G.; Perron-Sierra, F.; Tucker, G.C.; Lortat-Jacob, H. Adenovirus dodecahedron cell attachment and entry are mediated by heparan sulfate and integrins and vary along the cell cycle. Virology 2008, 371, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Grigorov, B.; Reungoat, E.; Gentil dit Maurin, A.; Varbanov, M.; Blaising, J.; Michelet, M.; Manuel, R.; Parent, R.; Bartosch, B.; Zoulim, F.; et al. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell. Microbiol. 2017, 19, e12711. [Google Scholar] [CrossRef]
- Zhang, F.; Sodroski, C.; Cha, H.; Li, Q.; Liang, T.J. Infection of Hepatocytes with HCV Increases Cell Surface Levels of Heparan Sulfate Proteoglycans, Uptake of Cholesterol and Lipoprotein, and Virus Entry by Up-regulating SMAD6 and SMAD7. Gastroenterology 2017, 152, 257–270.e7. [Google Scholar] [CrossRef]
- Cerezo-Magaña, M.; Bång-Rudenstam, A.; Belting, M. Proteoglycans: A common portal for SARS-CoV-2 and extracellular vesicle uptake. Am. J. Physiol. Cell Physiol. 2023, 324, C76–C84. [Google Scholar] [CrossRef]
- Bell, T.J.; Brand, O.J.; Morgan, D.J.; Salek-Ardakani, S.; Jagger, C.; Fujimori, T.; Cholewa, L.; Tilakaratna, V.; Östling, J.; Thomas, M.; et al. Defective lung function following influenza virus is due to prolonged, reversible hyaluronan synthesis. Matrix Biol. 2019, 80, 14–28. [Google Scholar] [CrossRef]
- Pasupathy, S.; Tavella, R.; Grover, S.; Raman, B.; Procter, N.E.K.; Du, Y.T.; Mahadavan, G.; Stafford, I.; Heresztyn, T.; Holmes, A.; et al. Early use of N-acetylcysteine with nitrate therapy in patients undergoing primary percutaneous coronary intervention for ST-segment-elevation myocardial infarction reduces myocardial infarct size (the NACIAM trial [N-acetylcysteine in acute myocardial infarction]). Circulation 2017, 136, 894–903. [Google Scholar]
- Corrado, D.; Basso, C.; Thiene, G. Sudden cardiac death in young people with apparently normal heart. Cardiovasc. Res. 2001, 50, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Baksi, A.J.; Kanaganayagam, G.S.; Prasad, S.K. Arrhythmias in viral myocarditis and pericarditis. Card. Electrophysiol. Clin. 2015, 7, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Sala, S.; Rizzo, S.; Palmisano, A.; Esposito, A.; De Cobelli, F.; Campochiaro, C.; De Luca, G.; Foppoli, L.; Dagna, L.; et al. Ventricular Arrhythmias in Myocarditis: Characterization and Relationships with Myocardial Inflammation. J. Am. Coll. Cardiol. 2020, 75, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.M.; Piechnik, S.K.; Dall’Armellina, E.; Karamitsos, T.D.; Francis, J.M.; Ntusi, N.; Holloway, C.; Choudhury, R.P.; Kardos, A.; Robson, M.D.; et al. Native T1-mapping detects the location, extent and patterns of acute myocarditis without the need for gadolinium contrast agents. J. Cardiovasc. Magn. Reson. 2014, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Moroni, F.; Sormani, P.; Peritore, A.; Milazzo, A.; Quattrocchi, G.; Cipriani, M.; Oliva, F.; Giannattasio, C.; Frigerio, M.; et al. Quantitative changes in late gadolinium enhancement at cardiac magnetic resonance in the early phase of acute myocarditis. Int. J. Cardiol. 2017, 231, 216–221. [Google Scholar] [CrossRef]
- Harrison, J.L.; Jensen, H.K.; Peel, S.A.; Chiribiri, A.; Grondal, A.K.; Bloch, L.O.; Pedersen, S.F.; Bentzon, J.F.; Kolbitsch, C.; Karim, R.; et al. Cardiac magnetic resonance and electroanatomical mapping of acute and chronic atrial ablation injury: A histological validation study. Eur. Heart J. 2014, 35, 1486–1495. [Google Scholar] [CrossRef]
- Dickfeld, T.; Tian, J.; Ahmad, G.; Jimenez, A.; Turgeman, A.; Kuk, R.; Peters, M.; Saliaris, A.; Saba, M.; Shorofsky, S.; et al. MRI-Guided ventricular tachycardia ablation: Integration of late gadolinium-enhanced 3D scar in patients with implantable cardioverter-defibrillators. Circ. Arrhythm. Electrophysiol. 2011, 4, 172–184. [Google Scholar] [CrossRef]
- Fernández-Armenta, J.; Berruezo, A.; Andreu, D.; Camara, O.; Silva, E.; Serra, L.; Barbarito, V.; Carotenutto, L.; Evertz, R.; Ortiz-Pérez, J.T.; et al. Three-dimensional architecture of scar and conducting channels based on high resolution ce-CMR: Insights for ventricular tachycardia ablation. Circ. Arrhythm. Electrophysiol. 2013, 6, 528–537. [Google Scholar] [CrossRef]
- Perez-David, E.; Arenal, Á.; Rubio-Guivernau, J.L.; Del Castillo, R.; Atea, L.; Arbelo, E.; Caballero, E.; Celorrio, V.; Datino, T.; Gonzalez-Torrecilla, E.; et al. Noninvasive identification of ventricular tachycardia-related conducting channels using contrast-enhanced magnetic resonance imaging in patients with chronic myocardial infarction: Comparison of signal intensity scar mapping and endocardial voltage mappin. J. Am. Coll. Cardiol. 2011, 57, 184–194. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death Developed by the task force for the management of patients with death of the European Society of Cardiology (ESC) Endorsed by the. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Inomata, T.; Kohno, T.; Asaumi, Y.; Fujino, T.; Ikeda, Y.; Kubo, T.; Matsuura, H.; Nakamura, K.; Okumura, T.; et al. JCS 2023 Guideline on the Diagnosis and Treatment of Myocarditis. Circ. J. 2023, 87, 674–754. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Stolfo, D.; Collini, V.; Sinagra, G. Advanced Heart Failure in Special Population: Cardiomyopathies and Myocarditis. Heart Fail. Clin. 2021, 17, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, M.; Banfi, C.; Grinberg, D.; Koffel, C.; Bendjelid, K.; Robin, J.; Giraud, R.; Obadia, J.F. Veno-arterial extracorporeal membrane oxygenation for cardiogenic shock due to myocarditis in adult patients. J. Thorac. Dis. 2016, 8, E495. [Google Scholar] [CrossRef]
- Combes, A.; Price, S.; Slutsky, A.S.; Brodie, D. Temporary circulatory support for cardiogenic shock. Lancet 2020, 396, 199–212. [Google Scholar] [CrossRef]
- Means, R.T. The anaemia of infection. Baillieres. Best Pract. Res. Clin. Haematol. 2000, 13, 151–162. [Google Scholar] [CrossRef]
- Bellmann-Weiler, R.; Lanser, L.; Barket, R.; Rangger, L.; Schapfl, A.; Schaber, M.; Fritsche, G.; Wöll, E.; Weiss, G. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J. Clin. Med. 2020, 9, 2429. [Google Scholar] [CrossRef]
- Giustino, G.; Kirtane, A.J.; Baber, U.; Généreux, P.; Witzenbichler, B.; Neumann, F.J.; Weisz, G.; Maehara, A.; Rinaldi, M.J.; Metzger, C.; et al. Impact of Anemia on Platelet Reactivity and Ischemic and Bleeding Risk: From the Assessment of Dual Antiplatelet Therapy with Drug-Eluting Stents Study. Am. J. Cardiol. 2016, 117, 1877–1883. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Kopp, C.W.; Koppensteiner, R.; Lang, I.M.; Pultar, J.; Lee, S.; Weikert, C.; Panzer, S.; Gremmel, T. Decreased platelet inhibition by P2Y12 receptor blockers in anaemia. Eur. J. Clin. Investig. 2018, 48, e12861. [Google Scholar] [CrossRef]
- Ammirati, E.; Bizzi, E.; Veronese, G.; Groh, M.; Van de Heyning, C.M.; Lehtonen, J.; de Chambrun, M.P.; Cereda, A.; Picchi, C.; Trotta, L.; et al. Immunomodulating Therapies in Acute Myocarditis and Recurrent/Acute Pericarditis. Front. Med. 2022, 9, 838564. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Khl, U.; Cooper, L.T. The management of myocarditis. Eur. Heart J. 2011, 32, 2616–2625. [Google Scholar] [CrossRef] [PubMed]
- Lampejo, T.; Durkin, S.M.; Bhatt, N.; Guttmann, O. Acute myocarditis: Aetiology, diagnosis and management. Clin. Med. 2021, 21, e505. [Google Scholar] [CrossRef] [PubMed]
- Bang, V.; Ganatra, S.; Shah, S.P.; Dani, S.S.; Neilan, T.G.; Thavendiranathan, P.; Resnic, F.S.; Piemonte, T.C.; Barac, A.; Patel, R.; et al. Management of Patients with Giant Cell Myocarditis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 1122–1134. [Google Scholar] [CrossRef]
- Cremer, P.C.; Sheng, C.C.; Sahoo, D.; Dugar, S.; Prada, R.A.; Wang, T.K.M.; Abou Hassan, O.K.; Hernandez-Montfort, J.; Wolinsky, D.A.; Culver, D.A.; et al. Double-blind randomized proof-of-concept trial of canakinumab in patients with COVID-19 associated cardiac injury and heightened inflammation. Eur. Heart J. Open 2021, 1, oeab002. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, Z.; Zheng, Y.; Yang, S.; Huang, K.; Li, H. Roles of inflammasomes in viral myocarditis. Front. Cell. Infect. Microbiol. 2023, 13, 1149911. [Google Scholar] [CrossRef]
- van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.; Pickkers, P.; Derde, L.; Leavis, H.; van Crevel, R.; Engel, J.J.; Wiersinga, W.J.; Vlaar, A.P.J.; Shankar-Hari, M.; et al. A guide to immunotherapy for COVID-19. Nat. Med. 2022, 28, 39–50. [Google Scholar] [CrossRef]
- Ammirati, E.; Frigerio, M.; Adler, E.D.; Basso, C.; Birnie, D.H.; Brambatti, M.; Friedrich, M.G.; Klingel, K.; Lehtonen, J.; Moslehi, J.J.; et al. Management of Acute Myocarditis and Chronic Inflammatory Cardiomyopathy: An Expert Consensus Document. Circ. Heart Fail. 2020, 13, e007405. [Google Scholar] [CrossRef]
- Merken, J.; Hazebroek, M.; Van Paassen, P.; Verdonschot, J.; Van Empel, V.; Knackstedt, C.; Abdul Hamid, M.; Seiler, M.; Kolb, J.; Hoermann, P.; et al. Immunosuppressive Therapy Improves Both Short- and Long-Term Prognosis in Patients with Virus-Negative Nonfulminant Inflammatory Cardiomyopathy. Circ. Heart Fail. 2018, 11, e004228. [Google Scholar] [CrossRef]
- Peretto, G.; Sala, S.; De Luca, G.; Marcolongo, R.; Campochiaro, C.; Sartorelli, S.; Tresoldi, M.; Foppoli, L.; Palmisano, A.; Esposito, A.; et al. Immunosuppressive Therapy and Risk Stratification of Patients with Myocarditis Presenting with Ventricular Arrhythmias. JACC Clin. Electrophysiol. 2020, 6, 1221–1234. [Google Scholar] [CrossRef]
- Abernethy, A.; Raza, S.; Sun, J.L.; Anstrom, K.J.; Tracy, R.; Steiner, J.; VanBuren, P.; LeWinter, M.M. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. Cardiovasc. Cerebrovasc. Dis. 2018, 7, e007385. [Google Scholar] [CrossRef] [PubMed]
- Wijk, S.S.-V.; Van Empel, V.; Davarzani, N.; Maeder, M.T.; Handschin, R.; Pfisterer, M.E.; Rocca, H.P.B.-L. Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, N.S.; Gupta, K.; Gharpure, N.; Pate, M.; Chopra, L.; Kalra, R.; Prabhu, S.D. Effect of immunomodulation on cardiac remodelling and outcomes in heart failure: A quantitative synthesis of the literature. ESC Heart Fail. 2020, 7, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted Anticytokine Therapy in Patients with Chronic Heart Failure. Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (AT). Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From systemic inflammation to myocardial fibrosis: The hfpef paradigm revisited. Circ. Res. 2021, 128, 1451. [Google Scholar] [CrossRef]
- Ning, N.; Guo, H.H.; Iagaru, A.; Mittra, E.; Fowler, M.; Witteles, R. Serial Cardiac FDG-PET for the Diagnosis and Therapeutic Guidance of Patients with Cardiac Sarcoidosis. J. Card. Fail. 2019, 25, 307–311. [Google Scholar] [CrossRef]
- Shelke, A.B.; Aurangabadkar, H.U.; Bradfield, J.S.; Ali, Z.; Kumar, K.S.; Narasimhan, C. Serial FDG-PET scans help to identify steroid resistance in cardiac sarcoidosis. Int. J. Cardiol. 2017, 228, 717–722. [Google Scholar] [CrossRef]
- Terasaki, F.; Azuma, A.; Anzai, T.; Ishizaka, N.; Ishida, Y.; Isobe, M.; Inomata, T.; Ishibashi-Ueda, H.; Eishi, Y.; Kitakaze, M.; et al. JCS 2016 Guideline on Diagnosis and Treatment of Cardiac Sarcoidosis-Digest Version. Circ. J. 2019, 83, 2329–2388. [Google Scholar] [CrossRef]
- Birnie, D.H.; Sauer, W.H.; Bogun, F.; Cooper, J.M.; Culver, D.A.; Duvernoy, C.S.; Judson, M.A.; Kron, J.; Mehta, D.; Nielsen, J.C.; et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. 2014, 11, 1304–1323. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, E895–E1032. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Delic, D.; Chu, C.; Xiong, Y.; Luo, T.; Chen, X.; Gaballa, M.M.S.; Xue, Y.; Chen, X.; Cao, Y.; et al. Antifibrotic effects of low dose SGLT2 Inhibition with empagliflozin in comparison to Ang II receptor blockade with telmisartan in 5/6 nephrectomised rats on high salt diet. Biomed. Pharmacother. 2022, 146, 112606. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef] [PubMed]
- Kosiborod, M.N.; Esterline, R.; Furtado, R.H.M.; Oscarsson, J.; Gasparyan, S.B.; Koch, G.G.; Martinez, F.; Mukhtar, O.; Verma, S.; Chopra, V.; et al. Dapagliflozin in patients with cardiometabolic risk factors hospitalised with COVID-19 (DARE-19): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 586–594. [Google Scholar] [CrossRef]
- European Society of Cardiology Press Office. SGLT2 Inhibitors not Linked with Improved Survival in Hospitalised COVID-19 Patients. Available online: https://www.escardio.org/The-ESC/Press-Office/Press-releases/SGLT2-inhibitors-not-linked-with-improved-survival-in-hospitalised-COVID-19-patients (accessed on 3 December 2023).
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, J.; Abbas, K.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abdallah, N.; et al. Empagliflozin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet Diabetes Endocrinol. 2023, 11, 905–914. [Google Scholar] [CrossRef]
- Pitt, B.; Agarwal, R.; Anker, S.D.; Ruilope, L.M.; Rossing, P.; Ahlers, C.; Brinker, M.; Joseph, A.; Lambelet, M.; Lawatscheck, R.; et al. Association of Finerenone Use with Reduction in Treatment-Emergent Pneumonia and COVID-19 Adverse Events Among Patients with Type 2 Diabetes and Chronic Kidney Disease: A FIDELITY Pooled Secondary Analysis. JAMA Netw. Open 2022, 5, e2236123. [Google Scholar] [CrossRef]
- Henri, O.; Pouehe, C.; Houssari, M.; Galas, L.; Nicol, L.; Edwards-Lévy, F.; Henry, J.P.; Dumesnil, A.; Boukhalfa, I.; Banquet, S.; et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 2016, 133, 1484–1497. [Google Scholar] [CrossRef]
- Schultheiss, H.P.; Bock, C.T.; Aleshcheva, G.; Baumeier, C.; Poller, W.; Escher, F. Interferon-β Suppresses Transcriptionally Active Parvovirus B19 Infection in Viral Cardiomyopathy: A Subgroup Analysis of the BICC-Trial. Viruses 2022, 14, 444. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Spillmann, F.; Bock, T.; Kühl, U.; Van Linthout, S.; Schultheiss, H.P.; Tschöpe, C.; Van Linthout, S.; Schultheiss, H.P.; Tschöpe, C. Interferon beta modulates endothelial damage in patients with cardiac persistence of human parvovirus b19 infection. J. Infect. Dis. 2010, 201, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Mindur, J.E.; Halle, L.; Sano, S.; Choi, J.L.; He, S.; McAlpine, C.S.; Chan, C.T.; Kahles, F.; Valet, C.; et al. Self-reactive CD4+ IL-3+ T cells amplify autoimmune inflammation in myocarditis by inciting monocyte chemotaxis. J. Exp. Med. 2019, 216, 369. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Varrenti, M.; Veronese, G.; Fanti, D.; Nava, A.; Cipriani, M.; Pedrotti, P.; Garascia, A.; Bottiroli, M.; Oliva, F.; et al. Prevalence and outcome of patients with acute myocarditis and positive viral search on nasopharyngeal swab. Eur. J. Heart Fail. 2021, 23, 1242–1245. [Google Scholar] [CrossRef] [PubMed]
- Gil-Cruz, C.; Perez-Shibayama, C.; de Martin, A.; Ronchi, F.; van der Borght, K.; Niederer, R.; Onder, L.; Lütge, M.; Novkovic, M.; Nindl, V.; et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 2019, 366, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Amdani, S.M.; Kim, H.S.; Orvedahl, A.; John, A.O.; Said, A.; Simpson, K. Successful treatment of fulminant neonatal enteroviral myocarditis in monochorionic diamniotic twins with cardiopulmonary support, intravenous immunoglobulin and pocapavir. BMJ Case Rep. 2018, 2018, bcr-2017-224133. [Google Scholar] [CrossRef]
- Abzug, M.J.; Michaels, M.G.; Wald, E.; Jacobs, R.F.; Romero, J.R.; Sánchez, P.J.; Wilson, G.; Krogstad, P.; Storch, G.A.; Lawrence, R.; et al. A Randomized, Double-Blind, Placebo-Controlled Trial of Pleconaril for the Treatment of Neonates with Enterovirus Sepsis. J. Pediatr. Infect. Dis. Soc. 2016, 5, 53–62. [Google Scholar] [CrossRef]
- Yen, M.H.; Huang, Y.C.; Chen, M.C.; Liu, C.C.; Chiu, N.C.; Lien, R.; Chang, L.Y.; Chiu, C.H.; Tsao, K.C.; Lin, T.Y. Effect of intravenous immunoglobulin for neonates with severe enteroviral infections with emphasis on the timing of administration. J. Clin. Virol. 2015, 64, 92–96. [Google Scholar] [CrossRef]
- Kühl, U.; Lassner, D.; Wallaschek, N.; Gross, U.M.; Krueger, G.R.F.; Seeberg, B.; Kaufer, B.B.; Escher, F.; Poller, W.; Schultheiss, H.P. Chromosomally integrated human herpesvirus 6 in heart failure: Prevalence and treatment. Eur. J. Heart Fail. 2015, 17, 9–19. [Google Scholar] [CrossRef]
- Sanchez, M.J.; Bergasa, N. V Hepatitis C associated cardiomyopathy: Potential pathogenic mechanisms and clinical implications. Med. Sci. Monit. 2008, 14, RA55–RA63. [Google Scholar]
- Baik, S.H.; Jeong, H.S.; Kim, S.J.; Yoon, Y.K.; Sohn, J.W.; Kim, M.J. A Case of Influenza Associated Fulminant Myocarditis Successfully Treated with Intravenous Peramivir. Infect. Chemother. 2015, 47, 272–277. [Google Scholar] [CrossRef]
- Ito, N.; Sato, M.; Momoi, N.; Aoyagi, Y.; Endo, K.; Chishiki, M.; Kawasaki, Y.; Hosoya, M. Influenza A H1N1 pdm09-associated myocarditis during zanamivir therapy. Pediatr. Int. 2015, 57, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Manaresi, E.; Gallinella, G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses 2019, 11, 659. [Google Scholar] [CrossRef] [PubMed]
- Düngen, H.D.; Dordevic, A.; Felix, S.B.; Pieske, B.; Voors, A.A.; McMurray, J.J.V.; Butler, J. β1-Adrenoreceptor Autoantibodies in Heart Failure: Physiology and Therapeutic Implications. Circ. Heart Fail. 2020, 13, e006155. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hilgenfeld, R.; Whitley, R.; Clercq, E. De Therapeutic strategies for COVID-19: Progress and lessons learned. Nat. Rev. Drug Discov. 2023, 22, 449. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef]
- Zhou, D.; Dai, S.M.; Tong, Q. COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, 75, 1667–1670. [Google Scholar] [CrossRef]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef]
- Bruegger, D.; Rehm, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Conzen, P.; Becker, B.F. Exogenous nitric oxide requires an endothelial glycocalyx to prevent postischemic coronary vascular leak in guinea pig hearts. Crit. Care 2008, 12, R73. [Google Scholar] [CrossRef]
- Koch, E.; Malek, F.A. Standardized Extracts from Hawthorn Leaves and Flowers in the Treatment of Cardiovascular Disorders–Preclinical and Clinical Studies. Planta Medica 2011, 77, 1123–1128. [Google Scholar] [CrossRef]
- Ebong, E.E.; Lopez-Quintero, S.V.; Rizzo, V.; Spray, D.C.; Tarbell, J.M. Shear-induced endothelial NOS activation and remodeling via heparan sulfate, glypican-1, and syndecan-1. Integr. Biol. 2014, 6, 338–347. [Google Scholar] [CrossRef]
- Peters, W.; Drueppel, V.; Kusche-Vihrog, K.; Schubert, C.; Oberleithner, H. Nanomechanics and Sodium Permeability of Endothelial Surface Layer Modulated by Hawthorn Extract WS 1442. PLoS ONE 2012, 7, e29972. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.; Schmid, A.I.; Keilani, M.; Doberer, D.; Bartko, J.; Crevenna, R.; Moser, E.; Wolzt, M. Effect of ischemic preconditioning in skeletal muscle measured by functional magnetic resonance imaging and spectroscopy: A randomized crossover trial. J. Cardiovasc. Magn. Reson. 2011, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.G.G.; Vink, H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef] [PubMed]
- Illien-Junger, S.; Grosjean, F.; Laudier, D.M.; Vlassara, H.; Striker, G.E.; Iatridis, J.C. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLoS ONE 2013, 8, e64302. [Google Scholar] [CrossRef] [PubMed]
- Salmon, A.H.J.; Ferguson, J.K.; Burford, J.L.; Gevorgyan, H.; Nakano, D.; Harper, S.J.; Bates, D.O.; Peti-Peterd, J. Loss of the endothelial glycocalyx links albuminuria and vascular dysfunction. J. Am. Soc. Nephrol. 2012, 23, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Maeda, M. Chemical structure of antithrombin-active rhamnan sulfate from monostrom nitidum. Biosci. Biotechnol. Biochem. 1998, 62, 1647–1652. [Google Scholar] [CrossRef]
- Giantsos-Adams, K.; Lopez-Quintero, V.; Kopeckova, P.; Kopecek, J.; Tarbell, J.M.; Dull, R. Study of the therapeutic benefit of cationic copolymer administration to vascular endothelium under mechanical stress. Biomaterials 2011, 32, 288–294. [Google Scholar] [CrossRef]
- Masola, V.; Zaza, G.; Onisto, M.; Lupo, A.; Gambaro, G. Glycosaminoglycans, proteoglycans and sulodexide and the endothelium: Biological roles and pharmacological effects. Int. Angiol. 2014, 33, 243–254. [Google Scholar]
- Frati Munari, A.C. Medical significance of endothelial glycocalyx. Part 2: Its role in vascular diseases and in diabetic complications. Arch. Cardiol. Mex. 2014, 84, 110–116. [Google Scholar] [CrossRef]
- Taneja, R. Current status of oral pentosan polysulphate in bladder pain syndrome/interstitial cystitis. Int. Urogynecol. J. 2021, 32, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Cancel, L.M.; Tarbell, J.M. Rhamnan sulfate enhances the endothelial glycocalyx and decreases the LDL permeability of human coronary artery endothelial cells in vitro. FASEB J. 2013, 27, 896.3. [Google Scholar] [CrossRef]
- Merchant, S.H.; Gurule, D.M.; Larson, R.S. Amelioration of ischemia-reperfusion injury with cyclic peptide blockade of ICAM-1. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1260–H1268. [Google Scholar] [CrossRef] [PubMed]
- Kozar, R.A.; Peng, Z.; Zhang, R.; Holcomb, J.B.; Pati, S.; Park, P.; Ko, T.C.; Paredes, A. Plasma restoration of endothelial glycocalyx in a rodent model of hemorrhagic shock. Anesth. Analg. 2011, 112, 1289–1295. [Google Scholar] [CrossRef]
- Suarez, J.I.; Martin, R.H.; Calvillo, E.; Bershad, E.M.; Venkatasubba Rao, C.P. Effect of human albumin on TCD vasospasm, DCI, and cerebral infarction in subarachnoid hemorrhage: The ALISAH study. Acta Neurochir. Suppl. 2015, 120, 287–290. [Google Scholar] [CrossRef]
- Curry, F.-R.E.; Adamson, R.H. Sphingosine-1-phosphate and the “albumin effect” on rat venular microvessels. FASEB J. 2013, 27, 896.2. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef]
- Carnevale, R.; Cammisotto, V.; Bartimoccia, S.; Nocella, C.; Castellani, V.; Bufano, M.; Loffredo, L.; Sciarretta, S.; Frati, G.; Coluccia, A.; et al. Toll-Like Receptor 4-Dependent Platelet-Related Thrombosis in SARS-CoV-2 Infection. Circ. Res. 2023, 132, 290–305. [Google Scholar] [CrossRef]
- Zhang, H.; Lao, Q.; Zhang, J.; Zhu, J. Coagulopathy in COVID-19 and anticoagulation clinical trials. Best Pract. Res. Clin. Haematol. 2022, 35, 101377. [Google Scholar] [CrossRef]
- Chandra, A.; Chakraborty, U.; Ghosh, S.; Dasgupta, S. Anticoagulation in COVID-19: Current concepts and controversies. Postgrad. Med. J. 2022, 98, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Kumar, D.; Meena, D.S.; Naresh, K.M.; Bohra, G.K.; Garg, M.K.; Purohit, A.H.L. Post-Discharge Prophylactic Anticoagulation in COVID-19 Patients: A Clinical Dilemma. Cardiovasc. Hematol. Disord. Drug Targets 2021, 21, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; Farkouh, M.E.; Lala, A.; Tinuoye, E.; Dressler, O.; Moreno, P.R.; Palacios, I.F.; Goodman, S.G.; Esper, R.B.; Abizaid, A.; et al. Randomized Trial of Anticoagulation Strategies for Noncritically Ill Patients Hospitalized with COVID-19. J. Am. Coll. Cardiol. 2023, 81, 1747. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Vivarini Áislan de, C.; Calegari-Silva, T.C.; Saliba, A.M.; Boaventura, V.S.; França-Costa, J.; Khouri, R.; Dierckx, T.; Dias-Teixeira, K.L.; Fasel, N.; Barral, A.M.P.; et al. Systems approach reveals nuclear factor erythroid 2-related factor 2/protein kinase r crosstalk in human cutaneous leishmaniasis. Front. Immunol. 2017, 8, 283936. [Google Scholar]
- Andreas, M.; Schmid, A.I.; Doberer, D.; Schewzow, K.; Weisshaar, S.; Heinze, G.; Bilban, M.; Moser, E.; Wolzt, M. Heme arginate improves reperfusion patterns after ischemia: A randomized, placebo-controlled trial in healthy male subjects. J. Cardiovasc. Magn. Reson. 2012, 14, 55. [Google Scholar] [CrossRef]
- Hangaishi, M.; Ishizaka, N.; Aizawa, T.; Kurihara, Y.; Taguchi, J.I.; Nagai, R.; Kimura, S.; Ohno, M. Induction of heme oxygenase-1 can act protectively against cardiac ischemia/reperfusion in vivo. Biochem. Biophys. Res. Commun. 2000, 279, 582–588. [Google Scholar] [CrossRef]
- Li, W.; Zhang, B.; Wei, X.; Cui, X.; Kobayashi, T. Effects of heme oxygenase 1 on brain edema and neurologic outcome after cardiopulmonary resuscitation in rats. Anesthesiology 2008, 109, 260–268. [Google Scholar] [CrossRef]
- Ligi, D.; Lo Sasso, B.; Giglio, R.V.; Maniscalco, R.; DellaFranca, C.; Agnello, L.; Ciaccio, M.; Mannello, F. Circulating histones contribute to monocyte and MDW alterations as common mediators in classical and COVID-19 sepsis. Crit. Care 2022, 26, 260. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Eichelberger, B.; Kopp, C.W.; Pultar, J.; Seidinger, D.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Disaggregation Following Agonist-Induced Platelet Activation in Patients on Dual Antiplatelet Therapy. J. Cardiovasc. Transl. Res. 2017, 10, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Panzer, B.; Wadowski, P.P.; Huber, K.; Panzer, S.; Gremmel, T. Protease-activated receptor-mediated platelet aggregation in patients with type 2 diabetes on potent P2Y12 inhibitors. Diabet. Med. 2022, 39, e14868. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Pultar, J.; Weikert, C.; Eichelberger, B.; Panzer, B.; Huber, K.; Lang, I.M.; Koppensteiner, R.; Panzer, S.; Gremmel, T. Protease-activated receptor-mediated platelet aggregation in acute coronary syndrome patients on potent P2Y12 inhibitors. Res. Pract. Thromb. Haemost. 2019, 3, 383–390. [Google Scholar] [CrossRef]
- Ofosu, F.A.; Dewar, L.; Craven, S.J.; Song, Y.; Cedrone, A.; Freedman, J.; Fenton, J.W. Coordinate activation of human platelet protease-activated receptor-1 and -4 in response to subnanomolar alpha-thrombin. J. Biol. Chem. 2008, 283, 26886–26893. [Google Scholar] [CrossRef]
- Gremmel, T.; Michelson, A.D.; Wadowski, P.P.; Pultar, J.; Weikert, C.; Tscharre, M.; Lee, S.; Panzer, S.; Frelinger, A.L. Sex-specific platelet activation through protease-activated receptor-1 in patients undergoing cardiac catheterization. Atherosclerosis 2021, 339, 12–19. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Weikert, C.; Pultar, J.; Lee, S.; Eichelberger, B.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Ticagrelor Inhibits Toll-Like and Protease-Activated Receptor Mediated Platelet Activation in Acute Coronary Syndromes. Cardiovasc. Drugs Ther. 2020, 34, 53–63. [Google Scholar] [CrossRef]
- Van Der Heijden, M.; Van Nieuw Amerongen, G.P.; Koolwijk, P.; Van Hinsbergh, V.W.M.; Groeneveld, A.B.J. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 2008, 63, 903–909. [Google Scholar] [CrossRef]
- Rau, A.; Schroeter, N.; Blazhenets, G.; Dressing, A.; Walter, L.I.; Kellner, E.; Bormann, T.; Mast, H.; Wagner, D.; Urbach, H.; et al. Widespread white matter oedema in subacute COVID-19 patients with neurological symptoms. Brain 2022, 145, 3203–3213. [Google Scholar] [CrossRef]
- Bocci, M.; Oudenaarden, C.; Sàenz-sardà, X.; Simrén, J.; Edén, A.; Sjölund, J.; Möller, C.; Gisslén, M.; Zetterberg, H.; Englund, E.; et al. Infection of brain pericytes underlying neuropathology of covid-19 patients. Int. J. Mol. Sci. 2021, 22, 11622. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Panagiotides, N.G.; Zimprich, F.; Machold, K.; Schlager, O.; Müller, M.; Ertl, S.; Löffler-Stastka, H.; Koppensteiner, R.; Wadowski, P.P. A Case of Autoimmune Small Fiber Neuropathy as Possible Post COVID Sequelae. Int. J. Environ. Res. Public Health 2023, 20, 4918. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.Y.; Yeralan, S. Is Long COVID a State of Systemic Pericyte Disarray? J. Clin. Med. 2022, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Abdi, A.; AlOtaiby, S.; Al Badarin, F.; Khraibi, A.; Hamdan, H.; Nader, M. Interaction of SARS-CoV-2 with cardiomyocytes: Insight into the underlying molecular mechanisms of cardiac injury and pharmacotherapy. Biomed. Pharmacother. 2022, 146, 112518. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.K.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellulartargets. Nature 2021, 595, 107. [Google Scholar] [CrossRef]
- Katwa, L.C.; Mendoza, C.; Clements, M. CVD and COVID-19: Emerging Roles of Cardiac Fibroblasts and Myofibroblasts. Cells 2022, 11, 1316. [Google Scholar] [CrossRef]
- Daems, M.; Liesenborghs, L.; Boudewijns, R.; Simmonds, S.J.; Kraisin, S.; Van Wauwe, J.; Cuijpers, I.; Raman, J.; Geuens, N.; Van Buyten, T.; et al. SARS-CoV-2 infection causes prolonged cardiomyocyte swelling and inhibition of HIF1α translocation in an animal model COVID-19. Front. Cardiovasc. Med. 2022, 9, 964512. [Google Scholar] [CrossRef]
- Avolio, E.; Carrabba, M.; Milligan, R.; Williamson, M.K.; Beltrami, A.P.; Gupta, K.; Elvers, K.T.; Gamez, M.; Foster, R.R.; Gillespie, K.; et al. The SARS-CoV-2 Spike protein disrupts human cardiac pericytes function through CD147 receptor-mediated signalling: A potential non-infective mechanism of COVID-19 microvascular disease. Clin. Sci. 2021, 135, 2667–2689. [Google Scholar] [CrossRef]
| JV = LPS[(PC-PI)-σ(ΠC-ΠG)] |
|---|
| LPS: ~0.35 mL min−1 mmHg−1 100 g−1 [31,32] |
| S: 500 cm2 g−1 [32,33] |
| PC: end-diastolic 20–30 mmHg [34,35] |
| PI: ~120 mmHg during systole, 15 mmHg during diastole [36,37] |
| σ: 0.51–0.67 for plasma proteins; 0.41–0.59 for albumin [36,38,39] |
| ΠC: 21–24 mmHg [36,40,41] |
| ΠG: 13–20 mmHg [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagiotides, N.G.; Poledniczek, M.; Andreas, M.; Hülsmann, M.; Kocher, A.A.; Kopp, C.W.; Piechota-Polanczyk, A.; Weidenhammer, A.; Pavo, N.; Wadowski, P.P. Myocardial Oedema as a Consequence of Viral Infection and Persistence—A Narrative Review with Focus on COVID-19 and Post COVID Sequelae. Viruses 2024, 16, 121. https://doi.org/10.3390/v16010121
Panagiotides NG, Poledniczek M, Andreas M, Hülsmann M, Kocher AA, Kopp CW, Piechota-Polanczyk A, Weidenhammer A, Pavo N, Wadowski PP. Myocardial Oedema as a Consequence of Viral Infection and Persistence—A Narrative Review with Focus on COVID-19 and Post COVID Sequelae. Viruses. 2024; 16(1):121. https://doi.org/10.3390/v16010121
Chicago/Turabian StylePanagiotides, Noel G., Michael Poledniczek, Martin Andreas, Martin Hülsmann, Alfred A. Kocher, Christoph W. Kopp, Aleksandra Piechota-Polanczyk, Annika Weidenhammer, Noemi Pavo, and Patricia P. Wadowski. 2024. "Myocardial Oedema as a Consequence of Viral Infection and Persistence—A Narrative Review with Focus on COVID-19 and Post COVID Sequelae" Viruses 16, no. 1: 121. https://doi.org/10.3390/v16010121