
Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection

 ,
,
Abstract
:1. Introduction
2. Autophagy—A Brief Overview during Mycobacterial Infection
2.1. Autophagy
2.2. Autophagy during Mycobacterial Infection
2.3. Mycobacteria Defense Mechanisms Against Host Immune Response
3. Measuring Autophagic Activity
3.1. Monitoring Autophagic Structures
3.2. LC3—A Lipidated Autophagic Protein Marker
3.3. p62/SQSTM1—An Autophagic Substrate Marker
3.4. Other Markers to Monitor Autophagic Activity
4. Potential Autophagy Activating Drugs for Host Directed Therapy against Mycobacterial Infection in Pre-Clinical Trials
4.1. Small-Molecules
4.2. Immunosuppressants
4.3. Immunomodulators
4.4. Plant Compounds
4.5. Antibiotics
4.6. Steroids
4.7. Anti-Cancer Drugs
4.8. Anti-Diabetic Drugs
4.9. Anti-Diarrheal Drugs
4.10. Anti-Protozoal Drug
4.11. Anti-Seizure Drugs
4.12. Lipid Lowering Drugs
4.13. Mucoactive Drugs
4.14. Psychotropic Drugs
5. Host Directed Therapeutic Drugs Tested as Adjuncts for Tuberculosis in Clinical Trials
6. Challenges in Studying Autophagy Activating Host Directed Therapeutic Drugs to Improve TB Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Global Tuberculosis Report. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports (accessed on 10 April 2021).
- Ehrt, S.; Schnappinger, D. Mycobacterial survival strategies in the phagosome: Defence against host stresses. Cell Microbiol. 2009, 11, 1170–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, C.; Walzl, G.; Du Plessis, N. Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol. 2020, 13, 190–204. [Google Scholar] [CrossRef] [Green Version]
- Kolloli, A.; Subbian, S. Host-Directed Therapeutic Strategies for Tuberculosis. Front. Med. 2017, 4, 171. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Wang, L.; Liu, C.H.; Ge, B. New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 2020, 17, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, I.; Amano, A.; Mizushima, N.; Yamamoto, A.; Yamaguchi, H.; Kamimoto, T.; Nara, A.; Funao, J.; Nakata, M.; Tsuda, K.; et al. Autophagy defends cells against invading group A Streptococcus. Science 2004, 306, 1037–1040. [Google Scholar] [CrossRef]
- Bento, C.F.; Empadinhas, N.; Mendes, V. Autophagy in the fight against tuberculosis. DNA Cell Biol. 2015, 34, 228–242. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Selective types of autophagy. Int. J. Cell Biol. 2012, 2012, 156272. [Google Scholar] [CrossRef] [Green Version]
- Cooper, K.F. Till Death Do Us Part: The Marriage of Autophagy and Apoptosis. Oxidative Med. Cell. Longev. 2018, 2018, 4701275. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.L.; Reggiori, F.; Baba, M.; Kovacs, A.L.; Seglen, P.O. Seeing is believing: The impact of electron microscopy on autophagy research. Autophagy 2011, 7, 935–956. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cuervo, A.M.; Dunn, W.A., Jr.; Levine, B.; van der Klei, I.; Seglen, P.O. How shall I eat thee? Autophagy 2007, 3, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Abdrakhmanov, A.; Gogvadze, V.; Zhivotovsky, B. To Eat or to Die: Deciphering Selective Forms of Autophagy. Trends Biochem. Sci. 2020, 45, 347–364. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Fullgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, L.C.; Dikic, I. Autophagy in antimicrobial immunity. Mol. Cell 2014, 54, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V. Autophagy in tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 4, a018481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.J. Amino acid regulation of autophagosome formation. Methods Mol. Biol. 2008, 445, 89–109. [Google Scholar] [CrossRef]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.J.; Codogno, P. AMP-activated protein kinase and autophagy. Autophagy 2007, 3, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romagnoli, A.; Etna, M.P.; Giacomini, E.; Pardini, M.; Remoli, M.E.; Corazzari, M.; Falasca, L.; Goletti, D.; Gafa, V.; Simeone, R.; et al. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 2012, 8, 1357–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, T.L.; Wandel, M.P.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.H.; Nair, V.R.; Scharn, C.R.; Xavier, R.J.; Torrealba, J.R.; Shiloh, M.U.; Levine, B. The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium tuberculosis and Anti-tuberculous Host Defense. Cell Host Microbe 2017, 21, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef]
- Jo, E.K. Autophagy as an innate defense against mycobacteria. Pathog. Dis. 2013, 67, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, E.; Carranza, C.; Hernandez-Sanchez, F.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Escobedo, D.; Torres, M.; Sada, E. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur. J. Immunol. 2012, 42, 880–889. [Google Scholar] [CrossRef]
- Singh, V.; Jamwal, S.; Jain, R.; Verma, P.; Gokhale, R.; Rao, K.V. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 2012, 12, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Zullo, A.J.; Lee, S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J. Biol. Chem. 2012, 287, 12668–12678. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; An, D.R.; Song, J.; Yoon, J.Y.; Kim, H.S.; Yoon, H.J.; Im, H.N.; Kim, J.; Kim, D.J.; Lee, S.J.; et al. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. USA 2012, 109, 7729–7734. [Google Scholar] [CrossRef] [Green Version]
- Shui, W.; Petzold, C.J.; Redding, A.; Liu, J.; Pitcher, A.; Sheu, L.; Hsieh, T.Y.; Keasling, J.D.; Bertozzi, C.R. Organelle membrane proteomics reveals differential influence of mycobacterial lipoglycans on macrophage phagosome maturation and autophagosome accumulation. J. Proteome Res. 2011, 10, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Koster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, H.E.; de Lartigue, J.; Rigden, D.J.; Reedijk, M.; Urbe, S.; Clague, M.J.; Tooze, S.A. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010, 6, 506–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, P.K.; Fahrner, A.; Vats, S.; Seranova, E.; Sharma, V.; Chipara, M.; Desai, P.; Torresi, J.; Rosenstock, T.; Kumar, D.; et al. Chemical Screening Approaches Enabling Drug Discovery of Autophagy Modulators for Biomedical Applications in Human Diseases. Front. Cell Dev. Biol. 2019, 7, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Murphy, L.O. Autophagy Assays for Biological Discovery and Therapeutic Development. Trends Biochem. Sci. 2020, 45, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Matsui, M.; Mizushima, N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy 2007, 3, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shvets, E.; Fass, E.; Elazar, Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy 2008, 4, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Koepke, L.; Winter, B.; Grenzner, A.; Regensburger, K.; Engelhart, S.; van der Merwe, J.A.; Krebs, S.; Blum, H.; Kirchhoff, F.; Sparrer, K.M.J. An improved method for high-throughput quantification of autophagy in mammalian cells. Sci. Rep. 2020, 10, 12241. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef] [Green Version]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An Autophagic Flux Probe that Releases an Internal Control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Maloyan, A.; Montes, A.M.C. Erratum. Autophagy 2016, 12, 443. [Google Scholar] [CrossRef] [Green Version]
- Larsen, K.B.; Lamark, T.; Overvatn, A.; Harneshaug, I.; Johansen, T.; Bjorkoy, G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010, 6, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Min, Z.; Ting, Y.; Mingtao, G.; Xiaofei, T.; Dong, Y.; Chenguang, Z.; Wei, D. Monitoring autophagic flux using p62/SQSTM1 based luciferase reporters in glioma cells. Exp. Cell Res. 2018, 363, 84–94. [Google Scholar] [CrossRef]
- Bresciani, A.; Spiezia, M.C.; Boggio, R.; Cariulo, C.; Nordheim, A.; Altobelli, R.; Kuhlbrodt, K.; Dominguez, C.; Munoz-Sanjuan, I.; Wityak, J.; et al. Quantifying autophagy using novel LC3B and p62 TR-FRET assays. PLoS ONE 2018, 13, e0194423. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proikas-Cezanne, T.; Pfisterer, S.G. Assessing mammalian autophagy by WIPI-1/Atg18 puncta formation. Methods Enzymol. 2009, 452, 247–260. [Google Scholar] [CrossRef]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Alsaadi, R.; Guo, Z.; Kalinina, A.; Carrier, M.; Tremblay, M.E.; Lacoste, B.; Lagace, D.; Russell, R.C. An antibody for analysis of autophagy induction. Nat. Methods 2020, 17, 232–239. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P. Untangling autophagy measurements: All fluxed up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Meyer, H. Detection and Clearance of Damaged Lysosomes by the Endo-Lysosomal Damage Response and Lysophagy. Curr. Biol. 2017, 27, R1330–R1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Matsuura, A.; Itakura, E. Identification of a factor controlling lysosomal homeostasis using a novel lysosomal trafficking probe. Sci. Rep. 2019, 9, 11635. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floto, R.A.; Sarkar, S.; Perlstein, E.O.; Kampmann, B.; Schreiber, S.L.; Rubinsztein, D.C. Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 2007, 3, 620–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.S.; Kim, J.J.; Lee, H.M.; Jin, H.S.; Lee, S.H.; Park, J.H.; Kim, S.J.; Kim, J.M.; Han, Y.M.; Lee, M.S.; et al. The AMPK-PPARGC1A pathway is required for antimicrobial host defense through activation of autophagy. Autophagy 2014, 10, 785–802. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Bhagyaraj, E.; Nanduri, R.; Ahuja, N.; Gupta, P. NR1D1 ameliorates Mycobacterium tuberculosis clearance through regulation of autophagy. Autophagy 2015, 11, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Lee, H.M.; Kim, J.K.; Yang, C.S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.Y.; Gutierrez, N.M.; Marzuki, M.B.; Lu, X.; Foreman, T.W.; Paleja, B.; Lee, B.; Balachander, A.; Chen, J.; Tsenova, L.; et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2017, 2, eaaj1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, G.; Gagandeep; Behura, A.; Gosain, T.P.; Shaliwal, R.P.; Kidwai, S.; Singh, P.; Kandi, S.K.; Dhiman, R.; Rawat, D.S.; et al. NSC 18725, a Pyrazole Derivative Inhibits Growth of Intracellular Mycobacterium tuberculosis by Induction of Autophagy. Front. Microbiol. 2019, 10, 3051. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Kim, Y.S.; Lee, H.M.; Jin, H.S.; Neupane, C.; Kim, S.; Lee, S.H.; Min, J.J.; Sasai, M.; Jeong, J.H.; et al. GABAergic signaling linked to autophagy enhances host protection against intracellular bacterial infections. Nat. Commun. 2018, 9, 4184. [Google Scholar] [CrossRef] [Green Version]
- Sivangala Thandi, R.; Radhakrishnan, R.K.; Tripathi, D.; Paidipally, P.; Azad, A.K.; Schlesinger, L.S.; Samten, B.; Mulik, S.; Vankayalapati, R. Ornithine—A urea cycle metabolite enhances autophagy and controls Mycobacterium tuberculosis infection. Nat. Commun. 2020, 11, 3535. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Makhdoomi, M.; Singh, L.; Kumar, P.; Khan, N.; Singh, S.; Verma, H.N.; Luthra, K.; Sarkar, S.; Kumar, D. Trehalose limits opportunistic mycobacterial survival during HIV co-infection by reversing HIV-mediated autophagy block. Autophagy 2021, 17, 476–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, J.E.; Blaikley, J.; Beesley, S.; Matthews, L.; Simpson, K.D.; Boyce, S.H.; Farrow, S.N.; Else, K.J.; Singh, D.; Ray, D.W.; et al. The nuclear receptor REV-ERBalpha mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerni, S.; Shafer, D.; To, K.; Venketaraman, V. Investigating the Role of Everolimus in mTOR Inhibition and Autophagy Promotion as a Potential Host-Directed Therapeutic Target in Mycobacterium tuberculosis Infection. J. Clin. Med. 2019, 8, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Yang, C.S.; Jin, H.S.; Kim, K.K.; Lee, Z.W.; Lee, S.H.; Kim, J.M.; Jo, E.K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabri, M.; Stenger, S.; Shin, D.M.; Yuk, J.M.; Liu, P.T.; Realegeno, S.; Lee, H.M.; Krutzik, S.R.; Schenk, M.; Sieling, P.A.; et al. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci. Transl. Med. 2011, 3, 104ra102. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kang, S.J.; Woo, Y.; Hahn, T.W.; Ko, H.J.; Jung, Y.J. TLR7 Stimulation With Imiquimod Induces Selective Autophagy and Controls Mycobacterium tuberculosis Growth in Mouse Macrophages. Front. Microbiol. 2020, 11, 1684. [Google Scholar] [CrossRef]
- Fang, F.; Ge, Q.; Li, R.; Lv, J.; Zhang, Y.; Feng, A.; Kelly, G.T.; Wang, H.; Wang, X.; Song, C.; et al. LPS restores protective immunity in macrophages against Mycobacterium tuberculosis via autophagy. Mol. Immunol. 2020, 124, 18–24. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, J.; Wang, Y.; He, W.; Wang, L.; Zheng, Y.; Wu, J.; Zhang, Y.; Jiang, X. Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium tuberculosis. Front. Microbiol. 2017, 8, 2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Ko, H.J.; Kim, S.H.; Jung, Y.J. Pasakbumin A controls the growth of Mycobacterium tuberculosis by enhancing the autophagy and production of antibacterial mediators in mouse macrophages. PLoS ONE 2019, 14, e0199799. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Vaghasiya, K.; Ray, E.; Gupta, P.; Gupta, U.D.; Singh, A.K.; Verma, R.K. Targeted Pulmonary Delivery of the Green Tea Polyphenol Epigallocatechin Gallate Controls the Growth of Mycobacterium tuberculosis by Enhancing the Autophagy and Suppressing Bacterial Burden. ACS Biomater. Sci. Eng. 2020, 6, 4126–4140. [Google Scholar] [CrossRef]
- Kim, T.S.; Jin, Y.B.; Kim, Y.S.; Kim, S.; Kim, J.K.; Lee, H.M.; Suh, H.W.; Choe, J.H.; Kim, Y.J.; Koo, B.S.; et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy 2019, 15, 1356–1375. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Behura, A.; Kumar, A.; Ghosh, A.; Naik, L.; Mawatwal, S.; Mohanty, S.S.; Saha, S.; Bhutia, S.K.; Singh, R.; et al. Soybean lectin induces autophagy through P2RX7 dependent activation of NF-kappaB-ROS pathway to kill intracellular mycobacteria. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129806. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, H.M.; Shin, D.M.; Kim, W.; Yuk, J.M.; Jin, H.S.; Lee, S.H.; Cha, G.H.; Kim, J.M.; Lee, Z.W.; et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012, 11, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Wang, Q.; Wang, S.; Wu, J.; Gao, Q.; Liu, W. Thiopeptide Antibiotics Exhibit a Dual Mode of Action against Intracellular Pathogens by Affecting Both Host and Microbe. Chem. Biol. 2015, 22, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawatwal, S.; Behura, A.; Mishra, A.; Singh, R.; Dhiman, R. Calcimycin induced IL-12 production inhibits intracellular mycobacterial growth by enhancing autophagy. Cytokine 2018, 111, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mawatwal, S.; Behura, A.; Ghosh, A.; Kidwai, S.; Mishra, A.; Deep, A.; Agarwal, S.; Saha, S.; Singh, R.; Dhiman, R. Calcimycin mediates mycobacterial killing by inducing intracellular calcium-regulated autophagy in a P2RX7 dependent manner. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3190–3200. [Google Scholar] [CrossRef]
- Bongiovanni, B.; Mata-Espinosa, D.; D’Attilio, L.; Leon-Contreras, J.C.; Marquez-Velasco, R.; Bottasso, O.; Hernandez-Pando, R.; Bay, M.L. Effect of cortisol and/or DHEA on THP1-derived macrophages infected with Mycobacterium tuberculosis. Tuberculosis 2015, 95, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Barczak, A.K.; Silvis, M.R.; Luo, S.S.; Sogi, K.; Vokes, M.; Bray, M.A.; Carpenter, A.E.; Moore, C.B.; Siddiqi, N.; et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014, 10, e1003946. [Google Scholar] [CrossRef] [Green Version]
- Rekha, R.S.; Rao Muvva, S.S.; Wan, M.; Raqib, R.; Bergman, P.; Brighenti, S.; Gudmundsson, G.H.; Agerberth, B. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy 2015, 11, 1688–1699. [Google Scholar] [CrossRef] [Green Version]
- Bruns, H.; Stegelmann, F.; Fabri, M.; Dohner, K.; van Zandbergen, G.; Wagner, M.; Skinner, M.; Modlin, R.L.; Stenger, S. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J. Immunol. 2012, 189, 4069–4078. [Google Scholar] [CrossRef] [Green Version]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Dong, H.; Hussain Mangi, M.; Ni, J.; et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells 2019, 8, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Wen, Z.; Liu, S.; Cai, Y.; Guo, J.; Xu, Y.; Lin, D.; Zhu, J.; Li, D.; Chen, X. Ibrutinib suppresses intracellular mycobacterium tuberculosis growth by inducing macrophage autophagy. J. Infect. 2020, 80, e19–e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Q.; Zhang, K.; Lin, D.; Feng, C.G.; Cai, Y.; Chen, X. Bazedoxifene Suppresses Intracellular Mycobacterium tuberculosis Growth by Enhancing Autophagy. mSphere 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef]
- Juarez, E.; Carranza, C.; Sanchez, G.; Gonzalez, M.; Chavez, J.; Sarabia, C.; Torres, M.; Sada, E. Loperamide Restricts Intracellular Growth of Mycobacterium tuberculosis in Lung Macrophages. Am. J. Respir. Cell Mol. Biol. 2016, 55, 837–847. [Google Scholar] [CrossRef]
- Lam, K.K.; Zheng, X.; Forestieri, R.; Balgi, A.D.; Nodwell, M.; Vollett, S.; Anderson, H.J.; Andersen, R.J.; Av-Gay, Y.; Roberge, M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012, 8, e1002691. [Google Scholar] [CrossRef] [Green Version]
- Schiebler, M.; Brown, K.; Hegyi, K.; Newton, S.M.; Renna, M.; Hepburn, L.; Klapholz, C.; Coulter, S.; Obregon-Henao, A.; Henao Tamayo, M.; et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 2015, 7, 127–139. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 2014, 209, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Guerra-De-Blas, P.D.C.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Estrada-Garcia, I.; Torres-Gonzalez, P.; Lopez-Saavedra, A.; Guzman-Beltran, S.; Ponce-de-Leon, A.; Sifuentes-Osornio, J. Simvastatin Enhances the Immune Response Against Mycobacterium tuberculosis. Front. Microbiol. 2019, 10, 2097. [Google Scholar] [CrossRef] [PubMed]
- Bruiners, N.; Dutta, N.K.; Guerrini, V.; Salamon, H.; Yamaguchi, K.D.; Karakousis, P.C.; Gennaro, M.L. The anti-tubercular activity of simvastatin is mediated by cholesterol-driven autophagy via the AMPK-mTORC1-TFEB axis. J. Lipid Res. 2020, 61, 1617–1628. [Google Scholar] [CrossRef]
- Lobato, L.S.; Rosa, P.S.; Ferreira Jda, S.; Neumann Ada, S.; da Silva, M.G.; do Nascimento, D.C.; Soares, C.T.; Pedrini, S.C.; Oliveira, D.S.; Monteiro, C.P.; et al. Statins increase rifampin mycobactericidal effect. Antimicrob. Agents Chemother. 2014, 58, 5766–5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.W.; Gu, Y.; Peters, R.S.; Salgame, P.; Ellner, J.J.; Timmins, G.S.; Deretic, V. Ambroxol Induces Autophagy and Potentiates Rifampin Antimycobacterial Activity. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaramurthy, V.; Barsacchi, R.; Samusik, N.; Marsico, G.; Gilleron, J.; Kalaidzidis, I.; Meyenhofer, F.; Bickle, M.; Kalaidzidis, Y.; Zerial, M. Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe 2013, 13, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Sauve, A.A. NAD metabolism and sirtuins: Metabolic regulation of protein deacetylation in stress and toxicity. AAPS J. 2006, 8, E632–E643. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.W. Escherichia coli acid resistance: Tales of an amateur acidophile. Nat. Rev. Microbiol. 2004, 2, 898–907. [Google Scholar] [CrossRef]
- Dong, X.P.; Shen, D.; Wang, X.; Dawson, T.; Li, X.; Zhang, Q.; Cheng, X.; Zhang, Y.; Weisman, L.S.; Delling, M.; et al. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat. Commun. 2010, 1, 38. [Google Scholar] [CrossRef] [Green Version]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, Hormone, and Immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Ezell, S.A.; Wang, S.; Bihani, T.; Lai, Z.; Grosskurth, S.E.; Tepsuporn, S.; Davies, B.R.; Huszar, D.; Byth, K.F. Differential regulation of mTOR signaling determines sensitivity to AKT inhibition in diffuse large B cell lymphoma. Oncotarget 2016, 7, 9163–9174. [Google Scholar] [CrossRef] [Green Version]
- Chaudhury, A.; Duvoor, C.; Reddy Dendi, V.S.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical Review of Antidiabetic Drugs: Implications for Type 2 Diabetes Mellitus Management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J. Pharmacologic Agents for Chronic Diarrhea. Intest. Res. 2015, 13, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spina, E.; Perugi, G. Antiepileptic drugs: Indications other than epilepsy. Epileptic Disord. 2004, 6, 57–75. [Google Scholar] [PubMed]
- Cascade, E.; Kalali, A.H.; Weisler, R.H. Varying uses of anticonvulsant medications. Psychiatry 2008, 5, 31–33. [Google Scholar]
- Pahan, K. Lipid-lowering drugs. Cell Mol. Life Sci. 2006, 63, 1165–1178. [Google Scholar] [CrossRef]
- Balsamo, R.; Lanata, L.; Egan, C.G. Mucoactive drugs. Eur. Respir. Rev. 2010, 19, 127–133. [Google Scholar] [CrossRef]
- Frank, R.G.; Conti, R.M.; Goldman, H.H. Mental health policy and psychotropic drugs. Milbank Q. 2005, 83, 271–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, N.K.; Bruiners, N.; Pinn, M.L.; Zimmerman, M.D.; Prideaux, B.; Dartois, V.; Gennaro, M.L.; Karakousis, P.C. Statin adjunctive therapy shortens the duration of TB treatment in mice. J. Antimicrob. Chemother. 2016, 71, 1570–1577. [Google Scholar] [CrossRef] [Green Version]
- Mily, A.; Rekha, R.S.; Kamal, S.M.; Arifuzzaman, A.S.; Rahim, Z.; Khan, L.; Haq, M.A.; Zaman, K.; Bergman, P.; Brighenti, S.; et al. Significant Effects of Oral Phenylbutyrate and Vitamin D3 Adjunctive Therapy in Pulmonary Tuberculosis: A Randomized Controlled Trial. PLoS ONE 2015, 10, e0138340. [Google Scholar] [CrossRef]
- Padmapriyadarsini, C.; Bhavani, P.K.; Natrajan, M.; Ponnuraja, C.; Kumar, H.; Gomathy, S.N.; Guleria, R.; Jawahar, S.M.; Singh, M.; Balganesh, T.; et al. Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF): Study protocol for a randomised clinical trial. BMJ Open 2019, 9, e024363. [Google Scholar] [CrossRef] [PubMed]
- Martineau, A.R.; Timms, P.M.; Bothamley, G.H.; Hanifa, Y.; Islam, K.; Claxton, A.P.; Packe, G.E.; Moore-Gillon, J.C.; Darmalingam, M.; Davidson, R.N.; et al. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: A double-blind randomised controlled trial. Lancet 2011, 377, 242–250. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Subbian, S. Harnessing the mTOR Pathway for Tuberculosis Treatment. Front. Microbiol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallis, R.S.; Ginindza, S.; Beattie, T.; Arjun, N.; Likoti, M.; Edward, V.A.; Rassool, M.; Ahmed, K.; Fielding, K.; Ahidjo, B.A.; et al. Adjunctive host-directed therapies for pulmonary tuberculosis: A prospective, open-label, phase 2, randomised controlled trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Rekha, R.S.; Mily, A.; Sultana, T.; Haq, A.; Ahmed, S.; Mostafa Kamal, S.M.; van Schadewijk, A.; Hiemstra, P.S.; Gudmundsson, G.H.; Agerberth, B.; et al. Immune responses in the treatment of drug-sensitive pulmonary tuberculosis with phenylbutyrate and vitamin D3 as host directed therapy. BMC Infect. Dis. 2018, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Tukvadze, N.; Sanikidze, E.; Kipiani, M.; Hebbar, G.; Easley, K.A.; Shenvi, N.; Kempker, R.R.; Frediani, J.K.; Mirtskhulava, V.; Alvarez, J.A.; et al. High-dose vitamin D3 in adults with pulmonary tuberculosis: A double-blind randomized controlled trial. Am. J. Clin. Nutr. 2015, 102, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Salahuddin, N.; Ali, F.; Hasan, Z.; Rao, N.; Aqeel, M.; Mahmood, F. Vitamin D accelerates clinical recovery from tuberculosis: Results of the SUCCINCT Study [Supplementary Cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis’. BMC Infect. Dis. 2013, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Ganmaa, D.; Giovannucci, E.; Bloom, B.R.; Fawzi, W.; Burr, W.; Batbaatar, D.; Sumberzul, N.; Holick, M.F.; Willett, W.C. Vitamin D, tuberculin skin test conversion, and latent tuberculosis in Mongolian school-age children: A randomized, double-blind, placebo-controlled feasibility trial. Am. J. Clin. Nutr. 2012, 96, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Ralph, A.P.; Waramori, G.; Pontororing, G.J.; Kenangalem, E.; Wiguna, A.; Tjitra, E.; Sandjaja; Lolong, D.B.; Yeo, T.W.; Chatfield, M.D.; et al. L-arginine and vitamin D adjunctive therapies in pulmonary tuberculosis: A randomised, double-blind, placebo-controlled trial. PLoS ONE 2013, 8, e70032. [Google Scholar] [CrossRef] [Green Version]
- Bekele, A.; Gebreselassie, N.; Ashenafi, S.; Kassa, E.; Aseffa, G.; Amogne, W.; Getachew, M.; Aseffa, A.; Worku, A.; Raqib, R.; et al. Daily adjunctive therapy with vitamin D3 and phenylbutyrate supports clinical recovery from pulmonary tuberculosis: A randomized controlled trial in Ethiopia. J. Intern. Med. 2018, 284, 292–306. [Google Scholar] [CrossRef]
- Ganmaa, D.; Munkhzul, B.; Fawzi, W.; Spiegelman, D.; Willett, W.C.; Bayasgalan, P.; Baasansuren, E.; Buyankhishig, B.; Oyun-Erdene, S.; Jolliffe, D.A.; et al. High-Dose Vitamin D3 during Tuberculosis Treatment in Mongolia. A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2017, 196, 628–637. [Google Scholar] [CrossRef]
- Daley, P.; Jagannathan, V.; John, K.R.; Sarojini, J.; Latha, A.; Vieth, R.; Suzana, S.; Jeyaseelan, L.; Christopher, D.J.; Smieja, M.; et al. Adjunctive vitamin D for treatment of active tuberculosis in India: A randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2015, 15, 528–534. [Google Scholar] [CrossRef]
- Ganmaa, D.; Uyanga, B.; Zhou, X.; Gantsetseg, G.; Delgerekh, B.; Enkhmaa, D.; Khulan, D.; Ariunzaya, S.; Sumiya, E.; Bolortuya, B.; et al. Vitamin D Supplements for Prevention of Tuberculosis Infection and Disease. N. Engl. J. Med. 2020, 383, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Martineau, A.R.; Wilkinson, R.J.; Wilkinson, K.A.; Newton, S.M.; Kampmann, B.; Hall, B.M.; Packe, G.E.; Davidson, R.N.; Eldridge, S.M.; Maunsell, Z.J.; et al. A single dose of vitamin D enhances immunity to mycobacteria. Am. J. Respir. Crit. Care Med. 2007, 176, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICMR. The Clinical Trials Registry- India (CTRI). Available online: http://www.ctri.nic.in/Clinicaltrials/showallp.php?mid1=20868&EncHid=&userName=011176 (accessed on 13 May 2021).
- NIH. Clinical Trials. Available online: https://clinicaltrials.gov/ct2/results?cond=Tuberculosis&term=Vitamin+D&cntry=&state=&city=&dist= (accessed on 13 May 2021).
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.K. Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 2019, 10, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Malherbe, S.T.; Shenai, S.; Ronacher, K.; Loxton, A.G.; Dolganov, G.; Kriel, M.; Van, T.; Chen, R.Y.; Warwick, J.; Via, L.E.; et al. Persisting positron emission tomography lesion activity and Mycobacterium tuberculosis mRNA after tuberculosis cure. Nat. Med. 2016, 22, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
 ): Activation, (
): Activation, (  ):inhibition, (
):inhibition, (  ): Drug induced activation, (
): Drug induced activation, (  ): Drug induced inhibition, (
): Drug induced inhibition, (  ): Upregulation, (
): Upregulation, (  ): Downregulation, (
): Downregulation, (  ): Drug induced upregulation, (
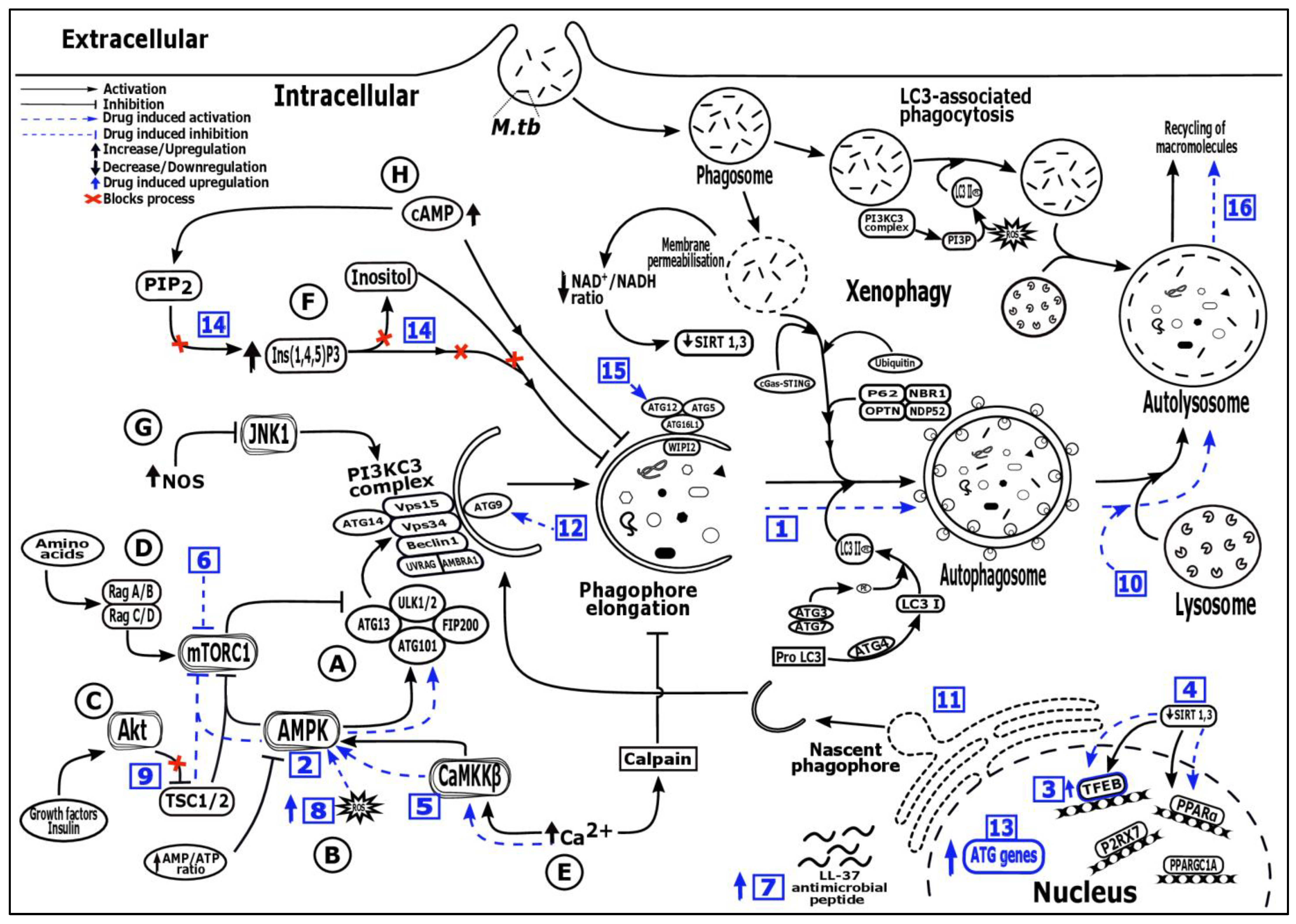
): Drug induced upregulation, (  ): Blocks activation process, Numbers in blue boxes indicate drug induced activation of the following processes—1: Increased autophagosome formation (SMER 18 & 28, nortriptyline); 2: AMPK activation: (AICAR, ornithine, metformin, statins); 3: TFEB signaling upregulation (GSK4112, GW7647, trehalose, Wy14643, honokiol, ambroxol); 4: SIRT 1, 3 activator (SRT 1720, resveratrol, honokiol); 5: Ca2+—AMPK signaling (GABA, vitamin D, soyabean lectin, isoniazid, pyrazinamide, calcimycin); 6: mTOR inhibition (Rapamycin, evorilimus, nitazoxanide); 7: LL-37 induced expression (Vitamin D, Interferon-γ, 4-phenylbutyrate); 8: Increased ROS production (Imiquimod); 9: PI3K/Akt/mTOR pathway inhibition (Baicalin, imatinib, nilotinib, ibrutinib, bazedoxifene); 10: Autophagic flux induction (Pasakbumin A, fluoxetine); 11: ER stress mediated autophagy induction (Thiostrepton); 12: Depletion of p38 MAPK mediated autophagy activation via p38IP and mATG9 (Gefitnib); 13: Autophagy genes (ATG16L1 and LC3) upregulation (Loperamide); 14: AMPK activation via inositol signaling pathway (Carbamazepine); 15: mTOR independent autophagy formation via ATG 12 (Valproic acid); 16: Slow autophagic flux and increased lysozyme acidification (Prochlorperazine edisylate), Alphabets (A-H) describes the different pathways—A: mTORC1/ULK/ATG13/FIP200 dependent pathway, B: AMPK/TSC/mTORC1 dependent pathway, C: PI3KC1a/Akt/TSC/mTORC1 dependent pathway, D: Rag/mTORC1 dependent pathway, E: mTOR independent Ca2+/calpain pathway, F: mTOR independent inositol signalling pathway, G: mTOR independent JNK1/Beclin1/PI3KC3 pathway, H: mTOR independent cAMP/Ins(1,4,5)P3 pathway, AMBRA1: Activating molecule in BECN1 regulated autophagy protein 1, AMPK: AMP-activated protein kinase, ATG: Autophagy-related genes or proteins, Ca2+: Calcium ions, CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase β, cAMP/Ins(1,4,5)P3: Cyclic adenosine monophosphate/inositol (1,4,5)-trisphosphate, cGAS-STING: Cyclic GMP-AMP Synthase—simulator of interferon genes, FIP200: focal adhesion kinase family-interacting protein of 200 kDa, JNK1: c-Jun N-terminal kinase 1, LC3: Microtubule associated protein 1 (MAP1) light chain 3, LL-37: Human cathelicidin, NAD+/NADH: Nicotinamide adenine dinucleotide (NAD) + hydrogen (H), NBR1: Neighbor of BRCA1 gene 1, NDP52: Nuclear domain 10 protein 52, NOS: Nitric oxide synthase, OPTN: Optineurin, PE: phosphatidylethanolamine, PI3KC3 complex: Class III phosphatidylinositol 3-kinase (PI3K) complex, PI3KC1a/Akt/TSC/mTORC1: Class 1a phosphoinositide 3-kinase/protein kinase B/tuberous sclerosis complex/mTORC1, PI3P: Phosphatidylinositol 3-phosphate, PIP2: Phosphatidylinositol bisphosphate, PPARα: Peroxisome proliferation factor-activated receptor α, PPARGC1A: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, P2RX7: Purinergic Receptor 7, ROS: Reactive oxygen species, Rag/mTORC1: Ras-related GTP-binding protein/mammalian target of rapamycin complex 1, SIRT: Sirtuin protein, TFEB: Transcription factors EB, ULK1/2: UNC-51-Like Ser/Thr kinase, VPS: Vacuolar protein sorting, WIPI2: WD repeat domain phosphoinositide-interacting protein 2.
): Activation, ( ):inhibition, ( ): Drug induced activation, ( ): Drug induced inhibition, ( ): Upregulation, ( ): Downregulation, ( ): Drug induced upregulation, ( ): Blocks activation process, Numbers in blue boxes indicate drug induced activation of the following processes—1: Increased autophagosome formation (SMER 18 & 28, nortriptyline); 2: AMPK activation: (AICAR, ornithine, metformin, statins); 3: TFEB signaling upregulation (GSK4112, GW7647, trehalose, Wy14643, honokiol, ambroxol); 4: SIRT 1, 3 activator (SRT 1720, resveratrol, honokiol); 5: Ca2+—AMPK signaling (GABA, vitamin D, soyabean lectin, isoniazid, pyrazinamide, calcimycin); 6: mTOR inhibition (Rapamycin, evorilimus, nitazoxanide); 7: LL-37 induced expression (Vitamin D, Interferon-γ, 4-phenylbutyrate); 8: Increased ROS production (Imiquimod); 9: PI3K/Akt/mTOR pathway inhibition (Baicalin, imatinib, nilotinib, ibrutinib, bazedoxifene); 10: Autophagic flux induction (Pasakbumin A, fluoxetine); 11: ER stress mediated autophagy induction (Thiostrepton); 12: Depletion of p38 MAPK mediated autophagy activation via p38IP and mATG9 (Gefitnib); 13: Autophagy genes (ATG16L1 and LC3) upregulation (Loperamide); 14: AMPK activation via inositol signaling pathway (Carbamazepine); 15: mTOR independent autophagy formation via ATG 12 (Valproic acid); 16: Slow autophagic flux and increased lysozyme acidification (Prochlorperazine edisylate), Alphabets (A-H) describes the different pathways—A: mTORC1/ULK/ATG13/FIP200 dependent pathway, B: AMPK/TSC/mTORC1 dependent pathway, C: PI3KC1a/Akt/TSC/mTORC1 dependent pathway, D: Rag/mTORC1 dependent pathway, E: mTOR independent Ca2+/calpain pathway, F: mTOR independent inositol signalling pathway, G: mTOR independent JNK1/Beclin1/PI3KC3 pathway, H: mTOR independent cAMP/Ins(1,4,5)P3 pathway, AMBRA1: Activating molecule in BECN1 regulated autophagy protein 1, AMPK: AMP-activated protein kinase, ATG: Autophagy-related genes or proteins, Ca2+: Calcium ions, CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase β, cAMP/Ins(1,4,5)P3: Cyclic adenosine monophosphate/inositol (1,4,5)-trisphosphate, cGAS-STING: Cyclic GMP-AMP Synthase—simulator of interferon genes, FIP200: focal adhesion kinase family-interacting protein of 200 kDa, JNK1: c-Jun N-terminal kinase 1, LC3: Microtubule associated protein 1 (MAP1) light chain 3, LL-37: Human cathelicidin, NAD+/NADH: Nicotinamide adenine dinucleotide (NAD) + hydrogen (H), NBR1: Neighbor of BRCA1 gene 1, NDP52: Nuclear domain 10 protein 52, NOS: Nitric oxide synthase, OPTN: Optineurin, PE: phosphatidylethanolamine, PI3KC3 complex: Class III phosphatidylinositol 3-kinase (PI3K) complex, PI3KC1a/Akt/TSC/mTORC1: Class 1a phosphoinositide 3-kinase/protein kinase B/tuberous sclerosis complex/mTORC1, PI3P: Phosphatidylinositol 3-phosphate, PIP2: Phosphatidylinositol bisphosphate, PPARα: Peroxisome proliferation factor-activated receptor α, PPARGC1A: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, P2RX7: Purinergic Receptor 7, ROS: Reactive oxygen species, Rag/mTORC1: Ras-related GTP-binding protein/mammalian target of rapamycin complex 1, SIRT: Sirtuin protein, TFEB: Transcription factors EB, ULK1/2: UNC-51-Like Ser/Thr kinase, VPS: Vacuolar protein sorting, WIPI2: WD repeat domain phosphoinositide-interacting protein 2.
): Blocks activation process, Numbers in blue boxes indicate drug induced activation of the following processes—1: Increased autophagosome formation (SMER 18 & 28, nortriptyline); 2: AMPK activation: (AICAR, ornithine, metformin, statins); 3: TFEB signaling upregulation (GSK4112, GW7647, trehalose, Wy14643, honokiol, ambroxol); 4: SIRT 1, 3 activator (SRT 1720, resveratrol, honokiol); 5: Ca2+—AMPK signaling (GABA, vitamin D, soyabean lectin, isoniazid, pyrazinamide, calcimycin); 6: mTOR inhibition (Rapamycin, evorilimus, nitazoxanide); 7: LL-37 induced expression (Vitamin D, Interferon-γ, 4-phenylbutyrate); 8: Increased ROS production (Imiquimod); 9: PI3K/Akt/mTOR pathway inhibition (Baicalin, imatinib, nilotinib, ibrutinib, bazedoxifene); 10: Autophagic flux induction (Pasakbumin A, fluoxetine); 11: ER stress mediated autophagy induction (Thiostrepton); 12: Depletion of p38 MAPK mediated autophagy activation via p38IP and mATG9 (Gefitnib); 13: Autophagy genes (ATG16L1 and LC3) upregulation (Loperamide); 14: AMPK activation via inositol signaling pathway (Carbamazepine); 15: mTOR independent autophagy formation via ATG 12 (Valproic acid); 16: Slow autophagic flux and increased lysozyme acidification (Prochlorperazine edisylate), Alphabets (A-H) describes the different pathways—A: mTORC1/ULK/ATG13/FIP200 dependent pathway, B: AMPK/TSC/mTORC1 dependent pathway, C: PI3KC1a/Akt/TSC/mTORC1 dependent pathway, D: Rag/mTORC1 dependent pathway, E: mTOR independent Ca2+/calpain pathway, F: mTOR independent inositol signalling pathway, G: mTOR independent JNK1/Beclin1/PI3KC3 pathway, H: mTOR independent cAMP/Ins(1,4,5)P3 pathway, AMBRA1: Activating molecule in BECN1 regulated autophagy protein 1, AMPK: AMP-activated protein kinase, ATG: Autophagy-related genes or proteins, Ca2+: Calcium ions, CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase β, cAMP/Ins(1,4,5)P3: Cyclic adenosine monophosphate/inositol (1,4,5)-trisphosphate, cGAS-STING: Cyclic GMP-AMP Synthase—simulator of interferon genes, FIP200: focal adhesion kinase family-interacting protein of 200 kDa, JNK1: c-Jun N-terminal kinase 1, LC3: Microtubule associated protein 1 (MAP1) light chain 3, LL-37: Human cathelicidin, NAD+/NADH: Nicotinamide adenine dinucleotide (NAD) + hydrogen (H), NBR1: Neighbor of BRCA1 gene 1, NDP52: Nuclear domain 10 protein 52, NOS: Nitric oxide synthase, OPTN: Optineurin, PE: phosphatidylethanolamine, PI3KC3 complex: Class III phosphatidylinositol 3-kinase (PI3K) complex, PI3KC1a/Akt/TSC/mTORC1: Class 1a phosphoinositide 3-kinase/protein kinase B/tuberous sclerosis complex/mTORC1, PI3P: Phosphatidylinositol 3-phosphate, PIP2: Phosphatidylinositol bisphosphate, PPARα: Peroxisome proliferation factor-activated receptor α, PPARGC1A: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, P2RX7: Purinergic Receptor 7, ROS: Reactive oxygen species, Rag/mTORC1: Ras-related GTP-binding protein/mammalian target of rapamycin complex 1, SIRT: Sirtuin protein, TFEB: Transcription factors EB, ULK1/2: UNC-51-Like Ser/Thr kinase, VPS: Vacuolar protein sorting, WIPI2: WD repeat domain phosphoinositide-interacting protein 2.
): Activation, ( ):inhibition, ( ): Drug induced activation, ( ): Drug induced inhibition, ( ): Upregulation, ( ): Downregulation, ( ): Drug induced upregulation, ( ): Blocks activation process, Numbers in blue boxes indicate drug induced activation of the following processes—1: Increased autophagosome formation (SMER 18 & 28, nortriptyline); 2: AMPK activation: (AICAR, ornithine, metformin, statins); 3: TFEB signaling upregulation (GSK4112, GW7647, trehalose, Wy14643, honokiol, ambroxol); 4: SIRT 1, 3 activator (SRT 1720, resveratrol, honokiol); 5: Ca2+—AMPK signaling (GABA, vitamin D, soyabean lectin, isoniazid, pyrazinamide, calcimycin); 6: mTOR inhibition (Rapamycin, evorilimus, nitazoxanide); 7: LL-37 induced expression (Vitamin D, Interferon-γ, 4-phenylbutyrate); 8: Increased ROS production (Imiquimod); 9: PI3K/Akt/mTOR pathway inhibition (Baicalin, imatinib, nilotinib, ibrutinib, bazedoxifene); 10: Autophagic flux induction (Pasakbumin A, fluoxetine); 11: ER stress mediated autophagy induction (Thiostrepton); 12: Depletion of p38 MAPK mediated autophagy activation via p38IP and mATG9 (Gefitnib); 13: Autophagy genes (ATG16L1 and LC3) upregulation (Loperamide); 14: AMPK activation via inositol signaling pathway (Carbamazepine); 15: mTOR independent autophagy formation via ATG 12 (Valproic acid); 16: Slow autophagic flux and increased lysozyme acidification (Prochlorperazine edisylate), Alphabets (A-H) describes the different pathways—A: mTORC1/ULK/ATG13/FIP200 dependent pathway, B: AMPK/TSC/mTORC1 dependent pathway, C: PI3KC1a/Akt/TSC/mTORC1 dependent pathway, D: Rag/mTORC1 dependent pathway, E: mTOR independent Ca2+/calpain pathway, F: mTOR independent inositol signalling pathway, G: mTOR independent JNK1/Beclin1/PI3KC3 pathway, H: mTOR independent cAMP/Ins(1,4,5)P3 pathway, AMBRA1: Activating molecule in BECN1 regulated autophagy protein 1, AMPK: AMP-activated protein kinase, ATG: Autophagy-related genes or proteins, Ca2+: Calcium ions, CaMKKβ: Ca2+/calmodulin-dependent protein kinase kinase β, cAMP/Ins(1,4,5)P3: Cyclic adenosine monophosphate/inositol (1,4,5)-trisphosphate, cGAS-STING: Cyclic GMP-AMP Synthase—simulator of interferon genes, FIP200: focal adhesion kinase family-interacting protein of 200 kDa, JNK1: c-Jun N-terminal kinase 1, LC3: Microtubule associated protein 1 (MAP1) light chain 3, LL-37: Human cathelicidin, NAD+/NADH: Nicotinamide adenine dinucleotide (NAD) + hydrogen (H), NBR1: Neighbor of BRCA1 gene 1, NDP52: Nuclear domain 10 protein 52, NOS: Nitric oxide synthase, OPTN: Optineurin, PE: phosphatidylethanolamine, PI3KC3 complex: Class III phosphatidylinositol 3-kinase (PI3K) complex, PI3KC1a/Akt/TSC/mTORC1: Class 1a phosphoinositide 3-kinase/protein kinase B/tuberous sclerosis complex/mTORC1, PI3P: Phosphatidylinositol 3-phosphate, PIP2: Phosphatidylinositol bisphosphate, PPARα: Peroxisome proliferation factor-activated receptor α, PPARGC1A: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha, P2RX7: Purinergic Receptor 7, ROS: Reactive oxygen species, Rag/mTORC1: Ras-related GTP-binding protein/mammalian target of rapamycin complex 1, SIRT: Sirtuin protein, TFEB: Transcription factors EB, ULK1/2: UNC-51-Like Ser/Thr kinase, VPS: Vacuolar protein sorting, WIPI2: WD repeat domain phosphoinositide-interacting protein 2.

{kind=link}
| Class | Drugs/Compounds | Drug Action | Mechanism of Autophagy Activation during Mycobacterial Infection | Model | Reference |
|---|---|---|---|---|---|
| Small Molecules | |||||
| SMER | SMER18 and 28 | - | Induced autophagosome formation | Human PBMCs | [93] |
| Analog of AMP | AICAR | Allosteric activation of AMPK kinase which plays a key function in cellular homeostasis | Activates AMPK-PPARGC1A pathway that upregulates CEBPB-dependent autophagy genes and enhances autophagy. | RAW264.7 cells, THP-1 cells (human monocytic cell line), BMDMs, mice and Drosophila | [94] |
| Synthetic small molecule | GSK4112 | Activates NR1D1 receptor | Increases autophagic flux via upregulation of TFEB signaling | THP-1 cells, primary human monocyte, murine macrophage cell line, RAW264.7, HEK293T and HepG2 cell lines. | [95] |
| GW7647 | Activates PPARα receptor | Increases autophagic flux via upregulation of TFEB signaling, and enhanced lipid catabolism | BMDMs | [96] | |
| SRT 1720 | SIRT 1 activator | Enhances autophagy by activating SIRT 1 | THP-1 cells, HMDMs and mice | [97] | |
| NSC 18725 | Anti-mycobacterial activity | Modulates autophagy, mechanism unknown | THP-1 cells | [98] | |
| Amino acid | Gamma amino Butyric acid | Neurotransmitter inhibitor | Increases autophagic flux via Ca2+-AMPK signaling pathway. Additionally, increases phagosomal maturation | Human PBMCs, HMDMs, RAW264.7 cells and BMDMs | [99] |
| Ornithine | Crucial role in disposing excess nitrogen (ammonia) via urea cycle | Increases autophagy by reducing ammonia levels there by upregulating AMPK phosphorylation | Mouse alveolar macrophage, peritoneal macrophages, kupffer cells and BMDMs | [100] | |
| Disaccharides | Trehalose | - | Induces autophagic flux by increasing PI(3,5)P2 levels that activates calcineurin triggered translocation of TFEB. Additionally, it causes a pseudo-starvation like response by inhibiting glucose transporters (GLUT 3 and 8) to induce autophagy | U937, U1.1 and HEK293T cell lines | [101] |
| Immunosuppressants | |||||
| Macrolide compound | Rapamycin | Forms an immunosuppressive complex by binding to the immunophilin and also a potent mTOR inhibitor | Autophagy induction via mTORC1 complex inhibitor | Raw264.7 cells, HMDMs, Human PBMCs and BMDMs | [6] |
| Rapamycin analog | Everolimus * | Inhibits the activation of mTOR by forming a complex with FKBP-12 protein | Autophagy induction via mTORC1 complex inhibitor | - | [103] |
| Immunomodulators | |||||
| Vitamin | Vitamin D * | Regulation of hormone secretion, cell proliferation, differentiation and immune response | Induces autophagic flux via a signaling cascade that is triggered by the induced expression of human cathelicidin (hCAP-18/LL-37) | Primary human monocytes, HMDMs, THP-1 cells and RAW 264.7 cells | [104,105] |
| Cytokine | Interferon-γ (IFN-γ) | Promotes macrophage activation | Activates autophagic flux through vitamin D dependent effector pathway | Human T cells, primary human monocytes and HMDMs | [106] |
| Nucleoside analog of imidazoquinoline, a synthetic tricyclic organic molecule | Imiquimod | TLR7 and 8 agonist | Induces Autophagy by increasing mitochondrial ROS that triggers selective autophagy. Additionally, upregulates NO Production via the MEK/ERK1/2 and GSK-3β mediated Pathways. | Raw264.7 cells and THP-1 cells | [107] |
| Endotoxin derived from the outer membrane of Gram-negative bacteria | Lipopolysaccharides (LPS) | TLR4 agonist | Activates autophagy and restores M.tb inhibited immune activity | THP-1 cells | [108] |
| Plant compounds | |||||
| Stilbene | Resveratrol | SIRT 1 activator | Enhances autophagy by activating SIRT 1 | THP-1 cells, HMDMs and mice | [97] |
| Flavone glycoside | Baicalin | - | Induces the activation of autophagy by inhibiting PI3K/Akt/mTOR pathway. Additionally, inhibits the PI3K/Akt/NF-kB signal pathway, thereby limiting the NLRP3 inflammasome and subsequent production of pro-inflammatory cytokine IL-1β | Mice, raw264.7 cells, murine macrophage | [109] |
| Eurycomanone | Pasakbumin A | - | Induces autophagic flux and TNF-α production via activation of the ERK1/2-signaling pathway and enhances phagosome maturation and lysosome fusion | Raw264.7 cells, and THP-1 cells | [110] |
| Polyphenolic compound | Epigallocatechin gallate | - | Induces autophagic flux | Raw264.7 cells and mice | [111] |
| Lignans (low molecular weight polyphenols) | Honokiol | SIRT 3 activator | Increases autophagic flux via upregulation of TFEB signaling | Mice, BMDMs, HMDMs and Human PBMCs | [112] |
| Legume Lectins | Soybean lectin | - | Induces autophagic flux by activating P2RX7 that triggers Ca2+/AMPK signaling pathway and ROS generation via P2RX7/NF-κB axis | THP-1 cells | [113] |
| Antibiotics | |||||
| Small molecule–Isonicotinic acid derivative | Isoniazid | Inhibits the enzyme inh A during mycolic acid synthesis | Induces autophagic flux via NOX- derived ROS and calcium, Ca2+ and AMPK dependent pathways | BMDMs and HMDMs | [114] |
| Small molecule—Nicotinamide analogue | Pyrazinamide | Disrupts membrane potential, interferes with energy production and inhibits trans-translation by binding to ribosomal protein S1 | Induces autophagic flux via NOX- derived ROS and calcium, Ca2+—dependent AMPK activation | BMDMs and HMDMs | [114] |
| Thiopeptide | Thiostrepton | Disrupts prokaryotic translation by inhibiting the dissociation of elongation factor G from ribosomes | ER stress mediated autophagy activation | Zebrafish and Raw264.7 cells | [115] |
| Polyether | Calcimycin | Forms stable complexes with divalent cations and helps in membrane transportation | Induces autophagic flux by activating P2RX7 that triggers Ca2+/AMPK signaling pathway and IL-12 generation via P2RX7/NF-κB axis | THP-1 cells | [116,117] |
| Steroids | |||||
| Hormones | Dehydroepiandrosterone | Inhibits voltage-gated T-type calcium channels and activates PPARα | Induction of autophagy | THP-1 cells | [118] |
| Anticancer drugs | |||||
| Signal transduction inhibitor | Gefitinib | EGFR inhibitor | Enhancing host autophagy by inhibiting EGFR-mediated phosphorylation of the downstream signaling molecule p38 MAPK. Depletion of p38 MAPK activates autophagy via p38IP and mATG9 | J774 macrophages and BMDMs | [119] |
| Histone deacetylase inhibitor | 4-phenylbutyrate * | Transcription activation via acetylation of histones | LL-37-mediated autophagy activation via P2RX7 receptor which in turn activates AMPK and PI3K downstream of the P2RX7 receptor together with enhanced cytosolic free Ca2+ | HMDMs, and THP-1 cells | [120] |
| Kinase inhibitor | Imatinib * | Tyrosine kinase inhibitor | Increases autophagic flux by activating cathepsin D and increasing phagolysosomal acidification via the inhibition of ABL tyrosine kinase | Human PBMCs, HMDMs, human alveolar macrophages | [121] |
| Nilotinib | Tyrosine kinase inhibitor | Promotes autophagy by inhibiting the ABL tyrosine kinase mediated PI3K/Akt/mTOR pathway | THP-1 cells, RAW264.7 cells and BMDMs | [122] | |
| Ibrutinib | Bruton’s tyrosine kinase (BTK) inhibitor | Induces autophagy through inhibition of BTK/Akt/mTOR pathway and also facilitates the completion of autophagic flux | THP-1 cells | [123] | |
| Estrogen agonists | Bazedoxifene | Selective estrogen receptor modulator | Enhances autophagosome formation via phosphorylation of Akt/mTOR signaling | THP-1 cells | [124] |
| Antidiabetic drugs | |||||
| Biguanides | Metformin * | Activates AMPK via inhibiting mitochondrial respiratory complex I which elevates 5’-adenosine monophosphate (AMP) levels | Increases autophagic flux via enhancing autophagosome—lysozome fusion and additionally increases mROS production | THP-1 cells, HMDMs and mice | [125] |
| Antidiarrheal drugs | |||||
| Synthetic opioid—phenylpiperidine derivative | Loperamide | Decreases peristaltic activity by binding to opiate receptors in gastrointestinal tract, blocks voltage-dependent calcium channel and calmodulin inhibitor | Increased autophagy induction by upregulating the expression of genes viz., ATG16L1 and LC3 | Mice, HMDMs, murine alveolar cells and Human alveolar macrophages | [126] |
| Antiprotozoal agents | |||||
| Antiprotozoals | Nitazoxanide | Inhibits pyruvate:ferredoxin oxidoreductase enzyme-dependent electron transport and disrupts metabolism in anaerobic microbes | Autophagy induction via mTORC1 complex inhibitor | THP-1 cells, MCF-7 cells, HEK 293T cells and MEF cells | [127] |
| Antiseizure drugs | |||||
| First-generation (classic) anticonvulsants | Carbamazepine | Inactivates Na+ channels and inhibits receptors of CNS | Induction of mTOR-independent autophagy through Ins(1,4,5)P3depletion and AMPK activation | RAW264.7 cells, HMDMs, human alveolar macrophages, zebrafish and mice | [128] |
| Valproic acid | Inhibits GABA transaminase and increases GABA levels in CNS. It also inhibits histone deacetylase | Induction of mTOR-independent autophagosome formation through ATG12 | RAW264.7 cells, HMDMs and human alveolar macrophages | [128] | |
| Lipid-lowering drugs | |||||
| Fibrate | Wy14643 | Activates PPARα receptor protein | Increases autophagic flux via upregulation of TFEB signaling, and enhanced lipid catabolism | Mice and BMDMs | [96] |
| Statins | Pravastatin *, Rosuvastatin *, Atorvastatin * and Simvastatin | HMG-CoA reductase inhibitors | Promotes autophagy via the AMPK/mTORC1/TFEB axis. Additionaly increases phagosome maturation and lysosome fusion | Human PBMCs, HMDMs, THP-1 cells and mice | [129,130,131,132] |
| Mucoactive drug | |||||
| Mucokinetics | Ambroxol | Suppresses excessive mucus secretion by inhibiting NO-dependent activation of soluble guanylate cyclase | Induction of autophagy via, the activation of TFEB nuclear translocation | Mice and BMDMs | [133] |
| Psychotropic Drugs | |||||
| Anti-depressant | Nortriptyline | Norephinephrine and sereotonin reuptake inhibitor | Induces the formation of autophagosomes | HeLa cells and HMDMs | [134] |
| Fluoxetine | Sereotonin reuptake inhibitor | Induces autophagy by increasing the secretion of TNF- α | THP-1 cells, RAW264.7 cells, J774 macrophages and BMDMs | [119] | |
| Antipsychotics | Prochlorperazine edisylate | D2 dopamine receptor inhibitor | Slows down autophagic flux and progressively increases the acidity of lysozymes | HeLa cells and HMDMs | [134] |
| S No | Trial ID | Host Directed Therapeutic Drugs | HDT Drug Doseage (mg) | HDT Treatment Duration | Anti-Tuberculosis Drugs (ATD) (Dose) | Study Title | Phase | No. of Participants | Ages Eligible for Study | Study Sponsor | Country | Status | Remarks/Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CTRI/2018/01/011176 | Metformin | 1000 mg | Given daily for the first 2 months together with ATD followed by another 4 months with only ATD | Rifampicin, isoniazid, ethambuthol and pyrazinamide (Standard doseage) | Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF) | 2 | 316 | 18–60 Years | National Institute for Research in Tuberculosis | India | Active | Not applicable |
| 2 | NCT 03891901 | Imatinib | 50 mg, 100 mg, 200 mg and 400 mg | Daily for 14 days followed by another 14 days together with ATD | Isoniazid (300 mg) and rifabutin (300 mg) | A Clinical Trial of the Safety, Pharmacokinetics and Hematologic Effects of Imatinib on Myelopoiesis in Adults When Given With and Without Isoniazid and Rifabutin (IMPACT-TB) | 2 | 72 | 18 to 55 Years | National Institute of Allergy and Infectious Diseases (NIAID) | United States, georgia | Recruiting | Not applicable |
| 3 | NCT 04721795 | Atorvastatin | 30–40 mg | Daily for 2 months together with ATD followed by another 4 months with only ATD | Rifampicin, isoniazid, ethambuthol and pyrazinamide (Standard doseage) | Treating Tuberculosis With the Lipid Lowering Drug Atorvastatin in Nigeria (ATORvastatin in Pulmonary TUBerculosis) (ATORTUB) | 2 | 150 | 18 to 65 Years | Obafemi Awolowo University Teaching Hospital | |||
| 4 | NCT 04504851 | Rosuvastatin | 10 mg | Daily for 2 months together with ATD followed by another 4 months with only ATD | Rifampicin (10 mg/Kg), isoniazid (5 mg/Kg), ethambuthol (25 mg/Kg) and pyrazinamide (15 mg/Kg) | Rosuvastatin Evaluation as a Tuberculosis Treatment Adjunct (ROSETTA) | 2 | 154 | 18 to 75 Years | National University Hospital, Singapore | Philippines, Singapore, Uganda and Vietnam | Not yet recruiting | Not applicable |
| 5 | NCT 03882177 | Pravastatin | 40 mg, 80 mg, 120 mg and 160 mg | Given alone on 1st day followed by another 14 days together with ATD | Rifampicin, isoniazid, ethambuthol and pyrazinamide (Standard doseage) | StAT-TB (Statin Adjunctive Therapy for TB): A Phase 2b Dose-finding Study of Pravastatin in Adults With Tuberculosis | 2 | 35 | 18 Years and older | National Institute of Allergy and Infectious Diseases (NIAID) | South Africa | Active, not recruiting | Not applicable |
| 6 | NCT 02968927 | Everolimus | 0.5 mg | Daily for 112 days together with ATD followed by another 68 days with only ATD | Rifabutin (Standard doseage) | A Ph2 Randomized Trial to Evaluate the Safety Preliminary Efficacy and Biomarker Response of Host Directed Therapies Added to Rifabutin-modified Standard Therapy in Adults With Drug-Sensitive Smear-Positive Pulmonary TB | 2 | 200 | 18 to 65 Years | The Aurum Institute NPC | South Africa | Active, not recruiting | Everolimus in adults as adjunctive therapy for tuberculosis was safe and also improved recovery [154] |
| 7 | NCT 00918086 | Vitamin D | 1.25 mg | Three times a week for a total of 8 weeks followed by another 8 weeks with the same dose given every other week as dietary supplement | Standard ATDs | Impact of Vitamin D Supplementation on Host Immunity to Mycobacterium Tuberculosis and Response to Treatment | 2 | 199 | 18 Years and older | Emory University | United States, Georgia | Completed | Vitamin D supplementation failed to improve the rate of sputum Mtb clearance [156] |
| 8 | NCT 01722396 | Vitamin D | 2.5 mg | Given 8 weeks apart at 8, 16 and 24 weeks as dietary supplement together with standard ATD | Standard ATDs | Pharmacogenetics of Vitamin D Supplementation in Tuberculosis | 3 | 62 | 16 Years and older | University of Birmingham | United Kingdom | Completed | Result awaited |
| 9 | NCT 00788320 | Vitamin D | 1.25 mg | Three times a week for a total of 8 weeks as dietary supplement | Standard ATDs | Antimicrobial Peptide LL-37 (Cathelicidin) Production in Active Tuberculosis Disease: Role of Vitamin D Supplementation | NA | 0 | 18 Years and older | Atlanta VA Medical Center | United States, Georgia | Withdrawn (Inadequate enrollment) | Not applicable |
| 10 | NCT 04593524 | Vitamin D | 0.025 mg | 4 weeks | Standard ATDs | The Role of Vitamin D, A, and Beta Carotene in Tuberculosis Patients With Vitamin D Receptor Gene Polymorphism | NA | 48 | 20 to 60 Years | Universitas Sumatera Utara | Indonesia | Completed | Vitamin D supplementation to patients with vitamin D receptor gene polymorphism showed increased sputum conversion rates. |
| 11 | NCT 00507000 | Cholecalciferol (vitamin D) | 1.5 mg | Given weekly for 2 months followed by the same dose per month for the next 4 months as dietary supplement | Standard ATDs | Role of Oral Vitamin D as an Adjunct Therapy in Category I Pulmonary Tuberculosis Along with Assessment of Immunological Parameters. | 3 | 150 | 18 to 60 Years | Indian Council of Medical Research | India | Unknown | Not applicable |
| 12 | NCT 01130311 | cholecalciferol (vitamin D) | 15 mg | Given at week 0 and week 4 as dietary supplement | Standard ATDs | Clinical Trial of Vitamin D Replacement in Patients With Pulmonary Tuberculosis (SUCCINCT) | NA | 259 | 15 Years and older | Aga Khan University | Pakistan | Completed | Vitamin D supplementation showed improved recovery in all TB patients. It also increased host immune activation in vitamin D deficient patients [157] |
| 13 | NCT 01244204 | Vitamin D | 0.020 mg | Daily dose of 800IU of vitamin D | Standard ATDs | Vitamin D Supplementations as Adjunct to Anti-tuberculosis Drugs | NA | 120 | 10 to 18 Years | Harvard School of Public Health | Mongolia | Completed | Vitamin D supplementation resulted in fewer tuberculin skin test conversions [158] |
| 14 | NCT 00677339 | Vitamin D | 1.25 mg | Given once per month as dietary supplement | Standard ATDs | L-arginine and Vitamin D Adjunctive Therapy in Pulmonary Tuberculosis (TB) (AVDAPT) | 3 | 200 | 15 Years and older | Menzies School of Health Research | Indonesia | Completed | Vitamin D supplementation showed no effect on TB outcomes [159] |
| 15 | NCT 01698476 | Vitamin D | 0.125 mg | Given twice daily for 16 weeks as dietary supplement | Standard ATDs | Immune Reconstitution in Tuberculosis Disease Using Antimicrobial Treatment With Vitamin D and Phenylbutyrate | 2 | 390 | 18 to 75 Years | Karolinska Institutet | Ethiopia | Completed | Daily supplementation along with PBA results in reduction of clinical TB symptoms while the intervention had no effect on sputum conversion [160] |
| 16 | NCT 01698476 | 4-phenylbutyrate(PBA) | 500 mg | Given twice daily for 16 weeks | NA | Immune Reconstitution in Tuberculosis Disease Using Antimicrobial Treatment With Vitamin D and Phenylbutyrate | 2 | 390 | 18 to 75 Years | Karolinska Institutet | Ethiopia | Completed | Daily supplementation together with vitamin D results in reduction of clinical TB symptoms while the intervention had no effect on sputum conversion [160] |
| 17 | NCT 02169570 | Vitamin D | 15 mg | Given at week 0, 4 and 12 as dietary supplement together with standard ATD | NA | Effect of Supplementary Vitamin D in Patients With Diabetes Mellitus and Pulmonary Tuberculosis (EVIDENT Study) | 4 | 435 | 30 to 60 Years | Dow University of Health Sciences | Pakistan | Unknown | Not applicable |
| 18 | NCT 01580007 | Vitamin D | 0.125 mg | Given once daily for 2 months | NA | Clinical Trial of Phenylbutyrate and Vitamin D in Tuberculosis (TB) | 2 | 288 | 18 to 60 Years | International Centre for Diarrhoeal Disease Research, Bangladesh | Bangladesh | Completed | Vitamin D supplementation together with standard short-course therapy showed improved clinical recovery and better sputum culture conversion [150,155] |
| 19 | NCT 01580007 | 4-phenylbutyrate(PBA) | 500 mg | Given twice daily for 2 months | NA | Clinical Trial of Phenylbutyrate and Vitamin D in Tuberculosis (TB) | 2 | 288 | 18 to 60 Years | International Centre for Diarrhoeal Disease Research, Bangladesh | Bangladesh | Completed | PBA supplementation together with vitamin D results in improved clinical recovery and better sputum culture conversion [150,155] |
| 20 | NCT 03011580 | Vitamin D3 | 0.240 mg | Given every day for 8 weeks as dietary supplement | NA | Vitamin D3 to Enhance Resolution of Residual Pulmonary Inflammation in Patients Completing Antituberculosis Treatment (ResolveD-TB) | 2 | 15 | 20 Years and older | Queen Mary University of London | United Kingdom | Completed | Result awaited |
| 21 | NCT 01657656 | Vitamin D | 3.5 mg | Given twice a week as dietary supplement together with standard ATD | NA | Vitamin D Supplementations as Adjunct to Anti-Tuberculosis Drugs in Mongolia | NA | 350 | 18 to 80 Years | Harvard School of Public Health | Mongolia | Completed | Vitamin D supplementation had no effect on sputum culture conversion [161] |
| 22 | NCT 01992263 | Vitamin D | 0.015 mg, 0.050 mg and 0.100 mg | Given Daily for 12 months as dietary supplement | NA | A Trial of Vitamin D Supplementation Among Tuberculosis Patients in South India | NA | 200 | 18 to 60 Years | Cornell University | United States and India | Not yet recruiting | Not applicable |
| 23 | NCT 00366470 | Vitamin D | 2.5 mg | Given once every two weeks for 2 Months as dietary supplement together with standard ATD | NA | A Clinical Trial to Study the Effect of the Addition of Vitamin D to Conventional Treatment in New Pulmonary Tuberculosis Patients | 3 | 250 | 18 to 75 years | Peter Daley | India | Completed | Vitamin D supplementation showed no reduction in time to sputum culture conversion [162]. |
| 24 | NCT 02276755 | Cholecalciferol (vitamin D3) | 0.35 mg | Given weekly for 3 years as dietary supplement | NA | Vitamin D in TB Prevention in School Age Children | 3 | 8851 | 6 to 13 Years | Harvard School of Public Health | Mongolia | Completed | Vitamin D supplementation failed to lower risk of tuberculosis infection [163] |
| 25 | NCT 02880982 | Cholecalciferol (vitamin D3) | 0.25 mg | Given weekly for 3 years as dietary supplement | NA | Trial of Vitamin D Supplementation in Cape Town Primary Schoolchildren (ViDiKids) | 3 | 1743 | 6 to 11 Years | Queen Mary University of London | South Africa | Active, not recruiting | Not applicable |
| 26 | NCT 00419068 | Cholecalciferol (vitamin D3) | 2.5 mg | Given at week 0, 2, 4 and 6 as dietary supplement together with standard ATD | NA | Trial of Adjunctive Vitamin D in Tuberculosis Treatment | 3 | 146 | 18 Years and older | Barts &The London NHS Trust | United Kingdom | Completed | Vitamin D supplementation showed no effect on time to sputum culture conversion in the whole study population. However, participants with the known vitamin D receptor polymorphism showed quicker sputum culture conversion [152] |
| 27 |
NCT 00157066 | Ergocalciferol (vitamin D) | 2.5 mg | Single dose as supplement | NA | Effects of Vitamin D Supplementation on Antimycobacterial Immunity | NA | 230 | 18 Years and older | Barts & The London NHS Trust | United Kingdom | Completed | Improved in vitro restriction of BCG-lux luminescence was observed. In addition, antigen-stimulated IFN-gamma was not affected [164] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adikesavalu, H.; Gopalaswamy, R.; Kumar, A.; Ranganathan, U.D.; Shanmugam, S. Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection. Medicina 2021, 57, 522. https://doi.org/10.3390/medicina57060522
Adikesavalu H, Gopalaswamy R, Kumar A, Ranganathan UD, Shanmugam S. Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection. Medicina. 2021; 57(6):522. https://doi.org/10.3390/medicina57060522
Chicago/Turabian StyleAdikesavalu, Harresh, Radha Gopalaswamy, Ashok Kumar, Uma Devi Ranganathan, and Sivakumar Shanmugam. 2021. "Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection" Medicina 57, no. 6: 522. https://doi.org/10.3390/medicina57060522