
Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches
 , , ,
, , ,
Abstract
:1. Introduction
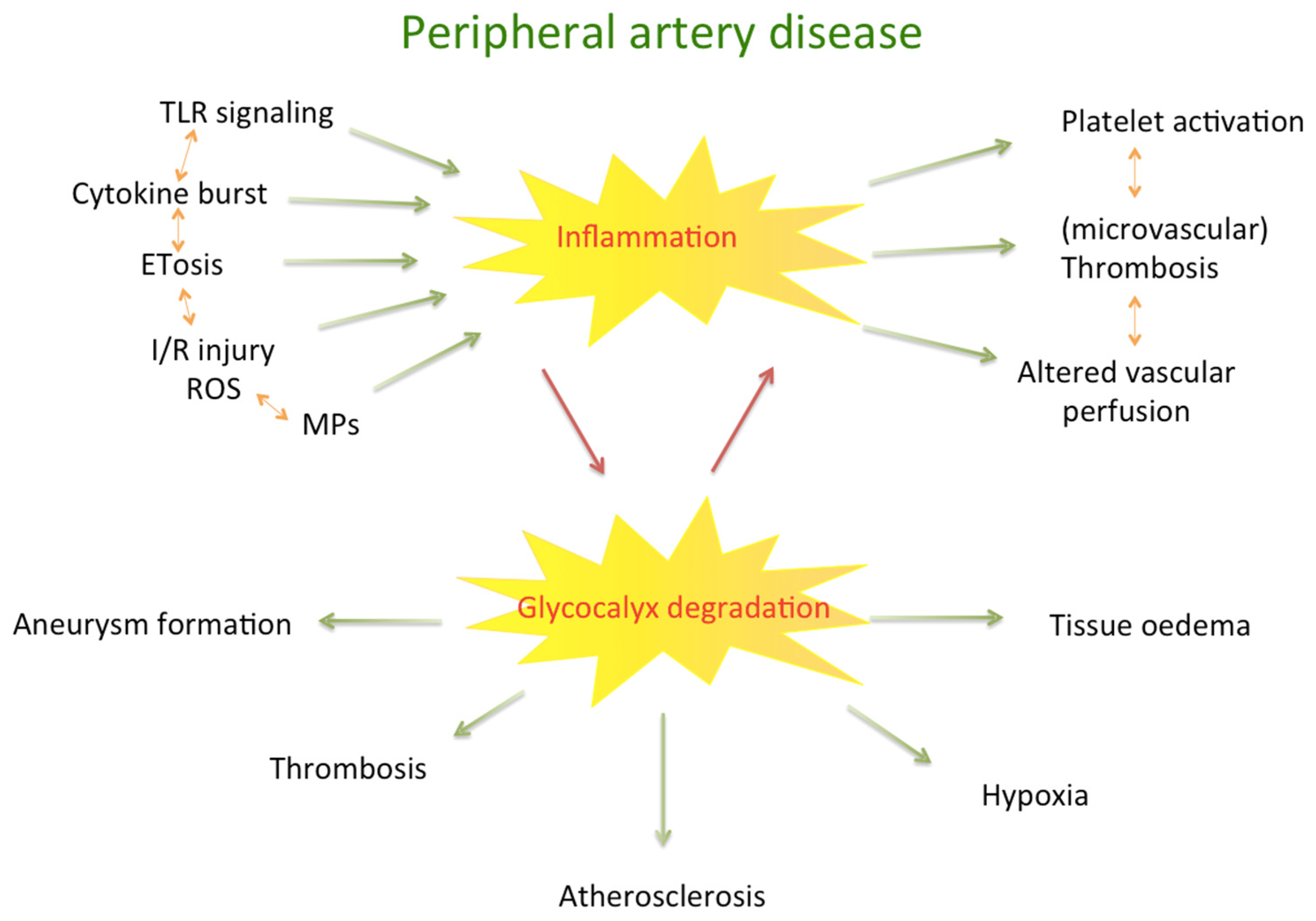
2. Pathophysiology
2.1. Inflammation and Endothelial Dysfunction
2.2. Microparticles
2.3. Neutrophil Extracellular Traps
2.4. The Role of Inflammation in Aneurysm Formation
2.5. Vasculitis
2.6. The Influence of Inflammation on Angiogenesis, Arteriogenesis, and Collateralisation
3. Current and Novel Therapeutic Targets and Strategies
3.1. Statins
3.2. Colchicine
3.3. Eicosapentaenoic Acid Ethyl Ester
3.4. Canakinumab and Anakinra
3.5. Glucocorticoids
3.6. Antidiabetic Drugs

{kind=link}
| Standard Application | Proposed Mechanism | Clinical Effect | Selected Evidence | |
|---|---|---|---|---|
| Statins | LDL reduction secondary prevention of CVD | NO synthesis ↑ leukocyte adhesion ↓ | cardiovascular events and death ↓ | Tawakol et al. [269] Ridker et al. [270] |
| Colchicine | gout familial Mediterranean fever | leukocyte chemotaxis ↓ TNF-α ↓ exocytosis of neutrophil granules ↓ NLRP3 activation ↓ | cardiovascular events and death following MI ↓ | Tardif et al. [279] Chen et al. [281] |
| Icosapent ethyl | no previous application | active metabolites (thromboxane A3, prostacyclin) ↑ biophysical effect on cell membranes TLP ↓ | cardiovascular events and death in established CVD or risk for CVD and hypertriglyceridemia plaque progression ↓ | Bhatt et al. [283] Budoff et al. [284] |
| Glucocorticoids | various inflammatory conditions | modulation of gene transcription | risk of CVD including CAD, PAD ↑ | Pujades-Rodriguez et al. [305] Macleod et al. [301] |
| IL-1β antagonists | cryopyrin-associated periodic syndromes gout familial Mediterranean fever macrophage activation syndrome recurrent pericarditis rheumatoid arthritis systemic juvenile idiopathic arthritis | endothelial activation ↓ adhesion molecule expression ↓ smooth muscle cell proliferation ↓ MCP-1 ↓ | cardiovascular events and death in patients with elevated CRP and MI | Ridker et al. [15] |
| SGLT-2 inhibitors | DM type 2 | NLRP3/IL-1β/MCP-1 pathway ↓ AMPK pathway ↑ cholesterol efflux and autophagy in macrophages ↑ | hospitalization and cardiovascular death in heart failure | McMurray et al. [308] Solomon et al. [359] Packer et al. [360] Anker et al. [311] |
| GLP-1 receptor agonists | DM type 2 | ROS generation ↓ NF-ĸB activation ↓ INF-γ, MMP, TNF-β, IL-1β, IL-2, IL-6 from macrophages ↓ IL-10 ↑ | CRP, TNF-α ↓ Trials inconclusive | Bethel et al. [325] |
| Metformin | DM type 2 | oxLDL phagocytosis ↓ scavenger receptor A, CD36 ↓ NLRP3, ROS, MCP-1, CRP, TNF-α NET formation ↓ NF-ĸB activation ↓ AMPK pathway ↑ | all-cause death in DM type 2 and atherothrombosis↓ | Roussel et al. [335] GLINT (ongoing) [340,341] |
| DDP4 inhibitors | DM type 2 | NO synthesis ↑ endothelin 1 ↓ MCP-1, TNF-α, IL-1β, IL-6 ↓ NF-ĸB activation ↓ AMPK and c-Jun N-terminal kinase pathway ↑ NLRP3 activation ↓ TLP ↓ | dyslipidaemia and hypertension in patients with DM type 2 ↓ cardiovascular death, MI, stroke in patients with DM type 2 ~ | Rosenstock et al. [355] Green et al. [356] Scirica et al. [357] White et al. [358] |
3.7. Antiplatelet Therapy
3.8. Attenuation of Ischaemia-Reperfusion Injury
3.9. Vascular Regeneration
3.10. Physical Exercise
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Townsend, N.; Wilson, L.; Bhatnagar, P.; Wickramasinghe, K.; Rayner, M.; Nichols, M. Cardiovascular Disease in Europe: Epidemiological Update 2016. Eur. Heart J. 2016, 37, 3232–3245. [Google Scholar] [CrossRef] [PubMed]
- Woolf, N. The Pathology of Atherosclerosis with Particular Reference to the Effects of Hyperlipidaemia. Eur. Heart J. 1987, 8 (Suppl. E), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hedin, U.; Matic, L.P. Recent Advances in Therapeutic Targeting of Inflammation in Atherosclerosis. J. Vasc. Surg. 2019, 69, 944–951. [Google Scholar] [CrossRef]
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of Inflammation in the Pathogenesis of Atherosclerosis and Therapeutic Interventions. Atherosclerosis 2018, 276, 98–108. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and Inflammation: Overview and Updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and Atherosclerosis: Signaling Pathways and Therapeutic Intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting Inflammation in Atherosclerosis—From Experimental Insights to the Clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Panzer, B.; Józkowicz, A.; Kopp, C.W.; Gremmel, T.; Panzer, S.; Koppensteiner, R. Microvascular Thrombosis as a Critical Factor in Severe COVID-19. Int. J. Mol. Sci. 2023, 24, 2492. [Google Scholar] [CrossRef]
- Steven, S.; Daiber, A.; Dopheide, J.F.; Münzel, T.; Espinola-Klein, C. Peripheral Artery Disease, Redox Signaling, Oxidative Stress—Basic and Clinical Aspects. Redox Biol. 2017, 12, 787–797. [Google Scholar] [CrossRef]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive Species-Induced Microvascular Dysfunction in Ischemia/Reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.C.; Libby, P. Cardiovascular Disease in Patients with Chronic Inflammation: Mechanisms Underlying Premature Cardiovascular Events in Rheumatologic Conditions. Eur. Heart J. 2015, 36, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Protogerou, A.; Kitas, G.; Sfikakis, P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef]
- Poledniczek, M.H. Coronary Artery Disease in Granulomatosis with Polyangiitis: A Review. SN Compr. Clin. Med. 2022, 4, 75. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar]
- Visseren, F.L.J.; MacH, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on Cardiovascular Disease Prevention in Clinical Practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Darabseh, M.Z.; Maden-Wilkinson, T.M.; Welbourne, G.; Wüst, R.C.I.; Ahmed, N.; Aushah, H.; Selfe, J.; Morse, C.I.; Degens, H. Fourteen Days of Smoking Cessation Improves Muscle Fatigue Resistance and Reverses Markers of Systemic Inflammation. Sci. Rep. 2021, 11, 12286. [Google Scholar] [CrossRef]
- McElroy, J.P.; Carmella, S.G.; Heskin, A.K.; Tang, M.K.; Murphy, S.E.; Reisinger, S.A.; Jensen, J.A.; Hatsukami, D.K.; Hecht, S.S.; Shields, P.G. Effects of Cessation of Cigarette Smoking on Eicosanoid Biomarkers of Inflammation and Oxidative Damage. PLoS ONE 2019, 14, e0218386. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-Inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in Collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Lu, Y.; Wei, J.; Wu, J.; Yang, J.; Cai, Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front. Immunol. 2020, 11, 609161. [Google Scholar] [CrossRef] [PubMed]
- Tilson, M.D. Decline of the Atherogenic Theory of the Etiology of the Abdominal Aortic Aneurysm and Rise of the Autoimmune Hypothesis. J. Vasc. Surg. 2016, 64, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Klopf, J.; Brostjan, C.; Neumayer, C.; Eilenberg, W. Neutrophils as Regulators and Biomarkers of Cardiovascular Inflammation in the Context of Abdominal Aortic Aneurysms. Biomedicines 2021, 9, 1236. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Oude Egbrink, M.G.; Heijnen, V.V.T.; Megens, R.T.A.; Engels, W.; Vink, H.; Slaaf, D.W.; van Zandvoort, M.A.M.J. Endothelial Glycocalyx Thickness and Platelet-Vessel Wall Interactions during Atherogenesis. Thromb. Haemost. 2011, 106, 939–946. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.M.J.; Oude Egbrink, M.G.A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Lipowsky, H.H. Protease Activity and the Role of the Endothelial Glycocalyx in Inflammation. Drug Discov. Today Dis. Models 2011, 8, 57. [Google Scholar] [CrossRef]
- van der Poll, T.; Parker, R.I. Platelet Activation and Endothelial Cell Dysfunction. Crit. Care Clin. 2020, 36, 233–253. [Google Scholar] [CrossRef]
- Dull, R.O.; Hahn, R.G. The Glycocalyx as a Permeability Barrier: Basic Science and Clinical Evidence. Crit. Care 2022, 26, 273. [Google Scholar] [CrossRef]
- Fels, B.; Kusche-Vihrog, K. It Takes More than Two to Tango: Mechanosignaling of the Endothelial Surface. Pflug. Arch. 2020, 472, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; O’Neil, G.L.; Harding, I.C.; Cheng, M.J.; Mensah, S.A.; Ebong, E.E. Glycocalyx in Atherosclerosis-Relevant Endothelium Function and as a Therapeutic Target. Curr. Atheroscler. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Cheng, Y.; Wu, W.; Yuan, L.; Liu, X. Glycocalyx Impairment in Vascular Disease: Focus on Inflammation. Front. Cell Dev. Biol. 2021, 9, 730621. [Google Scholar] [CrossRef]
- Panzer, B.; Kopp, C.W.; Neumayer, C.; Koppensteiner, R.; Jozkowicz, A.; Poledniczek, M.; Gremmel, T.; Jilma, B.; Wadowski, P.P. Toll-like Receptors as Pro-Thrombotic Drivers in Viral Infections: A Narrative Review. Cells 2023, 12, 1865. [Google Scholar] [CrossRef] [PubMed]
- Maschalidi, S.; Ravichandran, K.S. Phagocytosis: Sweet Repulsions via the Glycocalyx. Curr. Biol. 2021, 31, R20–R22. [Google Scholar] [CrossRef] [PubMed]
- Imbert, P.R.C.; Saric, A.; Pedram, K.; Bertozzi, C.R.; Grinstein, S.; Freeman, S.A. An Acquired and Endogenous Glycocalyx Forms a Bidirectional “Don’t Eat” and “Don’t Eat Me” Barrier to Phagocytosis. Curr. Biol. 2021, 31, 77–89.e5. [Google Scholar] [CrossRef] [PubMed]
- Marki, A.; Esko, J.D.; Pries, A.R.; Ley, K. Role of the Endothelial Surface Layer in Neutrophil Recruitment. J. Leukoc. Biol. 2015, 98, 503–515. [Google Scholar] [CrossRef]
- Möckl, L. The Emerging Role of the Mammalian Glycocalyx in Functional Membrane Organization and Immune System Regulation. Front. Cell Dev. Biol. 2020, 8, 253. [Google Scholar] [CrossRef]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef]
- Je, S.; Quan, H.; Yoon, Y.; Na, Y.; Kim, B.J.; Seok, S.H. Mycobacterium Massiliense Induces Macrophage Extracellular Traps with Facilitating Bacterial Growth. PLoS ONE 2016, 11, e0155685. [Google Scholar] [CrossRef]
- Fu, G.; Deng, M.; Neal, M.D.; Billiar, T.R.; Scott, M.J. Platelet-Monocyte Aggregates: Understanding Mechanisms and Functions in Sepsis. Shock 2021, 55, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Escaig, R.; Erber, J.; Nicolai, L. Neutrophil-Platelet Interactions as Novel Treatment Targets in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 8, 824112. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front. Immunol. 2017, 8, 928. [Google Scholar] [CrossRef]
- Banerjee, S.; Mwangi, J.G.; Stanley, T.K.; Mitra, R.; Ebong, E.E. Regeneration and Assessment of the Endothelial Glycocalyx to Address Cardiovascular Disease. Ind. Eng. Chem. Res. 2021, 60, 17328–17347. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [PubMed]
- Ofosu, F.A.; Dewar, L.; Craven, S.J.; Song, Y.; Cedrone, A.; Freedman, J.; Fenton, J.W. Coordinate Activation of Human Platelet Protease-Activated Receptor-1 and -4 in Response to Subnanomolar Alpha-Thrombin. J. Biol. Chem. 2008, 283, 26886–26893. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Pultar, J.; Weikert, C.; Eichelberger, B.; Panzer, B.; Huber, K.; Lang, I.M.; Koppensteiner, R.; Panzer, S.; Gremmel, T. Protease-Activated Receptor-Mediated Platelet Aggregation in Acute Coronary Syndrome Patients on Potent P2Y12 Inhibitors. Res. Pract. Thromb. Haemost. 2019, 3, 383–390. [Google Scholar] [CrossRef]
- Adam, F.; Guillin, M.C.; Jandrot-Perrus, M. Glycoprotein Ib-Mediated Platelet Activation. A Signalling Pathway Triggered by Thrombin. Eur. J. Biochem. 2003, 270, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-Like Receptors (TLRs): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef]
- Niklaus, M.; Klingler, P.; Weber, K.; Koessler, A.; Kuhn, S.; Boeck, M.; Kobsar, A.; Koessler, J. Platelet Toll-Like-Receptor-2 and -4 Mediate Different Immune-Related Responses to Bacterial Ligands. TH Open 2022, 6, e156–e167. [Google Scholar] [CrossRef]
- Salvador, B.; Arranz, A.; Francisco, S.; Córdoba, L.; Punzón, C.; Llamas, M.Á.; Fresno, M. Modulation of Endothelial Function by Toll like Receptors. Pharmacol. Res. 2016, 108, 46–56. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. DisSARMing Toll-like Receptor Signaling. Nat. Immunol. 2006, 7, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like Receptor Signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Schilling, D.; Thomas, K.; Nixdorff, K.; Vogel, S.N.; Fenton, M.J. Toll-Like Receptor 4 and Toll-IL-1 Receptor Domain-Containing Adapter Protein (TIRAP)/Myeloid Differentiation Protein 88 Adapter-Like (Mal) Contribute to Maximal IL-6 Expression in Macrophages. J. Immunol. 2002, 169, 5874–5880. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Goodbody, R.; Schröder, M.; Stack, J.; Moynagh, P.N.; Bowie, A.G. The Human Adaptor SARM Negatively Regulates Adaptor Protein TRIF–Dependent Toll-like Receptor Signaling. Nat. Immunol. 2006, 7, 1074–1081. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Rubio, D.; Xu, R.H.; Remakus, S.; Krouse, T.E.; Truckenmiller, M.E.; Thapa, R.J.; Balachandran, S.; Alcamí, A.; Norbury, C.C.; Sigal, L.J. Crosstalk between the Type 1 Interferon and Nuclear Factor Kappa B Pathways Confers Resistance to a Lethal Virus Infection. Cell Host Microbe 2013, 13, 701–710. [Google Scholar] [CrossRef]
- Ernst, O.; Vayttaden, S.J.; Fraser, I.D.C. Measurement of NF-ΚB Activation in TLR-Activated Macrophages. Methods Mol. Biol. 2018, 1714, 67–78. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-Induced NF-ΚB Activation Regulates NLRP3 Expression in Murine Macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Zimmer, J.; Sonawane, A.R.; Perez, K.; Turner, M.E.; Kuraoka, S.; Pham, T.; Li, F.; Aikawa, M.; Singh, S.; et al. NLRP3 Inflammasome Activation in Peripheral Arterial Disease. J. Am. Heart Assoc. 2023, 12, e026945. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.; Yu, T.; Chu, X.M. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Reilly, B.; Tan, C.; Wang, P.; Aziz, M. Extracellular CIRP Induces Macrophage Extracellular Trap Formation Via Gasdermin D Activation. Front. Immunol. 2021, 12, 780210. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; Bird, G.K.; la Flamme, A.C.; Harding, S.A.; Larsen, P.D. Platelets Modulate Multiple Markers of Neutrophil Function in Response to in Vitro Toll-like Receptor Stimulation. PLoS ONE 2019, 14, e0223444. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.E.; Scheiermann, C.; et al. Neutrophil Ageing Is Regulated by the Microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Mulivor, A.W.; Lipowsky, H.H. Inflammation- and Ischemia-Induced Shedding of Venular Glycocalyx. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1672–H1680. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arter. Thromb. Vasc. Biol. 2018, 38, 1427. [Google Scholar] [CrossRef]
- Lee, D.H.; Dane, M.J.C.; Van Den Berg, B.M.; Boels, M.G.S.; Van Teeffelen, J.W.; De Mutsert, R.; Den Heijer, M.; Rosendaal, F.R.; Van Der Vlag, J.; Van Zonneveld, A.J.; et al. Deeper Penetration of Erythrocytes into the Endothelial Glycocalyx Is Associated with Impaired Microvascular Perfusion. PLoS ONE 2014, 9, e96477. [Google Scholar] [CrossRef] [PubMed]
- Rabelink, T.J.; De Zeeuw, D. The Glycocalyx--Linking Albuminuria with Renal and Cardiovascular Disease. Nat. Rev. Nephrol. 2015, 11, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Liew, H.; Roberts, M.A.; MacGinley, R.; McMahon, L.P. Endothelial Glycocalyx in Health and Kidney Disease: Rising Star or False Dawn? Nephrology 2017, 22, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Kautzky-Willer, A.; Gremmel, T.; Koppensteiner, R.; Wolf, P.; Ertl, S.; Weikert, C.; Schörgenhofer, C.; Jilma, B. Sublingual Microvasculature in Diabetic Patients. Microvasc. Res. 2020, 129, 103971. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Pathophysiology of Diabetic Dyslipidemia. J. Atheroscler. Thromb. 2018, 25, 771–782. [Google Scholar] [CrossRef]
- Hagensen, M.K.; Mortensen, M.B.; Kjolby, M.; Palmfeldt, J.; Bentzon, J.F.; Gregersen, S. Increased Retention of LDL from Type 1 Diabetic Patients in Atherosclerosis-Prone Areas of the Murine Arterial Wall. Atherosclerosis 2019, 286, 156–162. [Google Scholar] [CrossRef]
- Singh, S.; Siva, B.V.; Ravichandiran, V. Advanced Glycation End Products: Key Player of the Pathogenesis of Atherosclerosis. Glycoconj. J. 2022, 39, 547–563. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric Oxide Orchestrates Metabolic Rewiring in M1 Macrophages by Targeting Aconitase 2 and Pyruvate Dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Koo, A.N.M.; Prag, H.A.; Krieg, T. Mitochondrial Redox and TCA Cycle Metabolite Signaling in the Heart. Free Radic. Biol. Med. 2021, 166, 287–296. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic Accumulation of Succinate Controls Reperfusion Injury through Mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-Deleon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K.; et al. M2 Macrophage Polarization Mediates Anti-Inflammatory Effects of Endothelial Nitric Oxide Signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Schaller, G.; Puttinger, H.; Födinger, M.; Kopp, C.W.; Seidinger, D.; Grisar, J.; Hörl, W.H.; Minar, E.; Vychytil, A.; et al. History of Cardiovascular Disease Is Associated with Endothelial Progenitor Cells in Peritoneal Dialysis Patients. Am. J. Kidney Dis. 2005, 46, 520–528. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Kohli, H.; Kumar, P. Multiple Therapeutic Effect of Endothelial Progenitor Cell Regulated by Drugs in Diabetes and Diabetes Related Disorder. J. Transl. Med. 2017, 15, 185. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Beck, E.B.; Adams, V.; Gielen, S.; Lenk, K.; Höllriegel, R.; Mangner, N.; Linke, A.; Erbs, S.; Möbius-Winkler, S.; et al. Maximal Exercise, Limb Ischemia, and Endothelial Progenitor Cells. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.P.; Meng, S. Vascular Regeneration in Peripheral Artery Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1627–1634. [Google Scholar] [CrossRef]
- Yamaguchi, J.I.; Kusano, K.F.; Masuo, O.; Kawamoto, A.; Silver, M.; Murasawa, S.; Bosch-Marce, M.; Masuda, H.; Losordo, D.W.; Isner, J.M.; et al. Stromal Cell-Derived Factor-1 Effects on Ex Vivo Expanded Endothelial Progenitor Cell Recruitment for Ischemic Neovascularization. Circulation 2003, 107, 1322–1328. [Google Scholar] [CrossRef]
- He, J.; Xiao, Z.; Chen, X.; Chen, M.; Fang, L.; Yang, M.; Lv, Q.; Li, Y.; Li, G.; Hu, J.; et al. The Expression of Functional Toll-like Receptor 4 Is Associated with Proliferation and Maintenance of Stem Cell Phenotype in Endothelial Progenitor Cells (EPCs). J. Cell Biochem. 2010, 111, 179–186. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Adams, V.; Walther, C.; Kleinecke, C.; Brugger, P.; Linke, A.; Walther, T.; Mohr, F.W.; Schuler, G. Reduced Number and Function of Endothelial Progenitor Cells in Patients with Aortic Valve Stenosis: A Novel Concept for Valvular Endothelial Cell Repair. Eur. Heart J. 2009, 30, 346–355. [Google Scholar] [CrossRef]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.D. The Role of Reactive Oxygen Species (ROS) in the Formation of Extracellular Traps (ETs) in Humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef]
- Ali, M.A.M.; Spinler, S.A. COVID-19 and Thrombosis: From Bench to Bedside. Trends Cardiovasc. Med. 2021, 31, 143–160. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Jilma, B.; Kopp, C.W.; Ertl, S.; Gremmel, T.; Koppensteiner, R. Glycocalyx as Possible Limiting Factor in COVID-19. Front. Immunol. 2021, 12, 607306. [Google Scholar] [CrossRef] [PubMed]
- Borrmann, M.; Brandes, F.; Kirchner, B.; Klein, M.; Billaud, J.N.; Reithmair, M.; Rehm, M.; Schelling, G.; Pfaffl, M.W.; Meidert, A.S. Extensive Blood Transcriptome Analysis Reveals Cellular Signaling Networks Activated by Circulating Glycocalyx Components Reflecting Vascular Injury in COVID-19. Front. Immunol. 2023, 14, 1129766. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial Dysfunction in COVID-19: An Overview of Evidence, Biomarkers, Mechanisms and Potential Therapies. Acta Pharmacol. Sin. 2023, 44, 695–709. [Google Scholar] [CrossRef]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-Derived Microvesicles: Important and Underappreciated Mediators of Cell-to-Cell Communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Chen, Y.T.; Yuan, H.X.; Ou, Z.J.; Ou, J.S. Microparticles (Exosomes) and Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 23. [Google Scholar] [CrossRef]
- Loyer, X.; Vion, A.C.; Tedgui, A.; Boulanger, C.M. Microvesicles as Cell-Cell Messengers in Cardiovascular Diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef]
- Février, B.; Raposo, G. Exosomes: Endosomal-Derived Vesicles Shipping Extracellular Messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, B.; Kuypers, S.; van den Akker, N.M.S.; Molin, D.G.M.; Michiels, L. Extracellular Vesicles Work as a Functional Inflammatory Mediator Between Vascular Endothelial Cells and Immune Cells. Front. Immunol. 2018, 9, 1789. [Google Scholar] [CrossRef]
- Wendt, S.; Goetzenich, A.; Goettsch, C.; Stoppe, C.; Bleilevens, C.; Kraemer, S.; Benstoem, C. Evaluation of the Cardioprotective Potential of Extracellular Vesicles—A Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 15702. [Google Scholar] [CrossRef]
- George, M.; Ganesh, M.R.; Sridhar, A.; Jena, A.; Rajaram, M.; Shanmugam, E.; Dhandapani, V.E. Evaluation of Endothelial and Platelet Derived Microparticles in Patients with Acute Coronary Syndrome. J. Clin. Diagn. Res. 2015, 9, OC09–OC13. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in Mice Lacking the Gene for Endothelial Nitric Oxide Synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural History and Histological Classification of Atherosclerotic Lesions: An Update. Arter. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal 2014, 20, 1126–1167. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated with Cardiovascular and Metabolic Diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial Reactive Oxygen Species Promote Production of Proinflammatory Cytokines and Are Elevated in TNFR1-Associated Periodic Syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef]
- Ryan, K.A.; Smith, M.F.; Sanders, M.K.; Ernst, P.B. Reactive Oxygen and Nitrogen Species Differentially Regulate Toll-Like Receptor 4-Mediated Activation of NF-ΚB and Interleukin-8 Expression. Infect. Immun. 2004, 72, 2123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Qiu, Q.; Yang, Z.; Cao, F.; Yang, C.; Hardy, P.; Yan, X.; Yang, S.; Xiong, W. Activation of NLRP3 Inflammasome by Lymphocytic Microparticles via TLR4 Pathway Contributes to Airway Inflammation. Exp. Cell Res. 2020, 386, 111737. [Google Scholar] [CrossRef] [PubMed]
- Jerez-Dolz, D.; Torramade-Moix, S.; Palomo, M.; Moreno-Castaño, A.; Lopez-Vilchez, I.; Hernandez, R.; Badimon, J.J.; Zafar, M.U.; Diaz-Ricart, M.; Escolar, G. Internalization of Microparticles by Platelets Is Partially Mediated by Toll-like Receptor 4 and Enhances Platelet Thrombogenicity. Atherosclerosis 2020, 294, 17–24. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, R.; Chen, Y.; Wang, M.; Du, J. Crosstalk between Oxidative Stress and Exosomes. Oxid. Med. Cell Longev. 2022, 2022, 3553617. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Franklin, B.S.; Hoelscher, M.; Schmitz, T.; Bedorf, J.; Nickenig, G.; Werner, N. High Glucose Condition Increases NADPH Oxidase Activity in Endothelial Microparticles That Promote Vascular Inflammation. Cardiovasc. Res. 2013, 98, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Mause, S.F.; Weber, C. Microparticles: Protagonists of a Novel Communication Network for Intercellular Information Exchange. Circ. Res. 2010, 107, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Ci, H.B.; Ou, Z.J.; Chang, F.J.; Liu, D.H.; He, G.W.; Xu, Z.; Yuan, H.Y.; Wang, Z.P.; Zhang, X.; Ou, J.S. Endothelial Microparticles Increase in Mitral Valve Disease and Impair Mitral Valve Endothelial Function. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E695–E702. [Google Scholar] [CrossRef] [PubMed]
- Densmore, J.C.; Signorino, P.R.; Ou, J.; Hatoum, O.A.; Rowe, J.J.; Shi, Y.; Kaul, S.; Jones, D.W.; Sabina, R.E.; Pritchard, K.A.; et al. Endothelium-Derived Microparticles Induce Endothelial Dysfunction and Acute Lung Injury. Shock 2006, 26, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, M.; Rozalski, M.; Luzak, B.; Michalak, M.; Ambrosius, W.; Watala, C.; Kozubski, W. Enhanced Platelet-Derived Microparticle Formation Is Associated with Carotid Atherosclerosis in Convalescent Stroke Patients. Platelets 2013, 24, 63–70. [Google Scholar] [CrossRef]
- Lin, Z.B.; Ci, H.B.; Li, Y.; Cheng, T.P.; Liu, D.H.; Wang, Y.S.; Xu, J.; Yuan, H.X.; Li, H.M.; Chen, J.; et al. Endothelial Microparticles Are Increased in Congenital Heart Diseases and Contribute to Endothelial Dysfunction. J. Transl. Med. 2017, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Wang, X.; Liu, X.; Du, H.; Sun, C.; Shao, X.; Tian, J.; Gu, X.; Wang, H.; Tian, J.; et al. Adipose-Derived Exosomes Exert Proatherogenic Effects by Regulating Macrophage Foam Cell Formation and Polarization. J. Am. Heart Assoc. 2018, 7, e007442. [Google Scholar] [CrossRef]
- Blaser, M.C.; Aikawa, E. Differential MiRNA Loading Underpins Dual Harmful and Protective Roles for Extracellular Vesicles in Atherogenesis. Circ. Res. 2019, 124, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, S.; Zhang, F.; Wu, M.; Liang, H.; Song, J.; Lee, C.; Chen, H. Endothelial Microparticles-Mediated Transfer of MicroRNA-19b Promotes Atherosclerosis via Activating Perivascular Adipose Tissue Inflammation in ApoE-/- Mice. Biochem. Biophys. Res. Commun. 2018, 495, 1922–1929. [Google Scholar] [CrossRef]
- Ceolotto, G.; Giannella, A.; Albiero, M.; Kuppusamy, M.; Radu, C.; Simioni, P.; Garlaschelli, K.; Baragetti, A.; Catapano, A.L.; Iori, E.; et al. MiR-30c-5p Regulates Macrophage-Mediated Inflammation and pro-Atherosclerosis Pathways. Cardiovasc. Res. 2017, 113, 1627–1638. [Google Scholar] [CrossRef]
- Pereira-Da-silva, T.; Napoleão, P.; Costa, M.C.; Gabriel, A.F.; Selas, M.; Silva, F.; Enguita, F.J.; Ferreira, R.C.; Carmo, M.M. Cigarette Smoking, MiR-27b Downregulation, and Peripheral Artery Disease: Insights into the Mechanisms of Smoking Toxicity. J. Clin. Med. 2021, 10, 890. [Google Scholar] [CrossRef]
- Badacz, R.; Kleczyński, P.; Legutko, J.; Żmudka, K.; Gacoń, J.; Przewłocki, T.; Kabłak-Ziembicka, A. Expression of MiR-1-3p, MiR-16-5p and MiR-122-5p as Possible Risk Factors of Secondary Cardiovascular Events. Biomedicines 2021, 9, 1055. [Google Scholar] [CrossRef]
- Wronska, A.; Kurkowska-Jastrzebska, I.; Santulli, G. Application of MicroRNAs in Diagnosis and Treatment of Cardiovascular Disease. Acta Physiol. 2015, 213, 60–83. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, W.; Zhang, L.; Wang, L.; Li, J.; Shu, C.; Li, X. Roles of MicroRNAs in Peripheral Artery In-Stent Restenosis after Endovascular Treatment. Biomed. Res. Int. 2021, 2021, 9935671. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 Complexes Carry a Population of Circulating MicroRNAs Independent of Vesicles in Human Plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs Are Transported in Plasma and Delivered to Recipient Cells by High-Density Lipoproteins. Nat. Cell Biol. 2011, 13, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Stojkovic, S.; Jurisic, M.; Kopp, C.W.; Koppensteiner, R.; Huber, K.; Wojta, J.; Gremmel, T. Circulating MicroRNAs Identify Patients at Increased Risk of In-Stent Restenosis after Peripheral Angioplasty with Stent Implantation. Atherosclerosis 2018, 269, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Badacz, R.; Przewłocki, T.; Legutko, J.; Żmudka, K.; Kabłak-Ziembicka, A. MicroRNAs Associated with Carotid Plaque Development and Vulnerability: The Clinician’s Perspective. Int. J. Mol. Sci. 2022, 23, 15645. [Google Scholar] [CrossRef]
- Stojkovic, S.; Wadowski, P.P.; Haider, P.; Weikert, C.; Pultar, J.; Lee, S.; Eichelberger, B.; Hengstenberg, C.; Wojta, J.; Panzer, S.; et al. Circulating MicroRNAs and Monocyte-Platelet Aggregate Formation in Acute Coronary Syndrome. Thromb. Haemost. 2021, 121, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Finn, N.A.; Eapen, D.; Manocha, P.; Al Kassem, H.; Lassegue, B.; Ghasemzadeh, N.; Quyyumi, A.; Searles, C.D. Coronary Heart Disease Alters Intercellular Communication by Modifying Microparticle-Mediated MicroRNA Transport. FEBS Lett. 2013, 587, 3456–3463. [Google Scholar] [CrossRef]
- Alexandru, N.; Andrei, E.; Niculescu, L.; Dragan, E.; Ristoiu, V.; Georgescu, A. Microparticles of Healthy Origins Improve Endothelial Progenitor Cell Dysfunction via MicroRNA Transfer in an Atherosclerotic Hamster Model. Acta Physiol. 2017, 221, 230–249. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Rada, B. Neutrophil Extracellular Traps. Methods Mol. Biol. 2019, 1982, 517–528. [Google Scholar] [CrossRef]
- Schönrich, G.; Raftery, M.J. Neutrophil Extracellular Traps Go Viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B.; Arneth, R. Neutrophil Extracellular Traps (NETs) and Vasculitis. Int. J. Med. Sci. 2021, 18, 1532–1540. [Google Scholar] [CrossRef]
- Nappi, F.; Bellomo, F.; Avtaar Singh, S.S. Worsening Thrombotic Complication of Atherosclerotic Plaques Due to Neutrophils Extracellular Traps: A Systematic Review. Biomedicines 2023, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated Endothelial Cells Induce Neutrophil Extracellular Traps and Are Susceptible to NETosis-Mediated Cell Death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase, Modified Lipoproteins, and Atherogenesis. J. Lipid Res. 2009, 50, S346–S351. [Google Scholar] [CrossRef]
- Alfaidi, M.; Wilson, H.; Daigneault, M.; Burnett, A.; Ridger, V.; Chamberlain, J.; Francis, S. Neutrophil Elastase Promotes Interleukin-1β Secretion from Human Coronary Endothelium. J. Biol. Chem. 2015, 290, 24067–24078. [Google Scholar] [CrossRef]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase Is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef]
- Brodsky, S.V.; Zhang, F.; Nasjletti, A.; Goligorsky, M.S. Endothelium-Derived Microparticles Impair Endothelial Function in Vitro. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1910–H1915. [Google Scholar] [CrossRef]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL Induced Extracellular Trap Formation in Human Neutrophils via TLR-PKC-IRAK-MAPK and NADPH-Oxidase Activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil Extracellular Traps License Macrophages for Cytokine Production in Atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil Extracellular Trap-Derived Enzymes Oxidize High-Density Lipoprotein: An Additional Proatherogenic Mechanism in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef]
- Sharma, S.; Hofbauer, T.M.; Ondracek, A.S.; Chausheva, S.; Alimohammadi, A.; Artner, T.; Panzenboeck, A.; Rinderer, J.; Shafran, I.; Mangold, A.; et al. Neutrophil Extracellular Traps Promote Fibrous Vascular Occlusions in Chronic Thrombosis. Blood 2021, 137, 1104–1116. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Gillum, R.F. Epidemiology of Aortic Aneurysm in the United States. J. Clin. Epidemiol. 1995, 48, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Al-Balah, A.; Goodall, R.; Salciccioli, J.D.; Marshall, D.C.; Shalhoub, J. Mortality from Abdominal Aortic Aneurysm: Trends in European Union 15+ Countries from 1990 to 2017. Br. J. Surg. 2020, 107, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Treska, V.; Kocova, J.; Boudova, L.; Neprasova, P.; Topolcan, O.; Pecen, L.; Tonar, Z. Inflammation in the Wall of Abdominal Aortic Aneurysm and Its Role in the Symptomatology of Aneurysm. Cytokines Cell Mol. Ther. 2002, 7, 91–97. [Google Scholar] [CrossRef]
- Beckman, E.N. Plasma Cell Infiltrates in Atherosclerotic Abdominal Aortic Aneurysms. Am. J. Clin. Pathol. 1986, 85, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Newmans, K.M.; Malon, A.M.; Shin, R.D.; Scholes, J.V.; Ramey, W.G.; Tilson, M.D. Matrix Metalloproteinases in Abdominal Aortic Aneurysm: Characterization, Purification, and Their Possible Sources. Connect. Tissue Res. 1994, 30, 265–276. [Google Scholar] [CrossRef]
- Reilly, J.M.; Brophy, C.M.; Tilson, M.D. Characterization of an Elastase from Aneurysmal Aorta Which Degrades Intact Aortic Elastin. Ann. Vasc. Surg. 1992, 6, 499–502. [Google Scholar] [CrossRef]
- Brown, P.M.; Zelt, D.T.; Sobolev, B.; Hallett, J.W.; Sternbach, Y. The Risk of Rupture in Untreated Aneurysms: The Impact of Size, Gender, and Expansion Rate. J. Vasc. Surg. 2003, 37, 280–284. [Google Scholar] [CrossRef]
- Chaikof, E.L.; Dalman, R.L.; Eskandari, M.K.; Jackson, B.M.; Lee, W.A.; Mansour, M.A.; Mastracci, T.M.; Mell, M.; Murad, M.H.; Nguyen, L.L.; et al. The Society for Vascular Surgery Practice Guidelines on the Care of Patients with an Abdominal Aortic Aneurysm. J. Vasc. Surg. 2018, 67, 2–77.e2. [Google Scholar] [CrossRef]
- Brady, A.R.; Thompson, S.G.; Fowkes, F.G.R.; Greenhalgh, R.M.; Powell, J.T. Abdominal Aortic Aneurysm Expansion: Risk Factors and Time Intervals for Surveillance. Circulation 2004, 110, 16–21. [Google Scholar] [CrossRef]
- Kronmal, R.A.; McClelland, R.L.; Detrano, R.; Shea, S.; Lima, J.A.; Cushman, M.; Bild, D.E.; Burke, G.L. Risk Factors for the Progression of Coronary Artery Calcification in Asymptomatic Subjects. Circulation 2007, 115, 2722–2730. [Google Scholar] [CrossRef]
- Liabeuf, S.; Olivier, B.; Vemeer, C.; Theuwissen, E.; Magdeleyns, E.; Aubert, C.E.; Brazier, M.; Mentaverri, R.; Hartemann, A.; Massy, Z.A. Vascular Calcification in Patients with Type 2 Diabetes: The Involvement of Matrix Gla Protein. Cardiovasc. Diabetol. 2014, 13, 85. [Google Scholar] [CrossRef]
- Leow, K.; Szulc, P.; Schousboe, J.T.; Kiel, D.P.; Teixeira-Pinto, A.; Shaikh, H.; Sawang, M.; Sim, M.; Bondonno, N.; Hodgson, J.M.; et al. Prognostic Value of Abdominal Aortic Calcification: A Systematic Review and Meta-Analysis of Observational Studies. J. Am. Heart Assoc. 2021, 10, e017205. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Targher, G.; Zoppini, G.; Cicoira, M.; Bonapace, S.; Negri, C.; Stoico, V.; Faggiano, P.; Vassanelli, C.; Bonora, E. Aortic and Mitral Annular Calcifications Are Predictive of All-Cause and Cardiovascular Mortality in Patients with Type 2 Diabetes. Diabetes Care 2012, 35, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Shao, J.; Yu, B.; Liu, G.; Wang, R.; Dong, H.; Che, H.; Li, L. Association Between Metformin and Abdominal Aortic Aneurysm: A Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 908747. [Google Scholar] [CrossRef] [PubMed]
- Limiting AAA with Metformin (LIMIT) Trial—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04500756?cond=abdominal+aortic+aneurysm+metformin&draw=2&rank=3 (accessed on 5 March 2023).
- Metformin Therapy in Non-Diabetic AAA Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03507413?cond=abdominal+aortic+aneurysm+metformin&draw=2&rank=1 (accessed on 5 March 2023).
- Metformin for Abdominal Aortic Aneurysm Growth Inhibition—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04224051?cond=abdominal+aortic+aneurysm+metformin&draw=2&rank=2 (accessed on 5 March 2023).
- Klopf, J.; Fuchs, L.; Schernthaner, R.; Domenig, C.M.; Gollackner, B.; Brostjan, C.; Neumayer, C.; Eilenberg, W. The Prognostic Impact of Vascular Calcification on Abdominal Aortic Aneurysm Progression. J. Vasc. Surg. 2022, 75, 1926–1934. [Google Scholar] [CrossRef]
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.J.; Gelijns, A.C.; Greco, G. Analysis of Risk Factors for Abdominal Aortic Aneurysm in a Cohort of More than 3 Million Individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Akutsu, K.; Tamori, Y.; Sakamoto, S.; Yoshimuta, T.; Hashimoto, H.; Takeshita, S. Differences in Atherosclerotic Profiles Between Patients with Thoracic and Abdominal Aortic Aneurysms. Am. J. Cardiol. 2008, 101, 696–699. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, J.; Gao, Y. Targeting Platelet Activation in Abdominal Aortic Aneurysm: Current Knowledge and Perspectives. Biomolecules 2022, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Clément, M.; Hassen-Khodja, R.; Chinetti, G.; Mallat, Z. Monocytes and Macrophages in Abdominal Aortic Aneurysm. Nat. Rev. Cardiol. 2017, 14, 457–471. [Google Scholar] [CrossRef]
- Houard, X.; Touat, Z.; Ollivier, V.; Louedec, L.; Philippe, M.; Sebbag, U.; Meilhac, O.; Rossignol, P.; Michel, J.B. Mediators of Neutrophil Recruitment in Human Abdominal Aortic Aneurysms. Cardiovasc. Res. 2009, 82, 532–541. [Google Scholar] [CrossRef]
- Thompson, R.W.; Curci, J.A.; Ennis, T.L.; Mao, D.; Pagano, M.B.; Pham, C.T.N. Pathophysiology of Abdominal Aortic Aneurysms: Insights from the Elastase-Induced Model in Mice with Different Genetic Backgrounds. Ann. N. Y. Acad. Sci. 2006, 1085, 59–73. [Google Scholar] [CrossRef]
- Rao, J.; Brown, B.N.; Weinbaum, J.S.; Ofstun, E.L.; Makaroun, M.S.; Humphrey, J.D.; Vorp, D.A. Distinct Macrophage Phenotype and Collagen Organization within the Intraluminal Thrombus of Abdominal Aortic Aneurysm. J. Vasc. Surg. 2015, 62, 585–593. [Google Scholar] [CrossRef]
- Dutertre, C.A.; Clement, M.; Morvan, M.; Schäkel, K.; Castier, Y.; Alsac, J.M.; Michel, J.B.; Nicoletti, A. Deciphering the Stromal and Hematopoietic Cell Network of the Adventitia from Non-Aneurysmal and Aneurysmal Human Aorta. PLoS ONE 2014, 9, e89983. [Google Scholar] [CrossRef]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A.; Brasier, A.R.; Tilton, R.G. Aortic Adventitial Fibroblasts Participate in Angiotensin-Induced Vascular Wall Inflammation and Remodeling. J. Vasc. Res. 2011, 48, 261–272. [Google Scholar] [CrossRef]
- Tieu, B.C.; Lee, C.; Sun, H.; LeJeune, W.; Recinos, A.; Ju, X.; Spratt, H.; Guo, D.C.; Milewicz, D.; Tilton, R.G.; et al. An Adventitial IL-6/MCP1 Amplification Loop Accelerates Macrophage-Mediated Vascular Inflammation Leading to Aortic Dissection in Mice. J. Clin. Investig. 2009, 119, 3637–3651. [Google Scholar] [CrossRef]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined Inhibition of CCL2, CX3CR1, and CCR5 Abrogates Ly6C(Hi) and Ly6C(Lo) Monocytosis and Almost Abolishes Atherosclerosis in Hypercholesterolemic Mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II Promotes Atherosclerotic Lesions and Aneurysms in Apolipoprotein E-Deficient Mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, K.; Chen, F.; Liu, Q.; Ni, J.; Cao, W.; Hua, Y.; He, F.; Liu, Z.; Li, L.; et al. Role of the CCL2-CCR2 Axis in Cardiovascular Disease: Pathogenesis and Clinical Implications. Front. Immunol. 2022, 13, 975367. [Google Scholar] [CrossRef]
- Roshan, M.H.K.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflamm. 2016, 2016, 1532832. [Google Scholar] [CrossRef]
- Yang, M.; Chen, Q.; Mei, L.; Wen, G.; An, W.; Zhou, X.; Niu, K.; Liu, C.; Ren, M.; Sun, K.; et al. Neutrophil Elastase Promotes Neointimal Hyperplasia by Targeting Toll-like Receptor 4 (TLR4)-NF-ΚB Signalling. Br. J. Pharmacol. 2021, 178, 4048–4068. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Dalli, J.; Montero-Melendez, T.; Norling, L.V.; Yin, X.; Hinds, C.; Haskard, D.; Mayr, M.; Perretti, M. Heterogeneity in Neutrophil Microparticles Reveals Distinct Proteome and Functional Properties. Mol. Cell Proteom. 2013, 12, 2205–2219. [Google Scholar] [CrossRef]
- Mortaz, E.; Alipoor, S.D.; Adcock, I.M.; Mumby, S.; Koenderman, L. Update on Neutrophil Function in Severe Inflammation. Front. Immunol. 2018, 9, 2171. [Google Scholar] [CrossRef]
- Klopf, J.; Brostjan, C.; Eilenberg, W.; Neumayer, C. Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int. J. Mol. Sci. 2021, 22, 559. [Google Scholar] [CrossRef]
- Arbănași, E.M.; Mureșan, A.V.; Coșarcă, C.M.; Arbănași, E.M.; Niculescu, R.; Voidăzan, S.T.; Ivănescu, A.D.; Hălmaciu, I.; Filep, R.C.; Mărginean, L.; et al. Computed Tomography Angiography Markers and Intraluminal Thrombus Morphology as Predictors of Abdominal Aortic Aneurysm Rupture. Int. J. Environ. Res. Public Health 2022, 19, 15961. [Google Scholar] [CrossRef] [PubMed]
- Behr-Rasmussen, C.; Grøndal, N.; Bramsen, M.B.; Thomsen, M.D.; Lindholt, J.S. Mural Thrombus and the Progression of Abdominal Aortic Aneurysms: A Large Population-Based Prospective Cohort Study. Eur. J. Vasc. Endovasc. Surg. 2014, 48, 301–307. [Google Scholar] [CrossRef]
- Kazi, M.; Thyberg, J.; Religa, P.; Roy, J.; Eriksson, P.; Hedin, U.; Swedenborg, J. Influence of Intraluminal Thrombus on Structural and Cellular Composition of Abdominal Aortic Aneurysm Wall. J. Vasc. Surg. 2003, 38, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Schrottmaier, W.C.; Mussbacher, M.; Salzmann, M.; Assinger, A. Platelet-Leukocyte Interplay during Vascular Disease. Atherosclerosis 2020, 307, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, D.A.; Yin, W. Platelet-Activation Mechanisms and Vascular Remodeling. Compr. Physiol. 2018, 8, 1117–1156. [Google Scholar] [CrossRef]
- Houard, X.; Ollivier, V.; Louedec, L.; Michel, J.; Back, M. Differential Inflammatory Activity across Human Abdominal Aortic Aneurysms Reveals Neutrophil-Derived Leukotriene B4 as a Major Chemotactic Factor Released from the Intraluminal Thrombus. FASEB J. 2009, 23, 1376–1383. [Google Scholar] [CrossRef]
- Karaolanis, G.; Moris, D.; Palla, V.V.; Karanikola, E.; Bakoyiannis, C.; Georgopoulos, S. Neutrophil Gelatinase Associated Lipocalin (NGAL) as a Biomarker. Does It Apply in Abdominal Aortic Aneurysms? A Review of Literature. Indian J. Surg. 2015, 77 (Suppl. S3), 1313–1317. [Google Scholar] [CrossRef]
- Petersen, E.; Wågberg, F.; Ängquist, K.A. Serum Concentrations of Elastin-Derived Peptides in Patients with Specific Manifestations of Atherosclerotic Disease. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 440–444. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals 2019, 12, 118. [Google Scholar] [CrossRef]
- Hendy, K.; Gunnarson, R.; Golledge, J. Growth Rates of Small Abdominal Aortic Aneurysms Assessed by Computerised Tomography—A Systematic Literature Review. Atherosclerosis 2014, 235, 182–188. [Google Scholar] [CrossRef]
- Selders, G.S.; Fetz, A.E.; Radic, M.Z.; Bowlin, G.L. An Overview of the Role of Neutrophils in Innate Immunity, Inflammation and Host-Biomaterial Integration. Regen. Biomater. 2017, 4, 55–68. [Google Scholar] [CrossRef]
- Yan, H.; Zhou, H.F.; Akk, A.; Hu, Y.; Springer, L.E.; Ennis, T.L.; Pham, C.T.N. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arter. Thromb. Vasc. Biol. 2016, 36, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Delbosc, S.; Alsac, J.M.; Journe, C.; Louedec, L.; Castier, Y.; Bonnaure-Mallet, M.; Ruimy, R.; Rossignol, P.; Bouchard, P.; Michel, J.B.; et al. Porphyromonas Gingivalis Participates in Pathogenesis of Human Abdominal Aortic Aneurysm by Neutrophil Activation. Proof of Concept in Rats. PLoS ONE 2011, 6, e18679. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska, A.; Zagrapan, B.; Paradowska, E.; Neumayer, C.; Eilenberg, W.; Brostjan, C.; Klinger, M.; Nanobachvili, J.; Huk, I. Abdominal Aortic Aneurysm and Virus Infection: A Potential Causative Role for Cytomegalovirus Infection? J. Med. Virol. 2021, 93, 5017–5024. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas Gingivalis: Major Periodontopathic Pathogen Overview. J. Immunol. Res. 2014, 2014, 476068. [Google Scholar] [CrossRef]
- Salhi, L.; Rijkschroeff, P.; Van Hede, D.; Laine, M.L.; Teughels, W.; Sakalihasan, N.; Lambert, F. Blood Biomarkers and Serologic Immunological Profiles Related to Periodontitis in Abdominal Aortic Aneurysm Patients. Front. Cell Infect. Microbiol. 2022, 11, 766462. [Google Scholar] [CrossRef] [PubMed]
- Salhi, L.; Sakalihasan, N.; Okroglic, A.G.; Labropoulos, N.; Seidel, L.; Albert, A.; Teughels, W.; Defraigne, J.O.; Lambert, F. Further Evidence on the Relationship between Abdominal Aortic Aneurysm and Periodontitis: A Cross-Sectional Study. J. Periodontol. 2020, 91, 1453–1464. [Google Scholar] [CrossRef]
- Salhi, L.; Rompen, E.; Sakalihasan, N.; Laleman, I.; Teughels, W.; Michel, J.B.; Lambert, F. Can Periodontitis Influence the Progression of Abdominal Aortic Aneurysm? A Systematic Review. Angiology 2019, 70, 479–491. [Google Scholar] [CrossRef]
- Gredmark-Russ, S.; Dzabic, M.; Rahbar, A.; Wanhainen, A.; Björck, M.; Larsson, E.; Michel, J.B.; Söderberg-Nauclér, C. Active Cytomegalovirus Infection in Aortic Smooth Muscle Cells from Patients with Abdominal Aortic Aneurysm. J. Mol. Med. 2009, 87, 347–356. [Google Scholar] [CrossRef]
- Pinard, A.; Jones, G.T.; Milewicz, D.M. Genetics of Thoracic and Abdominal Aortic Diseases: Aneurysms, Dissections, and Ruptures. Circ. Res. 2019, 124, 588. [Google Scholar] [CrossRef]
- La Rocca, G.; Del Frate, G.; Delvino, P.; Di Cianni, F.; Moretti, M.; Italiano, N.; Treppo, E.; Monti, S.; Talarico, R.; Ferro, F.; et al. Systemic Vasculitis: One Year in Review 2022. Clin. Exp. Rheumatol. 2022, 40, 673–687. [Google Scholar] [CrossRef]
- Phillip, R.; Luqmani, R. Mortality in Systemic Vasculitis: A Systematic Review. Clin. Exp. Rheumatol. 2008, 26 (Suppl. S51), S94–S104. [Google Scholar]
- Wallace, Z.S.; Fu, X.; Harkness, T.; Stone, J.H.; Zhang, Y.; Choi, H. All-Cause and Cause-Specific Mortality in ANCA-Associated Vasculitis: Overall and According to ANCA Type. Rheumatology 2020, 59, 2308–2315. [Google Scholar] [CrossRef]
- Hill, C.L.; Black, R.J.; Nossent, J.C.; Ruediger, C.; Nguyen, L.; Ninan, J.V.; Lester, S. Risk of Mortality in Patients with Giant Cell Arteritis: A Systematic Review and Meta-Analysis. Semin. Arthritis Rheum. 2017, 46, 513–519. [Google Scholar] [CrossRef]
- Mueller, M.; Gschwandtner, M.E.; Gamper, J.; Giurgea, G.A.; Kiener, H.P.; Perkmann, T.; Koppensteiner, R.; Schlager, O. Chronic Inflammation Predicts Long-Term Mortality in Patients with Raynaud’s Phenomenon. J. Intern. Med. 2018, 283, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.; Pagnoux, C.; Simon, A.; Pasquinelli-Balice, M.; Del-Pino, M.; Gariepy, J.; Guillevin, L. Increased Prevalence of Subclinical Atherosclerosis in Patients with Small-Vessel Vasculitis. Heart 2007, 93, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Farrah, T.E.; Melville, V.; Czopek, A.; Fok, H.; Bruce, L.; Mills, N.L.; Bailey, M.A.; Webb, D.J.; Dear, J.W.; Dhaun, N. Arterial Stiffness, Endothelial Dysfunction and Impaired Fibrinolysis Are Pathogenic Mechanisms Contributing to Cardiovascular Risk in ANCA-Associated Vasculitis. Kidney Int. 2022, 102, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Clifford, A.H.; Cohen Tervaert, J.W. Cardiovascular Events and the Role of Accelerated Atherosclerosis in Systemic Vasculitis. Atherosclerosis 2021, 325, 8–15. [Google Scholar] [CrossRef]
- Hilhorst, M.; Winckers, K.; Wilde, B.; Van Oerle, R.; Ten Cate, H.; Tervaert, J.W.C. Patients with Antineutrophil Cytoplasmic Antibodies Associated Vasculitis in Remission Are Hypercoagulable. J. Rheumatol. 2013, 40, 2042–2046. [Google Scholar] [CrossRef]
- De Leeuw, K.; Sanders, J.S.; Stegeman, C.; Smit, A.; Kallenberg, C.G.; Bijl, M. Accelerated Atherosclerosis in Patients with Wegener’s Granulomatosis. Ann. Rheum. Dis. 2005, 64, 753–759. [Google Scholar] [CrossRef]
- Shirai, T.; Hilhorst, M.; Harrison, D.G.; Goronzy, J.J.; Weyand, C.M. Macrophages in Vascular Inflammation--From Atherosclerosis to Vasculitis. Autoimmunity 2015, 48, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Wallace, Z.S.; Fu, X.; Liao, K.; Kallenberg, C.G.M.; Langford, C.A.; Merkel, P.A.; Monach, P.; Seo, P.; Specks, U.; Spiera, R.; et al. Disease Activity, Antineutrophil Cytoplasmic Antibody Type, and Lipid Levels in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2019, 71, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Proven, A.; Gabriel, S.E.; Orces, C.; Michael O’Fallon, W.; Hunder, G.G. Glucocorticoid Therapy in Giant Cell Arteritis: Duration and Adverse Outcomes. Arthritis Rheum. 2003, 49, 703–708. [Google Scholar] [CrossRef]
- Bramlage, C.P.; Kröplin, J.; Wallbach, M.; Minguet, J.; Smith, K.H.; Lüders, S.; Schrader, J.; Patschan, S.; Gross, O.; Deutsch, C.; et al. Management of Cardiovascular Risk Factors in Patients with ANCA-Associated Vasculitis. J. Eval. Clin. Pract. 2017, 23, 747–754. [Google Scholar] [CrossRef]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2022 Update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef]
- Ertus, C.; Scailteux, L.-M.; Lescoat, A.; Berthe, P.; Auffret, V.; Dupuy, A.; Oger, E.; Droitcourt, C. Major Adverse Cardiovascular Events in Patients Treated with Oral Janus Kinase Inhibitors for Atopic Dermatitis: A Systematic Review and Meta-Analysis. Br. J. Dermatol. 2023, 6, ljad229. [Google Scholar] [CrossRef] [PubMed]
- Hoisnard, L.; Pina Vegas, L.; Dray-Spira, R.; Weill, A.; Zureik, M.; Sbidian, E. Risk of Major Adverse Cardiovascular and Venous Thromboembolism Events in Patients with Rheumatoid Arthritis Exposed to JAK Inhibitors versus Adalimumab: A Nationwide Cohort Study. Ann. Rheum. Dis. 2023, 82, 182–188. [Google Scholar] [CrossRef]
- Cooke, J.P. Inflammation and Its Role in Regeneration and Repair: A Caution for Novel Anti-Inflammatory Therapies. Circ. Res. 2019, 124, 1166. [Google Scholar] [CrossRef]
- Mantsounga, C.S.; Lee, C.; Neverson, J.; Sharma, S.; Healy, A.; Berus, J.M.; Parry, C.; Ceneri, N.M.; López-Giráldez, F.; Chun, H.J.; et al. Macrophage IL-1β Promotes Arteriogenesis by Autocrine STAT3- and NF-ΚB-Mediated Transcription of pro-Angiogenic VEGF-A. Cell Rep. 2022, 38, 110309. [Google Scholar] [CrossRef]
- Heil, M.; Eitenmüller, I.; Schmitz-Rixen, T.; Schaper, W. Arteriogenesis versus Angiogenesis: Similarities and Differences. J. Cell Mol. Med. 2006, 10, 45. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Heil, M.; Ziegelhoeffer, T.; Wagner, S.; Fernández, B.; Helisch, A.; Martin, S.; Tribulova, S.; Kuziel, W.A.; Bachmann, G.; Schaper, W. Collateral Artery Growth (Arteriogenesis) after Experimental Arterial Occlusion Is Impaired in Mice Lacking CC-Chemokine Receptor-2. Circ. Res. 2004, 94, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Heuslein, J.L.; Meisner, J.K.; Li, X.; Song, J.; Vincentelli, H.; Leiphart, R.J.; Ames, E.G.; Blackman, B.R.; Price, R.J. Mechanisms of Amplified Arteriogenesis in Collateral Artery Segments Exposed to Flow Direction Reversal. Arter. Thromb. Vasc. Biol. 2015, 35, 2354. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C.; Locati, M. Macrophage Diversity and Polarization in Atherosclerosis: A Question of Balance. Arter. Thromb. Vasc. Biol. 2009, 29, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 Is Required for Tumor Invasiveness and Angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Okigaki, M.; Adachi, Y.; Fujiyama, S.; Mori, Y.; Kosaki, A.; Iwasaka, T.; Matsubara, H. Mechanism for IL-1β-Mediated Neovascularization Unmasked by IL-1β Knock-out Mice. J. Mol. Cell Cardiol. 2004, 36, 469–480. [Google Scholar] [CrossRef]
- De Winther, M.P.J.; Van Dijk, K.W.; Havekes, L.M.; Hofker, M.H. Macrophage Scavenger Receptor Class A: A Multifunctional Receptor in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2000, 20, 290–297. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, a Scavenger Receptor Implicated in Atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, S.; Li, B.; Sun, A.; Zou, Y.; Ge, J. A Protective Role of Ciglitazone in Ox-LDL-Induced Rat Microvascular Endothelial Cells via Modulating PPARγ-Dependent AMPK/ENOS Pathway. J. Cell Mol. Med. 2015, 19, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Michell, B.J.; Chen, Z.P.; Tiganis, T.; Stapleton, D.; Katsis, F.; Power, D.A.; Sim, A.T.; Kemp, B.E. Coordinated Control of Endothelial Nitric-Oxide Synthase Phosphorylation by Protein Kinase C and the CAMP-Dependent Protein Kinase. J. Biol. Chem. 2001, 276, 17625–17628. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wei, J.; Wang, W.; Cui, G.; Zhao, Y.; Zhu, X.; Zhu, M.; Guo, W.; Yu, J. Identification of Signaling Pathways Involved in Aberrant Production of Adipokines in Adipocytes Undergoing Oxidative Stress. Arch. Med. Res. 2009, 40, 241–248. [Google Scholar] [CrossRef]
- Chen, H.; Montagnani, M.; Funahashi, T.; Shimomura, I.; Quon, M.J. Adiponectin Stimulates Production of Nitric Oxide in Vascular Endothelial Cells. J. Biol. Chem. 2003, 278, 45021–45026. [Google Scholar] [CrossRef]
- Motoshima, H.; Wu, X.; Mahadev, K.; Goldstein, B.J. Adiponectin Suppresses Proliferation and Superoxide Generation and Enhances ENOS Activity in Endothelial Cells Treated with Oxidized LDL. Biochem. Biophys. Res. Commun. 2004, 315, 264–271. [Google Scholar] [CrossRef]
- Badran, A.; Nasser, S.A.; Mesmar, J.; El-Yazbi, A.F.; Bitto, A.; Fardoun, M.M.; Baydoun, E.; Eid, A.H. Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 8764. [Google Scholar] [CrossRef]
- Lim, S.; Quon, M.J.; Koh, K.K. Modulation of Adiponectin as a Potential Therapeutic Strategy. Atherosclerosis 2014, 233, 721–728. [Google Scholar] [CrossRef]
- Jabłońska, A.; Neumayer, C.; Bolliger, M.; Burghuber, C.; Klinger, M.; Demyanets, S.; Nanobachvili, J.; Huk, I. Insight into the Expression of Toll-like Receptors 2 and 4 in Patients with Abdominal Aortic Aneurysm. Mol. Biol. Rep. 2020, 47, 2685–2692. [Google Scholar] [CrossRef]
- Neumann, F.J.; Sechtem, U.; Banning, A.P.; Bonaros, N.; Bueno, H.; Bugiardini, R.; Chieffo, A.; Crea, F.; Czerny, M.; Delgado, V.; et al. 2019 ESC Guidelines for the Diagnosis and Management of Chronic Coronary Syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Aboyans, V.; Bauersachs, R.; Mazzolai, L.; Brodmann, M.; Palomares, J.F.R.; Debus, S.; Collet, J.P.; Drexel, H.; Espinola-Klein, C.; Lewis, B.S.; et al. Antithrombotic Therapies in Aortic and Peripheral Arterial Diseases in 2021: A Consensus Document from the ESC Working Group on Aorta and Peripheral Vascular Diseases, the ESC Working Group on Thrombosis, and the ESC Working Group on Cardiovascular Pharmacotherapy. Eur. Heart J. 2021, 42, 4013–4024. [Google Scholar] [CrossRef] [PubMed]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.R.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients with Lower Extremity Peripheral Artery Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2017, 135, e686–e725. [Google Scholar] [CrossRef] [PubMed]
- Abramson, B.L.; Al-Omran, M.; Anand, S.S.; Albalawi, Z.; Coutinho, T.; de Mestral, C.; Dubois, L.; Gill, H.L.; Greco, E.; Guzman, R.; et al. Canadian Cardiovascular Society 2022 Guidelines for Peripheral Arterial Disease. Can. J. Cardiol. 2022, 38, 560–587. [Google Scholar] [CrossRef]
- Frank, U.; Nikol, S.; Belch, J.; Boc, V.; Brodmann, M.; Carpentier, P.H.; Chraim, A.; Canning, C.; Dimakakos, E.; Gottsäter, A.; et al. ESVM Guideline on Peripheral Arterial Disease. Vasa 2019, 48 (Suppl. S102), 1–80. [Google Scholar] [CrossRef]
- El Assar, M.; Álvarez-Bustos, A.; Sosa, P.; Angulo, J.; Rodríguez-Mañas, L. Effect of Physical Activity/Exercise on Oxidative Stress and Inflammation in Muscle and Vascular Aging. Int. J. Mol. Sci. 2022, 23, 8713. [Google Scholar] [CrossRef]
- Myette-Côté, É.; Durrer, C.; Neudorf, H.; Bammert, T.D.; Botezelli, J.D.; Johnson, J.D.; Desouza, C.A.; Little, J.P. The Effect of a Short-Term Low-Carbohydrate, High-Fat Diet with or without Postmeal Walks on Glycemic Control and Inflammation in Type 2 Diabetes: A Randomized Trial. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1210–R1219. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of Action and Effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Almeida, S.O.; Budoff, M. Effect of Statins on Atherosclerotic Plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef]
- Greenwood, J.; Mason, J.C. Statins and the Vascular Endothelial Inflammatory Response. Trends Immunol. 2007, 28, 88–98. [Google Scholar] [CrossRef]
- Piechota-Polanczyk, A.; Demyanets, S.; Nykonenko, O.; Huk, I.; Mittlboeck, M.; Domenig, C.M.; Neumayer, C.; Wojta, J.; Nanobachvili, J.; Klinger, M. Decreased Tissue Levels of Cyclophilin A, a Cyclosporine a Target and Phospho-ERK1/2 in Simvastatin Patients with Abdominal Aortic Aneurysm. Eur. J. Vasc. Endovasc. Surg. 2013, 45, 682–688. [Google Scholar] [CrossRef]
- Piechota-Polanczyk, A.; Goraca, A.; Demyanets, S.; Mittlboeck, M.; Domenig, C.; Neumayer, C.; Wojta, J.; Nanobachvili, J.; Huk, I.; Klinger, M. Simvastatin Decreases Free Radicals Formation in the Human Abdominal Aortic Aneurysm Wall via NF-ΚB. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Piechota-Polanczyk, A.; Demyanets, S.; Mittlboeck, M.; Hofmann, M.; Domenig, C.M.; Neumayer, C.; Wojta, J.; Klinger, M.; Nanobachvili, J.; Huk, I. The Influence of Simvastatin on NGAL, Matrix Metalloproteinases and Their Tissue Inhibitors in Human Intraluminal Thrombus and Abdominal Aortic Aneurysm Tissue. Eur. J. Vasc. Endovasc. Surg. 2015, 49, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Speidl, W.S.; Pleiner, J.; Seidinger, D.; Zorn, G.; Kaun, C.; Wojta, J.; Huber, K.; Minar, E.; Wolzt, M.; et al. Simvastatin Blunts Endotoxin-Induced Tissue Factor in Vivo. Circulation 2005, 111, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; Pfeffer, M.A.; Braunwald, E. C-Reactive Protein Levels and Outcomes after Statin Therapy. N. Engl. J. Med. 2005, 8, 8–9. [Google Scholar] [CrossRef]
- Tawakol, A.; Fayad, Z.A.; Mogg, R.; Alon, A.; Klimas, M.T.; Dansky, H.; Subramanian, S.S.; Abdelbaky, A.; Rudd, J.H.F.; Farkouh, M.E.; et al. Intensification of Statin Therapy Results in a Rapid Reduction in Atherosclerotic Inflammation: Results of a Multicenter Fluorodeoxyglucose-Positron Emission Tomography/Computed Tomography Feasibility Study. J. Am. Coll. Cardiol. 2013, 62, 909–917. [Google Scholar] [CrossRef]
- Ridker, P.M.; Bhatt, D.L.; Pradhan, A.D.; Glynn, R.J.; MacFadyen, J.G.; Nissen, S.E. Inflammation and Cholesterol as Predictors of Cardiovascular Events among Patients Receiving Statin Therapy: A Collaborative Analysis of Three Randomised Trials. Lancet 2023, 401, 1293–1301. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Z.; Qiu, Y.; Wu, L.; Wang, H.; Wu, L.; Zhao, L.; Xie, D. Statins Have an Anti-Inflammation in CKD Patients: A Meta-Analysis of Randomized Trials. Biomed. Res. Int. 2022, 2022, 4842699. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Zhang, H.F.; Lu, X.T.; Zhao, J.; Xu, Q. Statins Improve Asthma Symptoms by Suppressing Inflammation: A Meta-Analysis Based on RCTs. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 8401–8410. [Google Scholar] [CrossRef]
- Valerius, N.H. In Vitro Effect of Colchicine on Neutrophil Granulocyte Locomotion. Assessment of the Effect of Colchicine on Chemotaxis, Chemokinesis and Spontaneous Motility, Using a Modified Reversible Boyden Chamber. Acta Pathol. Microbiol. Scand. B 1978, 86, 149–154. [Google Scholar]
- Li, Z.; Davis, G.S.; Mohr, C.; Nain, M.; Gemsa, D. Inhibition of LPS-Induced Tumor Necrosis Factor-Alpha Production by Colchicine and Other Microtubule Disrupting Drugs. Immunobiology 1996, 195, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.G.; Malawista, S.E. Mobilization and Extracellular Release of Granular Enzymes from Human Leukocytes during Phagocytosis: Inhibition by Colchicine and Cortisol but Not by Salicylate. Arthritis Rheum. 1973, 16, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 Inflammasome and the Emerging Role of Colchicine to Inhibit Atherosclerosis-Associated Inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Bulnes, J.F.; Orellana, M.P.; Venturelli, P.M.; Rodriguez, G.M. The Role of Colchicine in Atherosclerosis: From Bench to Bedside. Pharmaceutics 2022, 14, 1395. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Thompson, P.L. Why Colchicine Should Be Considered for Secondary Prevention of Atherosclerosis: An Overview. Clin. Ther. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Wang, L.; Peng, Y.; Song, L.; Xia, D.; Li, C.; Li, Z.; Li, Q.; Yu, A.; Lu, C.; Wang, Y. Colchicine-Containing Nanoparticles Attenuates Acute Myocardial Infarction Injury by Inhibiting Inflammation. Cardiovasc. Drugs Ther. 2022, 36, 1075–1089. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Chen, Y.; Li, M.; Luo, W.; Liu, Y.; Fu, Y.; Xia, H.; Xu, C.; Jiang, Y.; et al. Colchicine May Become a New Cornerstone Therapy for Coronary Artery Disease: A Meta-Analysis of Randomized Controlled Trials. Clin. Rheumatol. 2022, 41, 1873–1887. [Google Scholar] [CrossRef]
- Bays, H.E.; Ballantyne, C.M.; Braeckman, R.A.; Stirtan, W.G.; Soni, P.N. Icosapent Ethyl, a Pure Ethyl Ester of Eicosapentaenoic Acid: Effects on Circulating Markers of Inflammation from the MARINE and ANCHOR Studies. Am. J. Cardiovasc. Drugs 2013, 13, 37–46. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Budoff, M.J.; Bhatt, D.L.; Kinninger, A.; Lakshmanan, S.; Muhlestein, J.B.; Le, V.T.; May, H.T.; Shaikh, K.; Shekar, C.; Roy, S.K.; et al. Effect of Icosapent Ethyl on Progression of Coronary Atherosclerosis in Patients with Elevated Triglycerides on Statin Therapy: Final Results of the EVAPORATE Trial. Eur. Heart J. 2020, 41, 3925–3932. [Google Scholar] [CrossRef]
- Mason, R.P.; Libby, P.; Bhatt, D.L. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arter. Thromb. Vasc. Biol. 2020, 40, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Braeckman, R.A.; Bays, H.E.; Kastelein, J.J.; Otvos, J.D.; Stirtan, W.G.; Doyle, R.T.; Soni, P.N.; Juliano, R.A. Effects of Icosapent Ethyl on Lipoprotein Particle Concentration and Size in Statin-Treated Patients with Persistent High Triglycerides (the ANCHOR Study). J. Clin. Lipidol. 2015, 9, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Onat, U.I.; Yildirim, A.D.; Tufanli, Ö.; Çimen, I.; Kocatürk, B.; Veli, Z.; Hamid, S.M.; Shimada, K.; Chen, S.; Sin, J.; et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2019, 73, 1149–1169. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.D.; Yalamanoglu, A.; Boyman, O. Systematic Review of Safety and Efficacy of IL-1-Targeted Biologics in Treating Immune-Mediated Disorders. Front. Immunol. 2022, 13, 888392. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Thuren, T.; Zalewski, A.; Libby, P. Interleukin-1β Inhibition and the Prevention of Recurrent Cardiovascular Events: Rationale and Design of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Am. Heart J. 2011, 162, 597–605. [Google Scholar] [CrossRef]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Ku, E.J.; Kim, B.R.; Lee, J.I.; Lee, Y.K.; Oh, T.J.; Jang, H.C.; Choi, S.H. The Anti-Atherosclerosis Effect of Anakinra, a Recombinant Human Interleukin-1 Receptor Antagonist, in Apolipoprotein E Knockout Mice. Int. J. Mol. Sci. 2022, 23, 4906. [Google Scholar] [CrossRef]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of Interleukin-1β Inhibition with Canakinumab on Hemoglobin A1c, Lipids, C-Reactive Protein, Interleukin-6, and Fibrinogen: A Phase IIb Randomized, Placebo-Controlled Trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef]
- Straub, R.H.; Cutolo, M. Glucocorticoids and Chronic Inflammation. Rheumatology 2016, 55 (Suppl. S2), ii6–ii14. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Williams-Dautovich, J.; Cummins, C.L. Minireview: New Molecular Mediators of Glucocorticoid Receptor Activity in Metabolic Tissues. Mol. Endocrinol. 2014, 28, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Cicala, M.V.; Mantero, F. Hypertension in Cushing’s Syndrome: From Pathogenesis to Treatment. Neuroendocrinology 2010, 92 (Suppl. S1), 44–49. [Google Scholar] [CrossRef] [PubMed]
- Akalestou, E.; Genser, L.; Rutter, G.A. Glucocorticoid Metabolism in Obesity and Following Weight Loss. Front. Endocrinol. 2020, 11, 59. [Google Scholar] [CrossRef]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of Dyslipidemia in Cushing’s Syndrome. Neuroendocrinology 2010, 92 (Suppl. S1), 86–90. [Google Scholar] [CrossRef]
- Coelho, M.C.A.; Santos, C.V.; Neto, L.V.; Gadelha, M.R. Adverse Effects of Glucocorticoids: Coagulopathy. Eur. J. Endocrinol. 2015, 173, M11–M21. [Google Scholar] [CrossRef]
- Faggiano, A.; Pivonello, R.; Spiezia, S.; De Martino, M.C.; Filippella, M.; Di Somma, C.; Lombardi, G.; Colao, A. Cardiovascular Risk Factors and Common Carotid Artery Caliber and Stiffness in Patients with Cushing’s Disease during Active Disease and 1 Year after Disease Remission. J. Clin. Endocrinol. Metab. 2003, 88, 2527–2533. [Google Scholar] [CrossRef]
- Macleod, C.; Hadoke, P.W.F.; Nixon, M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? Int. J. Mol. Sci. 2021, 22, 7622. [Google Scholar] [CrossRef]
- Petramala, L.; Lorenzo, D.; Iannucci, G.; Concistré, A.; Zinnamosca, L.; Marinelli, C.; De Vincentis, G.; Ciardi, A.; De Toma, G.; Letizia, C. Subclinical Atherosclerosis in Patients with Cushing Syndrome: Evaluation with Carotid Intima-Media Thickness and Ankle-Brachial Index. Endocrinol. Metab. 2015, 30, 488–493. [Google Scholar] [CrossRef]
- Souverein, P.C.; Berard, A.; Van Staa, T.P.; Cooper, C.; Egberts, A.C.G.; Leufkens, H.G.M.; Walker, B.R. Use of Oral Glucocorticoids and Risk of Cardiovascular and Cerebrovascular Disease in a Population Based Case-Control Study. Heart 2004, 90, 859–865. [Google Scholar] [CrossRef]
- Wei, L.; MacDonald, T.M.; Walker, B.R. Taking Glucocorticoids by Prescription Is Associated with Subsequent Cardiovascular Disease. Ann. Intern. Med. 2004, 141, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Pujades-Rodriguez, M.; Morgan, A.W.; Cubbon, R.M.; Wu, J. Dose-Dependent Oral Glucocorticoid Cardiovascular Risks in People with Immune-Mediated Inflammatory Diseases: A Population-Based Cohort Study. PLoS Med. 2020, 17, e1003432. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Ferrannini, E. Anti-Inflammatory Properties of Antidiabetic Drugs: A “Promised Land” in the COVID-19 Era? J. Diabetes Complicat. 2020, 34, 107723. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Docherty, K.F.; Claggett, B.L.; Jhund, P.S.; de Boer, R.A.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. SGLT-2 Inhibitors in Patients with Heart Failure: A Comprehensive Meta-Analysis of Five Randomised Controlled Trials. Lancet 2022, 400, 757–767. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Claggett, B.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; et al. Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N. Engl. J. Med. 2022, 387, 1089–1098. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, E895–E1032. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, X.; Ilyas, I.; Zheng, X.; Luo, S.; Little, P.J.; Kamato, D.; Sahebkar, A.; Wu, W.; Weng, J.; et al. Impact of Sodium Glucose Cotransporter 2 (SGLT2) Inhibitors on Atherosclerosis: From Pharmacology to Pre-Clinical and Clinical Therapeutics. Theranostics 2021, 11, 4502–4515. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Lu, J.H.; Lu, R.; Lundenberg, L.; Desjardins, E.M.; Green, A.E.; Lally, J.S.V.; Schertzer, J.D.; Steinberg, G.R. The SGLT2 Inhibitor Canagliflozin Suppresses Lipid Synthesis and Interleukin-1 Beta in ApoE Deficient Mice. Biochem. J. 2020, 477, 2347–2361. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, N.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin Attenuates the Progression of Atherosclerosis and Inflammation Process in APOE Knockout Mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.J.; Boyd, D.; Katwan, O.J.; Strembitska, A.; Almabrouk, T.A.; Kennedy, S.; Palmer, T.M.; Salt, I.P. Canagliflozin Inhibits Interleukin-1β-Stimulated Cytokine and Chemokine Secretion in Vascular Endothelial Cells by AMP-Activated Protein Kinase-Dependent and -Independent Mechanisms. Sci. Rep. 2018, 8, 5276. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 Inhibition Modulates NLRP3 Inflammasome Activity via Ketones and Insulin in Diabetes with Cardiovascular Disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Chen, H.; Teng, D.; Xu, B.; Wang, C.; Wang, H.; Jia, W.; Gong, L.; Dong, H.; Zhong, L.; Yang, J. The SGLT2 Inhibitor Canagliflozin Reduces Atherosclerosis by Enhancing Macrophage Autophagy. J. Cardiovasc. Transl. Res. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Ghanim, H.; Vora, M.; Sia, C.L.; Korzeniewski, K.; Dhindsa, S.; Makdissi, A.; Dandona, P. Exenatide Exerts a Potent Antiinflammatory Effect. J. Clin. Endocrinol. Metab. 2012, 97, 198–207. [Google Scholar] [CrossRef]
- Jonik, S.; Marchel, M.; Grabowski, M.; Opolski, G.; Mazurek, T. Gastrointestinal Incretins—Glucose-Dependent Insulinotropic Polypeptide (GIP) and Glucagon-like Peptide-1 (GLP-1) beyond Pleiotropic Physiological Effects Are Involved in Pathophysiology of Atherosclerosis and Coronary Artery Disease—State of the Art. Biology 2022, 11, 288. [Google Scholar] [CrossRef]
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 Analogs Liraglutide and Semaglutide Reduce Atherosclerosis in ApoE−/− and LDLr−/− Mice by a Mechanism That Includes Inflammatory Pathways. JACC Basic. Transl. Sci. 2018, 3, 844–857. [Google Scholar] [CrossRef]
- Bendotti, G.; Montefusco, L.; Lunati, M.E.; Usuelli, V.; Pastore, I.; Lazzaroni, E.; Assi, E.; Seelam, A.J.; El Essawy, B.; Jang, Y.; et al. The Anti-Inflammatory and Immunological Properties of GLP-1 Receptor Agonists. Pharmacol. Res. 2022, 182, 106320. [Google Scholar] [CrossRef]
- Bray, J.J.H.; Foster-Davies, H.; Salem, A.; Hoole, A.L.; Obaid, D.R.; Halcox, J.P.J.; Stephens, J.W. Glucagon-like Peptide-1 Receptor Agonists Improve Biomarkers of Inflammation and Oxidative Stress: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Diabetes Obes. Metab. 2021, 23, 1806–1822. [Google Scholar] [CrossRef]
- Bethel, M.A.; Patel, R.A.; Merrill, P.; Lokhnygina, Y.; Buse, J.B.; Mentz, R.J.; Pagidipati, N.J.; Chan, J.C.; Gustavson, S.M.; Iqbal, N.; et al. Cardiovascular Outcomes with Glucagon-like Peptide-1 Receptor Agonists in Patients with Type 2 Diabetes: A Meta-Analysis. Lancet Diabetes Endocrinol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Werkman, N.C.C.; Driessen, J.H.M.; Stehouwer, C.D.A.; Vestergaard, P.; Schaper, N.C.; van den Bergh, J.P.; Nielen, J.T.H. The Use of Sodium-Glucose Co-Transporter-2 Inhibitors or Glucagon-like Peptide-1 Receptor Agonists versus Sulfonylureas and the Risk of Lower Limb Amputations: A Nation-Wide Cohort Study. Cardiovasc. Diabetol. 2023, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.M. 60 Years of Metformin Use: A Glance at the Past and a Look to the Future. Diabetologia 2017, 60, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin Reduces All-Cause Mortality and Diseases of Ageing Independent of Its Effect on Diabetes Control: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, W.; Ni, X.; Little, P.J.; Xu, S.; Tang, L.; Weng, J. Metformin, Macrophage Dysfunction and Atherosclerosis. Front. Immunol. 2021, 12, 682853. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Litvinova, L.; Poggio, P.; Moschetta, D.; Sukhorukov, V.N.; Orekhov, A.N. From Diabetes to Atherosclerosis: Potential of Metformin for Management of Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 9738. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Y.; Tian, Y.; Zhou, Y. Metformin Suppresses Foam Cell Formation, Inflammation and Ferroptosis via the AMPK/ERK Signaling Pathway in Ox-LDL-induced THP-1 Monocytes. Exp. Ther. Med. 2022, 24, 636. [Google Scholar] [CrossRef]
- Yang, Q.; Yuan, H.; Chen, M.; Qu, J.; Wang, H.; Yu, B.; Chen, J.; Sun, S.; Tang, X.; Ren, W. Metformin Ameliorates the Progression of Atherosclerosis via Suppressing Macrophage Infiltration and Inflammatory Responses in Rabbits. Life Sci. 2018, 198, 56–64. [Google Scholar] [CrossRef]
- Kanigur Sultuybek, G.; Soydas, T.; Yenmis, G. NF-ΚB as the Mediator of Metformin’s Effect on Ageing and Ageing-Related Diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef]
- Gräni, C.; Eichhorn, C.; Bière, L.; Murthy, V.L.; Agarwal, V.; Kaneko, K.; Cuddy, S.; Aghayev, A.; Steigner, M.; Blankstein, R.; et al. Prognostic Value of Cardiac Magnetic Resonance Tissue Characterization in Risk Stratifying Patients with Suspected Myocarditis. J. Am. Coll. Cardiol. 2017, 70, 1964. [Google Scholar] [CrossRef]
- Roussel, R.; Travert, F.; Pasquet, B.; Wilson, P.W.F.; Smith, S.C.; Goto, S.; Ravaud, P.; Marre, M.; Porath, A.; Bhatt, D.L.; et al. Metformin Use and Mortality among Patients with Diabetes and Atherothrombosis. Arch. Intern. Med. 2010, 170, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Meaney, E.; Vela, A.; Samaniego, V.; Meaney, A.; Asbún, J.; Zempoalteca, J.C.; Elisa, Z.N.; Emma, M.N.; Guzman, M.; Hicks, J.; et al. Metformin, Arterial Function, Intima-Media Thickness and Nitroxidation in Metabolic Syndrome: The Mefisto Study. Clin. Exp. Pharmacol. Physiol. 2008, 35, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Ferrell, W.; Greer, I.A.; Petrie, J.R.; Cobbe, S.M.; Sattar, N. Effects of Metformin on Microvascular Function and Exercise Tolerance in Women with Angina and Normal Coronary Arteries: A Randomized, Double-Blind, Placebo-Controlled Study. J. Am. Coll. Cardiol. 2006, 48, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Preiss, D.; Lloyd, S.M.; Ford, I.; McMurray, J.J.; Holman, R.R.; Welsh, P.; Fisher, M.; Packard, C.J.; Sattar, N. Metformin for Non-Diabetic Patients with Coronary Heart Disease (the CAMERA Study): A Randomised Controlled Trial. Lancet Diabetes Endocrinol. 2014, 2, 116–124. [Google Scholar] [CrossRef]
- Luo, F.; Das, A.; Chen, J.; Wu, P.; Li, X.; Fang, Z. Metformin in Patients with and without Diabetes: A Paradigm Shift in Cardiovascular Disease Management. Cardiovasc. Diabetol. 2019, 18, 54. [Google Scholar] [CrossRef]
- Griffin, S.J.; Angelyn Bethel, M.; Holman, R.R.; Khunti, K.; Wareham, N.; Brierley, G.; Davies, M.; Dymond, A.; Eichenberger, R.; Evans, P.; et al. Metformin in Non-Diabetic Hyperglycaemia: The GLINT Feasibility RCT. Health Technol. Assess. 2018, 22, 1–64. [Google Scholar] [CrossRef]
- ISRCTN—ISRCTN34875079: The Glucose Lowering in Non-Diabetic hyperglycaemia Trial (GLINT)—Glucose Lowering in Those at Risk of Diabetes. Available online: https://www.isrctn.com/ISRCTN34875079 (accessed on 14 June 2023).
- National Institute of Diabetes and Digestive and Kidney Diseases. Dipeptidyl Peptidase-4 Inhibitors. Available online: https://pubmed.ncbi.nlm.nih.gov/31643671/ (accessed on 14 June 2023).
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration with the EASDThe Task Force for Diabetes, Pre-Diabetes, and Cardiovascular Diseases of the European Society of Cardiology (ESC) and the European Association for the Study of Diabetes (EASD). Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef]
- Liu, H.; Guo, L.; Xing, J.; Li, P.; Sang, H.; Hu, X.; Du, Y.; Zhao, L.; Song, R.; Gu, H. The Protective Role of DPP4 Inhibitors in Atherosclerosis. Eur. J. Pharmacol. 2020, 875, 173037. [Google Scholar] [CrossRef]
- Lee, D.S.; Lee, E.S.; Alam, M.M.; Jang, J.H.; Lee, H.S.; Oh, H.; Kim, Y.C.; Manzoor, Z.; Koh, Y.S.; Kang, D.G.; et al. Soluble DPP-4 up-Regulates Toll-like Receptors and Augments Inflammatory Reactions, Which Are Ameliorated by Vildagliptin or Mannose-6-Phosphate. Metabolism 2016, 65, 89–101. [Google Scholar] [CrossRef]
- Romacho, T.; Vallejo, S.; Villalobos, L.A.; Wronkowitz, N.; Indrakusuma, I.; Sell, H.; Eckel, J.; Sanchez-Ferrer, C.F.; Peiro, C. Soluble Dipeptidyl Peptidase-4 Induces Microvascular Endothelial Dysfunction through Proteinase-Activated Receptor-2 and Thromboxane A2 Release. J. Hypertens. 2016, 34, 869–876. [Google Scholar] [CrossRef]
- Tang, S.T.; Su, H.; Zhang, Q.; Tang, H.Q.; Wang, C.J.; Zhou, Q.; Wei, W.; Zhu, H.Q.; Wang, Y. Sitagliptin Inhibits Endothelin-1 Expression in the Aortic Endothelium of Rats with Streptozotocin-Induced Diabetes by Suppressing the Nuclear Factor-ΚB/IκBα System through the Activation of AMP-Activated Protein Kinase. Int. J. Mol. Med. 2016, 37, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.; Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A Dipeptidyl Peptidase-4 Inhibitor, Des-Fluoro-Sitagliptin, Improves Endothelial Function and Reduces Atherosclerotic Lesion Formation in Apolipoprotein E-Deficient Mice. J. Am. Coll. Cardiol. 2012, 59, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin Improves High Glucose-Induced Endothelial Mitochondrial Dysfunction via Inhibiting Mitochondrial Fission. J. Cell Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Boscaro, E.; Albiero, M.; Menegazzo, L.; Frison, V.; De Kreutzenberg, S.; Agostini, C.; Tiengo, A.; Avogaro, A. The Oral Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Increases Circulating Endothelial Progenitor Cells in Patients with Type 2 Diabetes: Possible Role of Stromal-Derived Factor-1alpha. Diabetes Care 2010, 33, 1607–1609. [Google Scholar] [CrossRef]
- Fadini, G.P.; Avogaro, A. Dipeptidyl Peptidase-4 Inhibition and Vascular Repair by Mobilization of Endogenous Stem Cells in Diabetes and Beyond. Atherosclerosis 2013, 229, 23–29. [Google Scholar] [CrossRef]
- Dai, Y.; Dai, D.; Wang, X.; Ding, Z.; Mehta, J.L. DPP-4 Inhibitors Repress NLRP3 Inflammasome and Interleukin-1beta via GLP-1 Receptor in Macrophages through Protein Kinase C Pathway. Cardiovasc. Drugs Ther. 2014, 28, 425–432. [Google Scholar] [CrossRef]
- Cha, S.A.; Park, Y.M.; Yun, J.S.; Lim, T.S.; Song, K.H.; Yoo, K.D.; Ahn, Y.B.; Ko, S.H. A Comparison of Effects of DPP-4 Inhibitor and SGLT2 Inhibitor on Lipid Profile in Patients with Type 2 Diabetes. Lipids Health Dis. 2017, 16, 58. [Google Scholar] [CrossRef]
- Boschmann, M.; Engeli, S.; Dobberstein, K.; Budziarek, P.; Strauss, A.; Boehnke, J.; Sweep, F.C.G.J.; Luft, F.C.; He, Y.L.; Foley, J.E.; et al. Dipeptidyl-Peptidase-IV Inhibition Augments Postprandial Lipid Mobilization and Oxidation in Type 2 Diabetic Patients. J. Clin. Endocrinol. Metab. 2009, 94, 846–852. [Google Scholar] [CrossRef]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs. Placebo on Major Cardiovascular Events in Adults with Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after Acute Coronary Syndrome in Patients with Type 2 Diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; Lindholm, D.; et al. Dapagliflozin in Heart Failure with Preserved and Mildly Reduced Ejection Fraction: Rationale and Design of the DELIVER Trial. Eur. J. Heart Fail. 2021, 23, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Butler, J.; Zannad, F.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Carson, P.; Anand, I.; Doehner, W.; Haass, M.; et al. Effect of Empagliflozin on Worsening Heart Failure Events in Patients with Heart Failure and Preserved Ejection Fraction: EMPEROR-Preserved Trial. Circulation 2021, 144, 1284–1294. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Weikert, C.; Pultar, J.; Lee, S.; Eichelberger, B.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Ticagrelor Inhibits Toll-Like and Protease-Activated Receptor Mediated Platelet Activation in Acute Coronary Syndromes. Cardiovasc. Drugs Ther. 2020, 34, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Panzer, B.; Wadowski, P.P.; Huber, K.; Panzer, S.; Gremmel, T. Protease-Activated Receptor-Mediated Platelet Aggregation in Patients with Type 2 Diabetes on Potent P2Y12 Inhibitors. Diabet. Med. 2022, 39, e14868. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Eichelberger, B.; Kopp, C.W.; Pultar, J.; Seidinger, D.; Koppensteiner, R.; Lang, I.M.; Panzer, S.; Gremmel, T. Disaggregation Following Agonist-Induced Platelet Activation in Patients on Dual Antiplatelet Therapy. J. Cardiovasc. Transl. Res. 2017, 10, 359–367. [Google Scholar] [CrossRef]
- Pultar, J.; Wadowski, P.P.; Panzer, S.; Gremmel, T. Oral Antiplatelet Agents in Cardiovascular Disease. Vasa 2019, 48, 291–302. [Google Scholar] [CrossRef]
- Li, W.; Liu, Q.; Tang, Y. Platelet to Lymphocyte Ratio in the Prediction of Adverse Outcomes after Acute Coronary Syndrome: A Meta-Analysis. Sci. Rep. 2017, 7, 40426. [Google Scholar] [CrossRef]
- Azab, B.; Shah, N.; Akerman, M.; McGinn, J.T. Value of Platelet/Lymphocyte Ratio as a Predictor of All-Cause Mortality after Non-ST-Elevation Myocardial Infarction. J. Thromb. Thrombolysis 2012, 34, 326–334. [Google Scholar] [CrossRef]
- Ugur, M.; Gul, M.; Bozbay, M.; Cicek, G.; Uyarel, H.; Koroglu, B.; Uluganyan, M.; Aslan, S.; Tusun, E.; Surgit, O.; et al. The Relationship between Platelet to Lymphocyte Ratio and the Clinical Outcomes in ST Elevation Myocardial Infarction Underwent Primary Coronary Intervention. Blood Coagul. Fibrinolysis 2014, 25, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hoberstorfer, T.; Wadowski, P.P.; Kopp, C.W.; Panzer, S.; Gremmel, T. Platelet-to-Lymphocyte and Neutrophil-to-Lymphocyte Ratios Predict Target Vessel Restenosis after Infrainguinal Angioplasty with Stent Implantation. J. Clin. Med. 2020, 9, 1729. [Google Scholar] [CrossRef] [PubMed]
- Gremmel, T.; Steiner, S.; Seidinger, D.; Koppensteiner, R.; Panzer, S.; Kopp, C.W. Adenosine Diphosphate-Inducible Platelet Reactivity Shows a Pronounced Age Dependency in the Initial Phase of Antiplatelet Therapy with Clopidogrel. J. Thromb. Haemost. 2010, 8, 37–42. [Google Scholar] [CrossRef]
- Gremmel, T.; Michelson, A.D.; Wadowski, P.P.; Pultar, J.; Weikert, C.; Tscharre, M.; Lee, S.; Panzer, S.; Frelinger, A.L. Sex-Specific Platelet Activation through Protease-Activated Receptor-1 in Patients Undergoing Cardiac Catheterization. Atherosclerosis 2021, 339, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wadowski, P.P.; Lee, S.; Kopp, C.W.; Koppensteiner, R.; Panzer, S.; Gremmel, T. Low Levels of High-Density Lipoprotein Cholesterol Are Linked to Impaired Clopidogrel-Mediated Platelet Inhibition. Angiology 2018, 69, 786–794. [Google Scholar] [CrossRef]
- Jäger, B.; Piackova, E.; Haller, P.M.; Andric, T.; Kahl, B.; Christ, G.; Geppert, A.; Wojta, J.; Huber, K. Increased Platelet Reactivity in Dyslipidemic Patients with Coronary Artery Disease on Dual Anti-Platelet Therapy. Arch. Med. Sci. 2019, 15, 65–71. [Google Scholar] [CrossRef]
- Gremmel, T.; Kopp, C.W.; Seidinger, D.; Koppensteiner, R.; Panzer, S.; Sunder-Plassmann, R.; Mannhalter, C.; Steiner, S. Differential Impact of Cytochrome 2C9 Allelic Variants on Clopidogrel-Mediated Platelet Inhibition Determined by Five Different Platelet Function Tests. Int. J. Cardiol. 2013, 166, 126–131. [Google Scholar] [CrossRef]
- Gremmel, T.; Kopp, C.W.; Moertl, D.; Seidinger, D.; Koppensteiner, R.; Panzer, S.; Mannhalter, C.; Steiner, S. Influence of Cytochrome 2C19 Allelic Variants on On-Treatment Platelet Reactivity Evaluated by Five Different Platelet Function Tests. Thromb. Res. 2012, 129, 616–622. [Google Scholar] [CrossRef]
- Giustino, G.; Kirtane, A.J.; Baber, U.; Généreux, P.; Witzenbichler, B.; Neumann, F.J.; Weisz, G.; Maehara, A.; Rinaldi, M.J.; Metzger, C.; et al. Impact of Anemia on Platelet Reactivity and Ischemic and Bleeding Risk: From the Assessment of Dual Antiplatelet Therapy with Drug-Eluting Stents Study. Am. J. Cardiol. 2016, 117, 1877–1883. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Kopp, C.W.; Koppensteiner, R.; Lang, I.M.; Pultar, J.; Lee, S.; Weikert, C.; Panzer, S.; Gremmel, T. Decreased Platelet Inhibition by P2Y12 Receptor Blockers in Anaemia. Eur. J. Clin. Investig. 2018, 48, e12861. [Google Scholar] [CrossRef]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of Inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Otaki, Y.; Watanabe, T.; Takahashi, H.; Sugai, T.; Yokoyama, M.; Tamura, H.; Kato, S.; Nishiyama, S.; Arimoto, T.; Shishido, T.; et al. Impact of Iron Deficiency on Peripheral Artery Disease After Endovascular Therapy. Circ. Rep. 2019, 1, 187–195. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.W.; Ibrahim, K.; Kickler, T.S.; Clarke, W.A.; Hasan, R.K.; Czarny, M.J.; Keramati, A.R.; Goli, R.R.; Gratton, T.P.; Brinker, J.A.; et al. Effect of Intravenous Fentanyl on Ticagrelor Absorption and Platelet Inhibition Among Patients Undergoing Percutaneous Coronary Intervention: The PACIFY Randomized Clinical Trial (Platelet Aggregation with Ticagrelor Inhibition and Fentanyl). Circulation 2018, 137, 307–309. [Google Scholar] [CrossRef]
- Kubica, J.; Adamski, P.; Ostrowska, M.; Sikora, J.; Kubica, J.M.; Sroka, W.D.; Stankowska, K.; Buszko, K.; Navarese, E.P.; Jilma, B.; et al. Morphine Delays and Attenuates Ticagrelor Exposure and Action in Patients with Myocardial Infarction: The Randomized, Double-Blind, Placebo-Controlled IMPRESSION Trial. Eur. Heart J. 2016, 37, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Hobl, E.L.; Reiter, B.; Schoergenhofer, C.; Schwameis, M.; Derhaschnig, U.; Lang, I.M.; Stimpfl, T.; Jilma, B. Morphine Interaction with Prasugrel: A Double-Blind, Cross-over Trial in Healthy Volunteers. Clin. Res. Cardiol. 2016, 105, 349–355. [Google Scholar] [CrossRef]
- Hobl, E.L.; Stimpfl, T.; Ebner, J.; Schoergenhofer, C.; Derhaschnig, U.; Sunder-Plassmann, R.; Jilma-Stohlawetz, P.; Mannhalter, C.; Posch, M.; Jilma, B. Morphine Decreases Clopidogrel Concentrations and Effects: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Coll. Cardiol. 2014, 63, 630–635. [Google Scholar] [CrossRef]
- Bartko, J.; Schoergenhofer, C.; Schwameis, M.; Wadowski, P.; Kubica, J.; Jilma, B.; Hobl, E.L. Morphine Interaction with Aspirin: A Double-Blind, Crossover Trial in Healthy Volunteers. J. Pharmacol. Exp. Ther. 2018, 365, 430–436. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Falconer, D.; Papageorgiou, N.; Salem, K.; Lim, W.Y.; Katsargyris, A.; Avgerinos, E.; Tousoulis, D. Nitric Oxide Donors for Peripheral Artery Disease. Curr. Opin. Pharmacol. 2018, 39, 77–85. [Google Scholar] [CrossRef]
- Park, S.Y.; Pekas, E.J.; Headid, R.J.; Son, W.M.; Wooden, T.K.; Song, J.; Layec, G.; Yadav, S.K.; Mishra, P.K.; Pipinos, I.I. Acute Mitochondrial Antioxidant Intake Improves Endothelial Function, Antioxidant Enzyme Activity, and Exercise Tolerance in Patients with Peripheral Artery Disease. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H456–H467. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. [Google Scholar] [CrossRef]
- Durante, W. Targeting Heme Oxygenase-1 in Vascular Disease. Curr. Drug Targets 2010, 11, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Dulak, J.; Deshane, J.; Jozkowicz, A.; Agarwal, A. Heme Oxygenase-1 and Carbon Monoxide in Vascular Pathobiology: Focus on Angiogenesis. Circulation 2008, 117, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.S.; Chang, C.C.; Zhu, Y.; Shyy, J.Y.J. Simvastatin Induces Heme Oxygenase-1: A Novel Mechanism of Vessel Protection. Circulation 2004, 110, 1296–1302. [Google Scholar] [CrossRef]
- Piechota-Polanczyk, A.; Kopacz, A.; Kloska, D.; Zagrapan, B.; Neumayer, C.; Grochot-Przeczek, A.; Huk, I.; Brostjan, C.; Dulak, J.; Jozkowicz, A. Simvastatin Treatment Upregulates HO-1 in Patients with Abdominal Aortic Aneurysm but Independently of Nrf2. Oxid. Med. Cell Longev. 2018, 2018, 2028936. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.; Oeser, C.; Kainz, F.M.; Shabanian, S.; Aref, T.; Bilban, M.; Messner, B.; Heidtmann, J.; Laufer, G.; Kocher, A.; et al. Intravenous Heme Arginate Induces HO-1 (Heme Oxygenase-1) in the Human Heart. Arter. Thromb. Vasc. Biol. 2018, 38, 2755–2762. [Google Scholar] [CrossRef] [PubMed]
- Doberer, D.; Haschemi, A.; Andreas, M.; Zapf, T.C.; Clive, B.; Jeitler, M.; Heinzl, H.; Wagner, O.; Wolzt, M.; Bilban, M. Haem Arginate Infusion Stimulates Haem Oxygenase-1 Expression in Healthy Subjects. Br. J. Pharmacol. 2010, 161, 1751–1762. [Google Scholar] [CrossRef]
- Andreas, M.; Schmid, A.I.; Doberer, D.; Schewzow, K.; Weisshaar, S.; Heinze, G.; Bilban, M.; Moser, E.; Wolzt, M. Heme Arginate Improves Reperfusion Patterns after Ischemia: A Randomized, Placebo-Controlled Trial in Healthy Male Subjects. J. Cardiovasc. Magn. Reson. 2012, 14, 55. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischaemia-Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Andreas, M.; Schmid, A.I.; Keilani, M.; Doberer, D.; Bartko, J.; Crevenna, R.; Moser, E.; Wolzt, M. Effect of Ischemic Preconditioning in Skeletal Muscle Measured by Functional Magnetic Resonance Imaging and Spectroscopy: A Randomized Crossover Trial. J. Cardiovasc. Magn. Reson. 2011, 13, 32. [Google Scholar] [CrossRef]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An Overview of Protective Strategies against Ischemia/Reperfusion Injury: The Role of Hyperbaric Oxygen Preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef] [PubMed]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted Pharmacotherapy for Ischemia Reperfusion Injury in Acute Myocardial Infarction. Expert. Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Belcher, D.A.; Williams, A.T.; Munoz, C.J.; Muller, C.R.; Walser, C.; Palmer, A.F.; Cabrales, P. Attenuating Ischemia-Reperfusion Injury with Polymerized Albumin. J. Appl. Physiol. 2022, 132, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.H.; Gurule, D.M.; Larson, R.S. Amelioration of Ischemia-Reperfusion Injury with Cyclic Peptide Blockade of ICAM-1. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1260–H1268. [Google Scholar] [CrossRef]
- O’Donnell, M.E.; Badger, S.A.; Sharif, M.A.; Makar, R.R.; McEneny, J.; Young, I.S.; Lee, B.; Soong, C.V. The Effects of Cilostazol on Exercise-Induced Ischaemia-Reperfusion Injury in Patients with Peripheral Arterial Disease. Eur. J. Vasc. Endovasc. Surg. 2009, 37, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Simard, M.J.; Huot, J. Endothelial MicroRNAs Regulating the NF-ΚB Pathway and Cell Adhesion Molecules during Inflammation. FASEB J. 2018, 32, 4070–4084. [Google Scholar] [CrossRef]
- Perkins, L.A.; Anderson, C.J.; Novelli, E.M. Targeting P-Selectin Adhesion Molecule in Molecular Imaging: P-Selectin Expression as a Valuable Imaging Biomarker of Inflammation in Cardiovascular Disease. J. Nucl. Med. 2019, 60, 1691–1697. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Sun, M.; Zhang, Z.; Cao, H.; Chen, X. NF-KB Activity-Dependent P-Selectin Involved in Ox-LDL-Induced Foam Cell Formation in U937 Cell. Biochem. Biophys. Res. Commun. 2011, 411, 543–548. [Google Scholar] [CrossRef]
- Chao, T.H.; Tseng, S.Y.; Chen, I.C.; Tsai, Y.S.; Huang, Y.Y.; Liu, P.Y.; Ou, H.Y.; Li, Y.H.; Wu, H.L.; Cho, C.L.; et al. Cilostazol Enhances Mobilization and Proliferation of Endothelial Progenitor Cells and Collateral Formation by Modifying Vasculo-Angiogenic Biomarkers in Peripheral Arterial Disease. Int. J. Cardiol. 2014, 172, e371–e374. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Rundback, J.; Bunte, M.; Hammad, T.A.; Miller, L.; Patel, P.D.; Sadanandan, S.; Fitzgerald, M.; Pastore, J.; Kashyap, V.; et al. SDF-1 Plasmid Treatment for Patients with Peripheral Artery Disease (STOP-PAD): Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Vasc. Med. 2019, 24, 200–207. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, Y.W.; Nasjletti, A.; Xu, X.; Wolin, M.S.; Hintze, T.H. ACE Inhibitors Promote Nitric Oxide Accumulation to Modulate Myocardial Oxygen Consumption. Circulation 1997, 95, 176–182. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 Inhibitor Empagliflozin Improves the Primary Diabetic Complications in ZDF Rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmanna Benedikt Preckel, M.W.; Koolwijk, P.; Van Hinsbergh, V.W.M.; Zuurbier, C.J.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, L.; Liu, S.; Liao, G.; Li, L.; Chen, Y.; Cheng, J.; Lu, Y.; Liu, J. GLP-1 Receptor Agonist Ameliorates Obesity-Induced Chronic Kidney Injury via Restoring Renal Metabolism Homeostasis. PLoS ONE 2018, 13, e0193473. [Google Scholar] [CrossRef] [PubMed]
- Herat, L.Y.; Ward, N.C.; Magno, A.L.; Rakoczy, E.P.; Kiuchi, M.G.; Schlaich, M.P.; Matthews, V.B. Sodium Glucose Co-Transporter 2 Inhibition Reduces Succinate Levels in Diabetic Mice. World J. Gastroenterol. 2020, 26, 3225–3235. [Google Scholar] [CrossRef]
- Van Royen, N.; Schirmer, S.H.; Atasever, B.; Behrens, C.Y.H.; Ubbink, D.; Buschmann, E.E.; Voskuil, M.; Bot, P.; Hoefer, I.; Schlingemann, R.O.; et al. START Trial: A Pilot Study on STimulation of ARTeriogenesis Using Subcutaneous Application of Granulocyte-Macrophage Colony-Stimulating Factor as a New Treatment for Peripheral Vascular Disease. Circulation 2005, 112, 1040–1046. [Google Scholar] [CrossRef]
- Cooper, L.T.; Hiatt, W.R.; Creager, M.A.; Regensteiner, J.G.; Casscells, W.; Isner, J.M.; Cooke, J.P.; Hirsch, A.T. Proteinuria in a Placebo-Controlled Study of Basic Fibroblast Growth Factor for Intermittent Claudication. Vasc. Med. 2001, 6, 235–239. [Google Scholar] [CrossRef]
- Zachman, A.L.; Wang, X.; Tucker-Schwartz, J.M.; Fitzpatrick, S.T.; Lee, S.H.; Guelcher, S.A.; Skala, M.C.; Sung, H.J. Uncoupling Angiogenesis and Inflammation in Peripheral Artery Disease with Therapeutic Peptide-Loaded Microgels. Biomaterials 2014, 35, 9635. [Google Scholar] [CrossRef]
- Caolo, V.; Vries, M.; Zupancich, J.; Houben, M.; Mihov, G.; Wagenaar, A.; Swennen, G.; Nossent, Y.; Quax, P.; Suylen, D.; et al. CXCL1 Microspheres: A Novel Tool to Stimulate Arteriogenesis. Drug Deliv. 2016, 23, 2919–2926. [Google Scholar] [CrossRef]
- Chang, T.T.; Lin, L.Y.; Chen, J.W. Inhibition of Macrophage Inflammatory Protein-1β Improves Endothelial Progenitor Cell Function and Ischemia-Induced Angiogenesis in Diabetes. Angiogenesis 2019, 22, 53–65. [Google Scholar] [CrossRef]
- Cooke, J.P. Therapeutic Transdifferentiation: A Novel Approach for Vascular Disease. Circ. Res. 2013, 112, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Wong, W.T.; Ospino, F.; Meng, S.; Lee, J.; Jha, A.; Dexheimer, P.; Aronow, B.J.; Cooke, J.P. Transdifferentiation of Human Fibroblasts to Endothelial Cells Role of Innate Immunity. Circulation 2015, 131, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.P.; Ryder, I.; Yang, N.; Lam, Y.T.; Santos, M.; Michael, P.L.; Robinson, D.A.; Ng, M.K.; Wise, S.G. Macrophage Polarization as a Novel Therapeutic Target for Endovascular Intervention in Peripheral Artery Disease. JACC Basic. Transl. Sci. 2021, 6, 693. [Google Scholar] [CrossRef] [PubMed]
- Schlager, O.; Giurgea, A.; Schuhfried, O.; Seidinger, D.; Hammer, A.; Gröger, M.; Fialka-Moser, V.; Gschwandtner, M.; Koppensteiner, R.; Steiner, S. Exercise Training Increases Endothelial Progenitor Cells and Decreases Asymmetric Dimethylarginine in Peripheral Arterial Disease: A Randomized Controlled Trial. Atherosclerosis 2011, 217, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, D. Effects of Aerobic Exercise on Lipids and Lipoproteins. Lipids Health Dis. 2017, 16, 132. [Google Scholar] [CrossRef]
- Sampath Kumar, A.; Maiya, A.G.; Shastry, B.A.; Vaishali, K.; Ravishankar, N.; Hazari, A.; Gundmi, S.; Jadhav, R. Exercise and Insulin Resistance in Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Ann. Phys. Rehabil. Med. 2019, 62, 98–103. [Google Scholar] [CrossRef]
- Sharman, J.E.; La Gerche, A.; Coombes, J.S. Exercise and Cardiovascular Risk in Patients with Hypertension. Am. J. Hypertens. 2015, 28, 147–158. [Google Scholar] [CrossRef]
- Palmefors, H.; DuttaRoy, S.; Rundqvist, B.; Börjesson, M. The Effect of Physical Activity or Exercise on Key Biomarkers in Atherosclerosis—A Systematic Review. Atherosclerosis 2014, 235, 150–161. [Google Scholar] [CrossRef]
- Carvalho de Arruda Veiga, E.; Ferreira Levy, R.; Sales Bocalini, D.; Maria Soares Junior, J.; Chada Baracat, E.; Carvalho Cavalli, R.; dos Santos, L. Exercise Training and Experimental Myocardial Ischemia and Reperfusion: A Systematic Review and Meta-Analysis. IJC Heart Vasc. 2023, 46, 101214. [Google Scholar] [CrossRef]
- Quindry, J.C.; Franklin, B.A. Exercise Preconditioning as a Cardioprotective Phenotype. Am. J. Cardiol. 2021, 148, 8–15. [Google Scholar] [CrossRef]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial Cell Metabolism in Health and Disease: Impact of Hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.R.; Quax, P.H.A. Plaque Angiogenesis and Its Relation to Inflammation and Atherosclerotic Plaque Destabilization. Curr. Opin. Lipidol. 2016, 27, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 Plays a Critical Role in Angiogenesis. Arter. Thromb. Vasc. Biol. 2014, 34, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, K.A.; Mäyränpää, M.I.; Kovanen, P.T. Mast Cells in Vulnerable Atherosclerotic Plaques—A View to a Kill. J. Cell Mol. Med. 2007, 11, 739–758. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Steinlechner, B.; Zimpfer, D.; Schlöglhofer, T.; Schima, H.; Hülsmann, M.; Lang, I.M.; Gremmel, T.; Koppensteiner, R.; Zehetmayer, S.; et al. Functional Capillary Impairment in Patients with Ventricular Assist Devices. Sci. Rep. 2019, 9, 5909. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Hülsmann, M.; Schörgenhofer, C.; Lang, I.M.; Wurm, R.; Gremmel, T.; Koppensteiner, R.; Steinlechner, B.; Schwameis, M.; Jilma, B. Sublingual Functional Capillary Rarefaction in Chronic Heart Failure. Eur. J. Clin. Investig. 2018, 48, e12869. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Schörgenhofer, C.; Rieder, T.; Ertl, S.; Pultar, J.; Serles, W.; Sycha, T.; Mayer, F.; Koppensteiner, R.; Gremmel, T.; et al. Microvascular Rarefaction in Patients with Cerebrovascular Events. Microvasc. Res. 2022, 140, 104300. [Google Scholar] [CrossRef]
- Pultar, J.; Ertl, S.; Weikert, C.; Gremmel, T.; Kopp, C.; Mitteregger, M.; Cenan, E.; Jilma, B.; Koppensteiner, R.; Wadowski, P. Systemic Capillary Rarefaction in Patients with Peripheral Arterial Disease. Wien. Klin. Wochenschr. 2022, 134, 211–227. [Google Scholar] [CrossRef]
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526. [Google Scholar] [CrossRef]
- Welten, G.M.; Schouten, O.; Hoeks, S.E.; Chonchol, M.; Vidakovic, R.; van Domburg, R.T.; Bax, J.J.; van Sambeek, M.R.; Poldermans, D. Long-Term Prognosis of Patients With Peripheral Arterial Disease: A Comparison in Patients With Coronary Artery Disease. J. Am. Coll. Cardiol. 2008, 51, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Abdoli, S.; Katz, S.; Ochoa, C. Long-Term Patency and Clinical Outcomes of Nitinol Stenting for Femoropopliteal Atherosclerotic Disease. Ann. Vasc. Surg. 2020, 66, 566–572. [Google Scholar] [CrossRef]
- Kaplovitch, E.; Eikelboom, J.W.; Dyal, L.; Aboyans, V.; Abola, M.T.; Verhamme, P.; Avezum, A.; Fox, K.A.A.; Berkowitz, S.D.; Bangdiwala, S.I.; et al. Rivaroxaban and Aspirin in Patients With Symptomatic Lower Extremity Peripheral Artery Disease: A Subanalysis of the COMPASS Randomized Clinical Trial. JAMA Cardiol. 2021, 6, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zanini, G.; Selleri, V.; Roncati, L.; Coppi, F.; Nasi, M.; Farinetti, A.; Manenti, A.; Pinti, M.; Mattioli, A.V. Vascular “Long COVID”: A New Vessel Disease? Angiology, 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Wadowski, P.P.; Piechota-Polańczyk, A.; Andreas, M.; Kopp, C.W. Cardiovascular Disease Management in the Context of Global Crisis. Int. J. Environ. Res. Public Health 2022, 20, 689. [Google Scholar] [CrossRef] [PubMed]
- Panagiotides, N.G.; Zimprich, F.; Machold, K.; Schlager, O.; Müller, M.; Ertl, S.; Löffler-Stastka, H.; Koppensteiner, R.; Wadowski, P.P. A Case of Autoimmune Small Fiber Neuropathy as Possible Post COVID Sequelae. Int. J. Environ. Res. Public Health 2023, 20, 4918. [Google Scholar] [CrossRef]
- Yue, C.; Zhang, C.; Ying, C.; Jiang, H. Reduced Serum Cholinesterase Is an Independent Risk Factor for All-Cause Mortality in the Pediatric Intensive Care Unit. Front. Nutr. 2022, 9, 809449. [Google Scholar] [CrossRef]
- Shao, X.; Yang, L.; Hu, K.; Shen, R.; Ye, Q.; Yuan, X.; Zhao, Q.; Shen, J. Serum Cholinesterases, a Novel Marker of Clinical Activity in Inflammatory Bowel Disease: A Retrospective Case-Control Study. Mediat. Inflamm. 2020, 2020, 4694090. [Google Scholar] [CrossRef]
- Gremmel, T.; Wadowski, P.P.; Mueller, M.; Kopp, C.W.; Koppensteiner, R.; Panzer, S. Serum Cholinesterase Levels Are Associated With 2-Year Ischemic Outcomes After Angioplasty and Stenting for Peripheral Artery Disease. J. Endovasc. Ther. 2016, 23, 738–743. [Google Scholar] [CrossRef]
- Turk, B.R.; Gschwandtner, M.E.; Mauerhofer, M.; Löffler-Stastka, H. Can We Clinically Recognize a Vascular Depression? The Role of Personality in an Expanded Threshold Model. Medicine 2015, 94, e743. [Google Scholar] [CrossRef]
- Balin, M.; Kivrak, T. Effect of Repeated Remote Ischemic Preconditioning on Peripheral Arterial Disease in Patients Suffering from Intermittent Claudication. Cardiovasc. Ther. 2019, 2019, 9592378. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. [Google Scholar] [CrossRef]
- Dardano, A.; Miccoli, R.; Bianchi, C.; Daniele, G.; Del Prato, S. Invited Review. Series: Implications of the Recent CVOTs in Type 2 Diabetes: Which Patients for GLP-1RA or SGLT-2 Inhibitor? Diabetes Res. Clin. Pract. 2020, 162, 108112. [Google Scholar] [CrossRef]
- Jaipersad, A.S.; Lip, G.Y.H.; Silverman, S.; Shantsila, E. The Role of Monocytes in Angiogenesis and Atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, A22. [Google Scholar] [CrossRef]
- Hurley, D.M.; Williams, E.R.; Cross, J.M.; Riedinger, B.R.; Meyer, R.A.; Abela, G.S.; Slade, J.M. Aerobic Exercise Improves Microvascular Function in Older Adults. Med. Sci. Sports Exerc. 2019, 51, 773–781. [Google Scholar] [CrossRef]
- Steiner, S.; Niessner, A.; Ziegler, S.; Richter, B.; Seidinger, D.; Pleiner, J.; Penka, M.; Wolzt, M.; Huber, K.; Wojta, J.; et al. Endurance Training Increases the Number of Endothelial Progenitor Cells in Patients with Cardiovascular Risk and Coronary Artery Disease. Atherosclerosis 2005, 181, 305–310. [Google Scholar] [CrossRef]

| Triggers | Involved Pathways | Resultant Effects | |
|---|---|---|---|
| Endothelial dysfunction | glycocalyx degradation [26] ROS [11] ETs [144,145] | eNOS ↓ [105] TLR/MyD88/MAPK/NF-ĸB ↑ [34] thrombin/PAR-1 and 4 ↑ [46] | endothelial permeability ↑ [30] leukocyte rolling and diapedesis ↑ [28,107] platelet adhesion and activation ↑ [29] binding of anticoagulant mediators ↓ [27] endothelium-induced vasodilation ↓ [120,121] macrophage M1 polarisation ↑ [83] |
| (Oxidised) LDL accumulation | endothelial dysfunction and glycocalyx degradation [32] hyperlipidaemia [3] | scavenger receptor A [242,243] TLR-2 and -6/MyD88/MAPK/NF-ĸB ↑ [151] NLRP3/IL-1β ↑ [61,152] PKC/IRAK/MAPK ↑ [151] | NET formation ↑ [151] ROS ↑ [151] endothelial dysfunction ↑ [61] SMC proliferation ↑ [61] leukocyte recruitment ↑ [61] MCP-1 ↑ [61] |
| Oxidative stress and ROS | I/R injury [10] microparticles [120,121] NETs [146,147] NF-ĸB [244] AGE [10] | NF-ĸB ↑ [244] PPARγ/AMPK/eNOS ↓ [245,246,247] NLRP3/IL-1β ↑ [11] adiponectin ↓ [247,248,249,250] | endothelial dysfunction ↑ [11,110] ET and MP formation ↑ [91,150] SMC proliferation ↑ [251] ICAM-1, VCAM-1 ↑ [252] |
| Ischaemia and reperfusion | Impaired perfusion and resolution | TCA dysfunction with succinate accumulation [79] HIF-1α/IL-1β ↑ [80] NO bioavailability ↓ [78] ROS ↑ [82] EPC function ↓ [85] | ROS ↑ [11] macrophage M1 polarisation ↑ [83] microvascular perfusion ↓ [11] regenerative potential ↓ [85] |
| TLR activation | oxLDL [151] DAMPs (oxLDL, HSP, fibronectin, fibrinogen, hyaluronic acid derivates) [34,189] PAMPs (bacterial, viral and fungal) [34,189] | caspase 3 ↑ [85,90] TLR/MyD88/MAPK/NF-ĸB ↑ [34] NLRP3/IL-1β ↑ [34] | EPC apoptosis ↑ [85,90] endothelial dysfunction ↑ [61] SMC proliferation ↑ [61] leukocyte recruitment ↑ [61] MCP-1 ↑ [61] aneurysm formation ↑ [253] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poledniczek, M.; Neumayer, C.; Kopp, C.W.; Schlager, O.; Gremmel, T.; Jozkowicz, A.; Gschwandtner, M.E.; Koppensteiner, R.; Wadowski, P.P. Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches. Biomedicines 2023, 11, 2284. https://doi.org/10.3390/biomedicines11082284
Poledniczek M, Neumayer C, Kopp CW, Schlager O, Gremmel T, Jozkowicz A, Gschwandtner ME, Koppensteiner R, Wadowski PP. Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches. Biomedicines. 2023; 11(8):2284. https://doi.org/10.3390/biomedicines11082284
Chicago/Turabian StylePoledniczek, Michael, Christoph Neumayer, Christoph W. Kopp, Oliver Schlager, Thomas Gremmel, Alicja Jozkowicz, Michael E. Gschwandtner, Renate Koppensteiner, and Patricia P. Wadowski. 2023. "Micro- and Macrovascular Effects of Inflammation in Peripheral Artery Disease—Pathophysiology and Translational Therapeutic Approaches" Biomedicines 11, no. 8: 2284. https://doi.org/10.3390/biomedicines11082284