
Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects
 , , ,
, , ,
Abstract
:1. Introduction

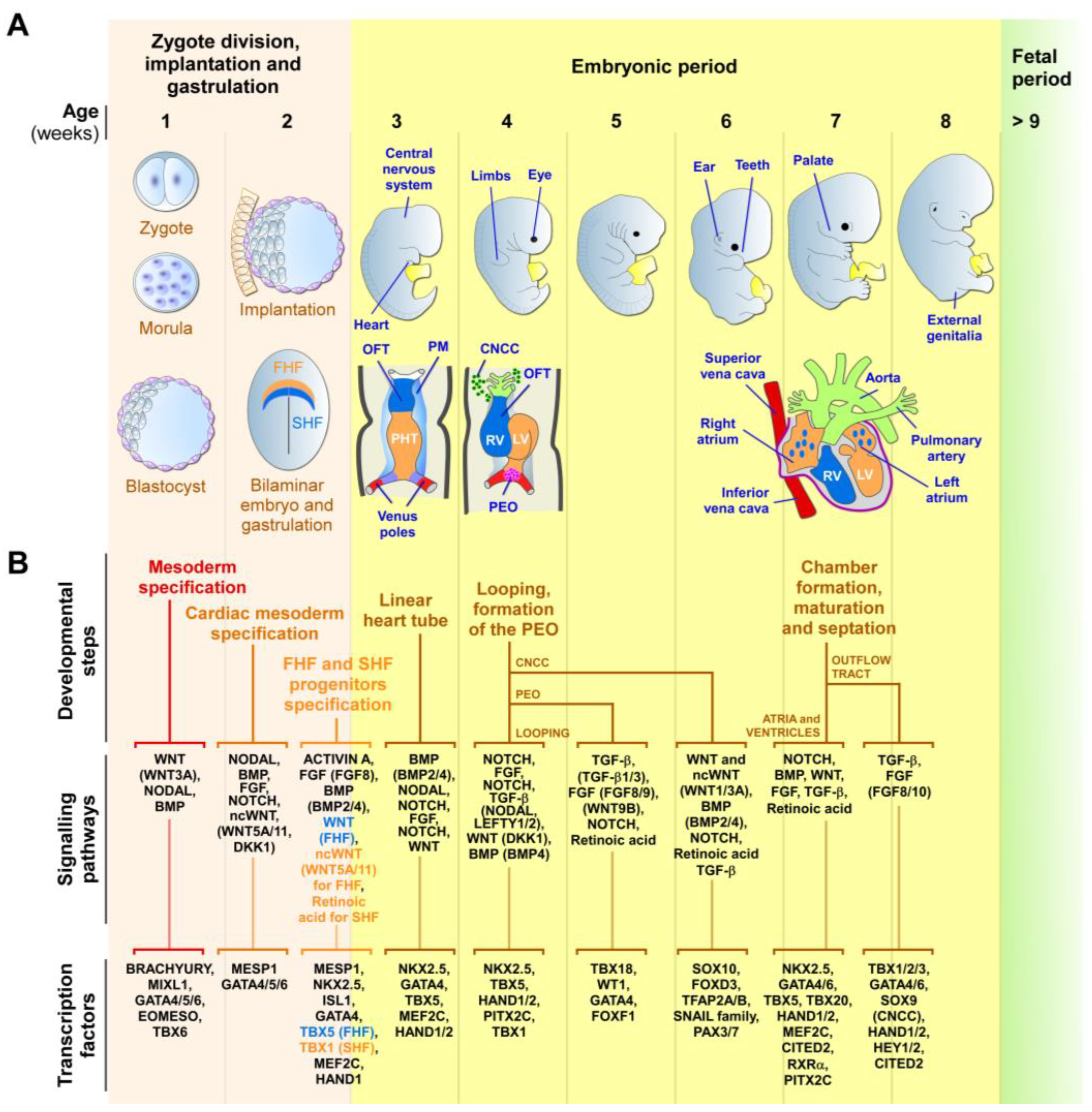{kind=link}
| Clinical Trial Number | Type and Status | Condition | Intervention or Treatment | Population | Evaluation | Results |
|---|---|---|---|---|---|---|
| NCT03944837 | Interventional Single-group assignment; Status: recruiting. | Severe Fetal CHD | Transient maternal oxygen gas administration during echocardio-graphic and MRI imaging. | Pregnant woman ≥18 years old, with fetal diagnosis of specific CHDs and intention of prenatal treatment. | Brain growth and maturation to birth, improvement of postnatal neurodevelopmental issues, identification of CHD types likely to benefit from chronic maternal hyperoxygenation. | Not available |
| NCT01736956 | Interventional, prospective, non-randomi-zed clinical trial; Status: complete. | Aortic stenosis and evolving HLHS | Fetal aortic transuterine valvuloplasty, periventricular approach. Control group with standard prenatal and postnatal care. | Pregnant woman ≥ 16 years old, with a fetus with normal heart anatomy and severe aortic stenosis. | Safety and efficacy of in utero percutaneous balloon dilation of fetal aortic valve with severe stenosis determined by fetal mitral valve and left ventricle growth, survival, and neurodevelopmental status. | No results posted |
| NCT03147014 | Interventional; Status: complete. | Fetal HLHS, atrial septal aneurysm, aortic coarctation. | Cardiovascular response to maternal hyperoxygenation in fetal CHDs. All singleton fetuses with CHDs at all gestational ages are eligible. | Pregnant women with a fetus harboring CHDs diagnosed at various gestational weeks. | Participants received 10–15 min hyperoxygenation, assessed for middle cerebral artery pulsation; myocardial diastolic function; flow patterns across tricuspid, mitral valves, and ductus venosus; changes in heart output and ratio of right–left ventricle flow; flow at the aortic isthmus if aortic coarctation. | No results posted |
| EudraCT 2016-003181-12 * | Prospective cohort study; Status: complete. | Pregnant women with fetus at risk of pulmonary hypoplasia due to VSD/AVSD. | Sonographic assessment of pulmonary vascular reactivity following maternal hyperoxygenation. | Pregnant woman ≥18 years old, with a fetus at risk for neonatal persistent pulmonary hypertension; non-pregnant control. | Fetal echocardiographic doppler within the first 48 h of life to assess pulmonary vasculature prior to and after maternal hyperoxygenation to predict development of neonatal pulmonary hypertension. | [49] |
2. Navigating Early Mammalian Embryonic Heart and Placental Development
2.1. Placental Development
2.2. Heart Development
3. Tracing the Roles of Signaling Pathways and Exosomes in Heart Development
3.1. Secretomes and Heart Development
3.2. Exosomes and Heart Development
4. Exploring the Impact of Diet and Medicinal Supplements on Heart Development
4.1. Diet Supplements and Heart Development
4.2. Cardiac Complications Due to Drugs Used to Treat Non-Cardiac Defects
5. Unveiling the Potential of Secretomes and Exosomes for CHD Prevention
5.1. Exogenous Instructive Molecules to Mitigate CHDs
5.2. Exosomes, a Non-Cellular Approach for Correcting Embryonic Cardiac Defects
6. Harnessing Secreted Factors in Embryogenesis for Protection against CHDs
7. Challenges and Ethical Considerations
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffman, J.I.E.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilboa, S.M.; Devine, O.J.; Kucik, J.E.; Oster, M.E.; Riehle-Colarusso, T.; Nembhard, W.N.; Xu, P.; Correa, A.; Jenkins, K.; Marelli, A.J. Congenital Heart Defects in the United States. Circulation 2016, 134, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.A.; Rajamarthandan, S.; Francis, B.; O’Leary-Kelly, M.K.; Sinha, P. Update on stem cell technologies in congenital heart disease. J. Card. Surg. 2020, 35, 174–179. [Google Scholar] [CrossRef]
- Chowdhury, D.; Johnson, J.N.; Baker-Smith, C.M.; Jaquiss, R.D.B.; Mahendran, A.K.; Curren, V.; Bhat, A.; Patel, A.; Marshall, A.C.; Fuller, S.; et al. Health Care Policy and Congenital Heart Disease: 2020 Focus on Our 2030 Future. J. Am. Heart Assoc. 2021, 10, e020605. [Google Scholar] [CrossRef]
- Jenkins, K.J.; Correa, A.; Feinstein, J.A.; Botto, L.; Britt, A.E.; Daniels, S.R.; Elixson, M.; Warnes, C.A.; Webb, C.L. Noninherited Risk Factors and Congenital Cardiovascular Defects: Current Knowledge. Circulation 2007, 115, 2995–3014. [Google Scholar] [CrossRef] [Green Version]
- Freud, L.R.; Tworetzky, W. Fetal interventions for congenital heart disease. Curr. Opin. Pediatr. 2016, 28, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Halder, V.; Ghosh, S.; Shrimanth, Y.S.; Manoj, R.K.; Mandal, B.; Thingnam, S.K.S.; Kumar, R. Fetal cardiac intervention and fetal cardiac surgery: Where are we in 21st century? Am. J. Cardiovasc. Dis. 2021, 11, 642–646. [Google Scholar]
- Friedman, K.G.; Tworetzky, W. Fetal cardiac interventions: Where do we stand? Arch. Cardiovasc. Dis. 2020, 113, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Gellis, L.; Tworetzky, W. The boundaries of fetal cardiac intervention: Expand or tighten? Semin. Fetal Neonatal Med. 2017, 22, 399–403. [Google Scholar] [CrossRef]
- Moon-Grady, A.J.; Morris, S.A.; Belfort, M.; Chmait, R.; Dangel, J.; Devlieger, R.; Emery, S.; Frommelt, M.; Galindo, A.; Gelehrter, S.; et al. International Fetal Cardiac Intervention Registry: A Worldwide Collaborative Description and Preliminary Outcomes. J. Am. Coll. Cardiol. 2015, 66, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Feng, Q.; Wang, L.; Zhou, B. Human Pluripotent Stem Cell-Derived Cardiac Cells: Application in Disease Modeling, Cell Therapy, and Drug Discovery. Front. Cell Dev. Biol. 2021, 9, 655161. [Google Scholar] [CrossRef]
- Terashvili, M.; Bosnjak, Z.J. Stem Cell Therapies in Cardiovascular Disease. J. Cardiothorac. Vasc. Anesth. 2019, 33, 209–222. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Oikonomou, E.K.; Moris, D.; Schizas, D.; Economopoulos, K.P.; Mylonas, K.S. Stem Cell Therapy for Congenital Heart Disease. Circulation 2017, 136, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Oh, H. Cell Therapy Trials in Congenital Heart Disease. Circ. Res. 2017, 120, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Yang, X.; Don, C.W.; Minami, E.; Liu, Y.-W.; Weyers, J.J.; Mahoney, W.M.; Van Biber, B.; Cook, S.M.; Palpant, N.J.; et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature 2014, 510, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Bragança, J.; Lopes, J.A.; Mendes-Silva, L.; Santos, J.M.A. Induced pluripotent stem cells, a giant leap for mankind therapeutic applications. World J. Stem Cells 2019, 11, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Correia, C.D.; Ferreira, A.; Fernandes, M.T.; Silva, B.M.; Esteves, F.; Leitão, H.S.; Bragança, J.; Calado, S.M. Human Stem Cells for Cardiac Disease Modeling and Pre-clinical and Clinical Applications—Are We on the Road to Success? Cells 2023, 12, 1727. [Google Scholar] [CrossRef] [PubMed]
- Trac, D.; Maxwell, J.T.; Brown, M.E.; Xu, C.; Davis, M.E. Aggregation of Child Cardiac Progenitor Cells Into Spheres Activates Notch Signaling and Improves Treatment of Right Ventricular Heart Failure. Circ. Res. 2019, 124, 526–538. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, S.; Shen, W.-B.; Kaushal, S.; Yang, P. The current status and future of cardiac stem/progenitor cell therapy for congenital heart defects from diabetic pregnancy. Pediatr. Res. 2018, 83, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, D.M.; Gallicchio, V.S. The effect of stem cells in congenital heart disease. Clin. Med. Investig. 2019, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Femminò, S.; Bonelli, F.; Brizzi, M.F. Extracellular vesicles in cardiac repair and regeneration: Beyond stem-cell-based approaches. Front. Cell Dev. Biol. 2022, 10, 996887. [Google Scholar] [CrossRef] [PubMed]
- de Abreu, R.C.; Fernandes, H.; da Costa Martins, P.A.; Sahoo, S.; Emanueli, C.; Ferreira, L. Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat. Rev. Cardiol. 2020, 17, 685–697. [Google Scholar] [CrossRef]
- Ibrahim, A.; Marbán, E. Exosomes: Fundamental Biology and Roles in Cardiovascular Physiology. Annu. Rev. Physiol. 2016, 78, 67–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.; Losordo, D.W. Exosomes and Cardiac Repair after Myocardial Infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef]
- Andriolo, G.; Provasi, E.; Lo Cicero, V.; Brambilla, A.; Soncin, S.; Torre, T.; Milano, G.; Biemmi, V.; Vassalli, G.; Turchetto, L.; et al. Exosomes from Human Cardiac Progenitor Cells for Therapeutic Applications: Development of a GMP-Grade Manufacturing Method. Front. Physiol. 2018, 9, 1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Ma, X.; Lin, S.; Wu, B.; Chen, Y.; Peng, C. Bone marrow mesenchymal stem cell-derived exosomes protect against myocardial infarction by promoting autophagy. Exp. Ther. Med. 2019, 18, 2574–2582. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Guo, Y.; Teng, L.; Nong, Y.; Tan, M.; Book, M.J.; Zhu, X.; Wang, X.L.; Du, J.; Wu, W.J.; et al. Preconditioning Human Cardiac Stem Cells with an HO-1 Inducer Exerts Beneficial Effects After Cell Transplantation in the Infarcted Murine Heart. Stem Cells 2015, 33, 3596–3607. [Google Scholar] [CrossRef]
- Diener, C.; Keller, A.; Meese, E. Emerging concepts of miRNA therapeutics: From cells to clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Li, T.; Gu, J.; Yang, O.; Wang, J.; Wang, Y.; Kong, J. Bone Marrow Mesenchymal Stem Cell-Derived Exosomal miRNA-29c Decreases Cardiac Ischemia/Reperfusion Injury Through Inhibition of Excessive Autophagy via the PTEN/Akt/mTOR Signaling Pathway. Circ. J. Off. J. Jpn. Circ. Soc. 2020, 84, 1304–1311. [Google Scholar] [CrossRef]
- Ibrahim, A.G.-E.; Cheng, K.; Marbán, E. Exosomes as Critical Agents of Cardiac Regeneration Triggered by Cell Therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Maleki, B.; Alani, B.; Tamehri Zadeh, S.S.; Saadat, S.; Rajabi, A.; Ayoubzadeh, S.M.J.; Verdi, J.; Farrokhian, A.; Ghanbarian, H.; Noureddini, M.; et al. MicroRNAs and exosomes: Cardiac stem cells in heart diseases. Pathol. Res. Pract. 2022, 229, 153701. [Google Scholar] [CrossRef]
- Xia, W.; Chen, H.; Xie, C.; Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 2020, 12, 8241–8260. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, X.; Zhang, X.; Sun, Y.; Yan, Y.; Shi, H.; Zhu, Y.; Wu, L.; Pan, Z.; Zhu, W.; et al. Human umbilical cord mesenchymal stem cell exosomes enhance angiogenesis through the Wnt4/β-catenin pathway. Stem Cells Transl. Med. 2015, 4, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, X.; Cao, W.; Ma, J.; Sun, L.; Qian, H.; Zhu, W.; Xu, W. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Relieve Acute Myocardial Ischemic Injury. Stem Cells Int. 2015, 2015, 761643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarà, M.; Amadio, P.; Campodonico, J.; Sandrini, L.; Barbieri, S.S. Exosomes in Cardiovascular Diseases. Diagnostics 2020, 10, 943. [Google Scholar] [CrossRef] [PubMed]
- Tseliou, E.; Fouad, J.; Reich, H.; Slipczuk, L.; de Couto, G.; Aminzadeh, M.; Middleton, R.; Valle, J.; Weixin, L.; Marbán, E. Fibroblasts Rendered Antifibrotic, Antiapoptotic, and Angiogenic by Priming with Cardiosphere-Derived Extracellular Membrane Vesicles. J. Am. Coll. Cardiol. 2015, 66, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.S.; Benedict, C.; et al. Embryonic Stem Cell–Derived Exosomes Promote Endogenous Repair Mechanisms and Enhance Cardiac Function Following Myocardial Infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Chen, K.; Liang, J.; Qin, T.; Zhang, Y.; Chen, X.; Wang, Z. The Role of Extracellular Vesicles in Embryo Implantation. Front. Endocrinol. 2022, 13, 809596. [Google Scholar] [CrossRef]
- Agarwal, U.; George, A.; Bhutani, S.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Mehta, Y.; Platt, M.O.; Liang, Y.; Sahoo, S.; et al. Experimental, Systems, and Computational Approaches to Understanding the MicroRNA-Mediated Reparative Potential of Cardiac Progenitor Cell–Derived Exosomes From Pediatric Patients. Circ. Res. 2017, 120, 701–712. [Google Scholar] [CrossRef]
- Hoffman, J.R.; Park, H.-J.; Bheri, S.; Jayaraman, A.R.; Davis, M.E. Comparative computational RNA analysis of cardiac-derived progenitor cells and their extracellular vesicles. Genomics 2022, 114, 110349. [Google Scholar] [CrossRef]
- Yin, X.; Jiang, L.-H. Extracellular vesicles: Targeting the heart. Front. Cardiovasc. Med. 2023, 9, 1041481. [Google Scholar] [CrossRef]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef]
- Kalisch-Smith, J.I.; Ved, N.; Sparrow, D.B. Environmental Risk Factors for Congenital Heart Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a037234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, T.B.; Foo, S.Y.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Spero, T.L.; Nolte, C.G.; Garcia, V.C.; Lin, Z.; Romitti, P.A.; Shaw, G.M.; Sheridan, S.C.; Feldkamp, M.L.; Woomert, A.; et al. Projected Changes in Maternal Heat Exposure During Early Pregnancy and the Associated Congenital Heart Defect Burden in the United States. J. Am. Heart Assoc. 2019, 8, e010995. [Google Scholar] [CrossRef] [Green Version]
- Morton, S.U.; Quiat, D.; Seidman, J.G.; Seidman, C.E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 2021, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Pierpont Mary, E.; Brueckner, M.; Chung Wendy, K.; Garg, V.; Lacro Ronald, V.; McGuire Amy, L.; Mital, S.; Priest James, R.; Pu William, T.; Roberts, A.; et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation 2018, 138, e653–e711. [Google Scholar] [CrossRef]
- Blue, G.M.; Kirk, E.P.; Sholler, G.F.; Harvey, R.P.; Winlaw, D.S. Congenital heart disease: Current knowledge about causes and inheritance. Med. J. Aust. 2012, 197, 155–159. [Google Scholar] [CrossRef]
- McHugh, A.; El-Khuffash, A.; Bussmann, N.; Doherty, A.; Franklin, O.; Breathnach, F. Hyperoxygenation in pregnancy exerts a more profound effect on cardiovascular hemodynamics than is observed in the nonpregnant state. Am. J. Obstet. Gynecol. 2019, 220, 397.e1–397.e8. [Google Scholar] [CrossRef]
- Courtney, J.A.; Cnota, J.F.; Jones, H.N. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Maslen, C.L. Recent Advances in Placenta–Heart Interactions. Front. Physiol. 2018, 9, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.A.; Rychik, J.; Savla, J.J. The placenta as the window to congenital heart disease. Curr. Opin. Cardiol. 2021, 36, 56–60. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Jongbloed, M.R.M.; Poelmann, R.E.; Bartelings, M.M. Structural Heart Disease: Embryology. In Fetal Therapy: Scientific Basis and Critical Appraisal of Clinical Benefits, 2nd ed.; Johnson, A., Oepkes, D., Kilby, M.D., Eds.; Cambridge University Press: Cambridge, UK, 2020; pp. 110–122. [Google Scholar] [CrossRef]
- Moreau, J.L.M.; Kesteven, S.; Martin, E.M.M.A.; Lau, K.S.; Yam, M.X.; O’Reilly, V.C.; del Monte-Nieto, G.; Baldini, A.; Feneley, M.P.; Moon, A.M.; et al. Gene-environment interaction impacts on heart development and embryo survival. Development 2019, 146, dev172957. [Google Scholar] [CrossRef] [Green Version]
- Bentham, J.; Michell, A.C.; Lockstone, H.; Andrew, D.; Schneider, J.E.; Brown, N.A.; Bhattacharya, S. Maternal high-fat diet interacts with embryonic Cited2 genotype to reduce Pitx2c expression and enhance penetrance of left-right patterning defects. Hum. Mol. Genet. 2010, 9, 3394–3401. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Sturzu, A.C.; Rajarajan, K.; Passer, D.; Plonowska, K.; Riley, A.; Tan, T.C.; Sharma, A.; Xu, A.F.; Engels, M.C.; Feistritzer, R.; et al. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation 2015, 132, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Ho, L.; Ford, G.H.; Chen, H.I.; Goldstone, A.B.; Woo, Y.J.; Quertermous, T.; Reversade, B.; Red-Horse, K. Alternative Progenitor Cells Compensate to Rebuild the Coronary Vasculature in Elabela- and Apj-Deficient Hearts. Dev. Cell 2017, 42, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Drenckhahn, J.-D.; Schwarz, Q.P.; Gray, S.; Laskowski, A.; Kiriazis, H.; Ming, Z.; Harvey, R.P.; Du, X.-J.; Thorburn, D.R.; Cox, T.C. Compensatory Growth of Healthy Cardiac Cells in the Presence of Diseased Cells Restores Tissue Homeostasis during Heart Development. Dev. Cell 2008, 15, 521–533. [Google Scholar] [CrossRef]
- Founta, K.-M.; Papanayotou, C. In Vivo Generation of Organs by Blastocyst Complementation: Advances and Challenges. Int. J. Stem Cells 2022, 15, 113–121. [Google Scholar] [CrossRef]
- Coppiello, G.; Barlabé, P.; Moya-Jódar, M.; Abizanda, G.; Barreda, C.; Iglesias, E.; Linares, J.; Arellano-Viera, E.; Ruiz-Villalba, A.; Larequi, E.; et al. In vivo generation of heart and vascular system by blastocyst complementation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kitajima, S.; Takagi, A.; Inoue, T.; Saga, Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development 2000, 127, 3215–3226. [Google Scholar] [CrossRef] [PubMed]
- Houyel, L.; Meilhac, S.M. Heart Development and Congenital Structural Heart Defects. Annu. Rev. Genom. Hum. Genet. 2021, 22, 257–284. [Google Scholar] [CrossRef]
- Tyser, R.C.V.; Ibarra-Soria, X.; McDole, K.; Arcot Jayaram, S.; Godwin, J.; van den Brand, T.A.H.; Miranda, A.M.A.; Scialdone, A.; Keller, P.J.; Marioni, J.C.; et al. Characterization of a common progenitor pool of the epicardium and myocardium. Science 2021, 371, eabb2986. [Google Scholar] [CrossRef] [PubMed]
- Swedlund, B.; Lescroart, F. Cardiopharyngeal Progenitor Specification: Multiple Roads to the Heart and Head Muscles. Cold Spring Harb. Perspect. Biol. 2020, 12, a036731. [Google Scholar] [CrossRef]
- Kloesel, B.; DiNardo, J.A.; Body, S.C. Cardiac Embryology and Molecular Mechanisms of Congenital Heart Disease—A Primer for Anesthesiologists. Anesth. Analg. 2016, 123, 551–569. [Google Scholar] [CrossRef]
- Lawson, K.A.; Meneses, J.J.; Pedersen, R.A. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 1991, 113, 891–911. [Google Scholar] [CrossRef]
- Scialdone, A.; Tanaka, Y.; Jawaid, W.; Moignard, V.; Wilson, N.K.; Macaulay, I.C.; Marioni, J.C.; GÃttgens, B. Resolving early mesoderm diversification through single-cell expression profiling. Nature 2016, 535, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, P.; Jahnel, S.M.; Mendjan, S. In vitro models of the human heart. Development 2021, 148, dev199672. [Google Scholar] [CrossRef]
- Martyn, I.; Kanno, T.Y.; Ruzo, A.; Siggia, E.D.; Brivanlou, A.H. Self-organization of a human organizer by combined Wnt and Nodal signalling. Nature 2018, 558, 132–135. [Google Scholar] [CrossRef]
- Deglincerti, A.; Croft, G.F.; Pietila, L.N.; Zernicka-Goetz, M.; Siggia, E.D.; Brivanlou, A.H. Self-organization of the in vitro attached human embryo. Nature 2016, 533, 251–254. [Google Scholar] [CrossRef]
- Shahbazi, M.N.; Jedrusik, A.; Vuoristo, S.; Recher, G.; Hupalowska, A.; Bolton, V.; Fogarty, N.M.E.; Campbell, A.; Devito, L.G.; Ilic, D.; et al. Self-organization of the human embryo in the absence of maternal tissues. Nat. Cell Biol. 2016, 18, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, H.; Javali, A.; Khoei, H.H.; Sommer, T.M.; Sestini, G.; Novatchkova, M.; Scholte op Reimer, Y.; Castel, G.; Bruneau, A.; Maenhoudt, N.; et al. Human blastoids model blastocyst development and implantation. Nature 2022, 601, 600–605. [Google Scholar] [CrossRef]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [Green Version]
- Huppertz, B. The anatomy of the normal placenta. J. Clin. Pathol. 2008, 61, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jaremek, A.; Jeyarajah, M.J.; Jaju Bhattad, G.; Renaud, S.J. Omics Approaches to Study Formation and Function of Human Placental Syncytiotrophoblast. Front. Cell Dev. Biol. 2021, 9, 674162. [Google Scholar] [CrossRef]
- Soma-Pillay, P.; Nelson-Piercy, C.; Tolppanen, H.; Mebazaa, A. Physiological changes in pregnancy. Cardiovasc. J. Afr. 2016, 27, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Sáez, M.A.; Álvarez-Mon, M.A.; Torres-Carranza, D.; Álvarez-Mon, M.; Bujan, J.; García-Honduvilla, N.; Bravo, C.; et al. The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease. Cells 2022, 11, 568. [Google Scholar] [CrossRef]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth-New Insights From Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, S.S.; Balasinor, N.H. Placental Defects: An Epigenetic Perspective. Reprod. Sci. 2018, 25, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, V.; Fineberg, E.; Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J.; et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef]
- Liu, S.; Joseph, K.S.; Lisonkova, S.; Rouleau, J.; Hof, M.V.d.; Sauve, R.; Kramer, M.S. Association between Maternal Chronic Conditions and Congenital Heart Defects. Circulation 2013, 128, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Camm, E.J.; Botting, K.J.; Sferruzzi-Perri, A.N. Near to One’s Heart: The Intimate Relationship Between the Placenta and Fetal Heart. Front. Physiol. 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. 2007, 113, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xu, A.; Xu, H. The roles of vascular endothelial growth factor gene polymorphisms in congenital heart diseases: A meta-analysis. Growth Factors 2018, 36, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Aplin, J.D.; Myers, J.E.; Timms, K.; Westwood, M. Tracking placental development in health and disease. Nat. Rev. Endocrinol. 2020, 16, 479–494. [Google Scholar] [CrossRef]
- Svensson-Arvelund, J.; Mehta, R.B.; Lindau, R.; Mirrasekhian, E.; Rodriguez-Martinez, H.; Berg, G.; Lash, G.E.; Jenmalm, M.C.; Ernerudh, J. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J. Immunol. 2015, 194, 1534–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Nakashima, A.; Shima, T.; Ito, M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am. J. Reprod. Immunol. 2010, 63, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Carlin, D.; Zhu, F.; Cattaneo, P.; Ideker, T.; Evans, S.M.; Bloomekatz, J.; Chi, N.C. Unveiling Complexity and Multipotentiality of Early Heart Fields. Circ. Res. 2021, 129, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The heart field transcriptional landscape at single-cell resolution. Dev. Cell 2023, 58, 257–266. [Google Scholar] [CrossRef]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovitch, K.; Soro-Barrio, P.; Chakravarty, P.; Jones, R.A.; Bell, D.M.; Mousavy Gharavy, S.N.; Stamataki, D.; Delile, J.; Smith, J.C.; Briscoe, J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLOS Biol. 2021, 19, e3001200. [Google Scholar] [CrossRef]
- Laugwitz, K.-L.; Moretti, A.; Caron, L.; Nakano, A.; Chien, K.R. Islet1 cardiovascular progenitors: A single source for heart lineages? Development 2008, 135, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, S.D.; Buckingham, M.E.; Peter, K. How to Make a Heart: The Origin and Regulation of Cardiac Progenitor Cells. Curr. Top Dev. Biol. 2010, 90, 1–41. [Google Scholar] [CrossRef] [PubMed]
- van Weerd, J.H.; Christoffels, V.M. The formation and function of the cardiac conduction system. Development 2016, 143, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Munshi, N.V. Development of the Cardiac Conduction System. Cold Spring Harb. Perspect. Biol. 2020, 12, a037408. [Google Scholar] [CrossRef]
- Mjaatvedt, C.H.; Yamamura, H.; Wessels, A.; Ramsdell, A.; Turner, D.; Markwald, R.R.; Rosenthal, N. 10—Mechanisms of Segmentation, Septation, and Remodeling of the Tubular Heart: Endocardial Cushion Fate and Cardiac Looping A2—Harvey, Richard P. In Heart Development; Academic Press: San Diego, CA, USA, 1999; pp. 159–177. [Google Scholar] [CrossRef]
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [Green Version]
- George, R.M.; Maldonado-Velez, G.; Firulli, A.B. The heart of the neural crest: Cardiac neural crest cells in development and regeneration. Development 2020, 147, dev188706. [Google Scholar] [CrossRef] [PubMed]
- Bardot, E.; Calderon, D.; Santoriello, F.; Han, S.; Cheung, K.; Jadhav, B.; Burtscher, I.; Artap, S.; Jain, R.; Epstein, J.; et al. Foxa2 identifies a cardiac progenitor population with ventricular differentiation potential. Nat. Commun. 2017, 8, 14428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct Compartments of the Proepicardial Organ Give Rise to Coronary Vascular Endothelial Cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Motoike, T.; Markham, D.W.; Rossant, J.; Sato, T.N. Evidence for novel fate of Flk1+ progenitor: Contribution to muscle lineage. Genesis 2003, 35, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Witman, N.; Zhou, C.; Grote Beverborg, N.; Sahara, M.; Chien, K.R. Cardiac progenitors and paracrine mediators in cardiogenesis and heart regeneration. Semin. Cell Dev. Biol. 2020, 100, 29–51. [Google Scholar] [CrossRef]
- Flaherty, M.P.; Dawn, B. Noncanonical Wnt11 Signaling and Cardiomyogenic Differentiation. Trends Cardiovasc. Med. 2008, 18, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Strate, I.; Tessadori, F.; Bakkers, J. Glypican4 promotes cardiac specification and differentiation by attenuating canonical Wnt and Bmp signaling. Development 2015, 142, 1767–1776. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Weidinger, G.; Osugi, T.; Kohn, A.D.; Golob, J.L.; Pabon, L.; Reinecke, H.; Moon, R.T.; Murry, C.E. Biphasic role for Wnt/β-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9685–9690. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Pfeiffer, M.J.; Frank, S.; Adachi, K.; Piccini, I.; Quaranta, R.; Arauzo-Bravo, M.; Schwarz, J.; Schade, D.; Leidel, S.; et al. Stepwise Clearance of Repressive Roadblocks Drives Cardiac Induction in Human ESCs. Cell Stem Cell 2016, 18, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buikema, J.W.; Zwetsloot, P.-P.M.; Doevendans, P.A.; Domian, I.J.; PG, S.J. Wnt/β-Catenin Signaling during Cardiac Development and Repair. J. Cardiovasc. Dev. Dis. 2014, 1, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Asakura, M.; Inoue, H.; Nakamura, T.; Sano, M.; Niu, Z.; Chen, M.; Schwartz, R.J.; Schneider, M.D. Sox17 is essential for the specification of cardiac mesoderm in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3859–3864. [Google Scholar] [CrossRef]
- Paige, S.L.; Osugi, T.; Afanasiev, O.K.; Pabon, L.; Reinecke, H.; Murry, C.E. Endogenous Wnt/beta-Catenin Signaling Is Required for Cardiac Differentiation in Human Embryonic Stem Cells. PLoS ONE 2010, 5, e11134. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jia, Z.; Wang, T.; Wang, W.; Zhang, C.; Chen, P.; Ma, K.; Zhou, C. Interaction of Wnt/β-catenin and notch signaling in the early stage of cardiac differentiation of P19CL6 cells. J. Cell. Biochem. 2012, 113, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Garside, V.; Chang, A.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Karsan, A. Notch Signaling in Cardiac Development. Circ. Res. 2008, 102, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Urbanek, K.; Nascimbene, A.; Hosoda, T.; Zheng, H.; Delucchi, F.; Amano, K.; Gonzalez, A.; Vitale, S.; Ojaimi, C.; et al. Notch1 regulates the fate of cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2008, 105, 15529–15534. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Kokubo, H.; Miyagawa-Tomita, S.; Endo, M.; Igarashi, K.; Aisaki, K.i.; Kanno, J.; Saga, Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development 2006, 133, 1625–1634. [Google Scholar] [CrossRef] [Green Version]
- Shamis, Y.; Cullen, D.; Liu, L.; Yang, G.; Ng, S.; Xiao, L.; Bell, F.; Ray, C.; Takikawa, S.; Moskowitz, I.; et al. Maternal and zygotic Zfp57 modulate NOTCH signaling in cardiac development. Proc. Natl. Acad. Sci. USA 2015, 112, E2020–E2029. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Hrstka, S.C.L.; Evans, J.M.; O’Byrne, M.M.; de Andrade, M.; O’Leary, P.W.; Nelson, T.J.; Olson, T.M. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum. Genet. 2015, 134, 1003–1011. [Google Scholar] [CrossRef]
- Luxan, G.; D’Amato, G.; MacGrogan, D.; de la Pompa, J.L. Endocardial Notch Signaling in Cardiac Development and Disease. Circ. Res. 2016, 118, e1–e18. [Google Scholar] [CrossRef]
- D’Amato, G.; Luxán, G.; de la Pompa, J.L. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J. 2016, 283, 4223–4237. [Google Scholar] [CrossRef]
- MacGrogan, D.; Luxán, G.; de la Pompa, J.L. Genetic and functional genomics approaches targeting the Notch pathway in cardiac development and congenital heart disease. Brief. Funct. Genom. 2014, 13, 15–27. [Google Scholar] [CrossRef] [Green Version]
- MacGrogan, D.; Münch, J.; de la Pompa, J.L. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 685–704. [Google Scholar] [CrossRef]
- Miao, L.; Lu, Y.; Nusrat, A.; Abdelnasser, H.Y.; Datta, S.; Zhou, B.; Schwartz, R.J.; Wu, M. The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis. Cells 2021, 10, 2192. [Google Scholar] [CrossRef]
- del Monte-Nieto, G.; Ramialison, M.; Adam, A.A.S.; Wu, B.; Aharonov, A.; D’Uva, G.; Bourke, L.M.; Pitulescu, M.E.; Chen, H.; de la Pompa, J.L.; et al. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 2018, 557, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Prall, O.W.J.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2-5/Bmp2/Smad1 Negative Feedback Loop Controls Heart Progenitor Specification and Proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Greene, S.B.; Bonilla-Claudio, M.; Tao, Y.; Zhang, J.; Bai, Y.; Huang, Z.; Black, B.L.; Wang, F.; Martin, J.F. Bmp Signaling Regulates Myocardial Differentiation from Cardiac Progenitors through a MicroRNA-Mediated Mechanism. Dev. Cell 2010, 19, 903–912. [Google Scholar] [CrossRef] [Green Version]
- van Wijk, B.; Moorman, A.F.M.; van den Hoff, M.J.B. Role of bone morphogenetic proteins in cardiac differentiation. Cardiovasc. Res. 2007, 74, 244–255. [Google Scholar] [CrossRef]
- Yuasa, S.; Fukuda, K. Multiple roles for BMP signaling in cardiac development. Drug Discov. Today Ther. Strateg. 2008, 5, 209–214. [Google Scholar] [CrossRef]
- McCulley, D.J.; Kang, J.-O.; Martin, J.F.; Black, B.L. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 2008, 237, 3200–3209. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Albini, S.; Wei, K.; Willems, E.; Guzzo, R.M.; Tsuda, M.; Giordani, L.; Spiering, S.; Kurian, L.; Yeo, G.W.; et al. Coordinate Nodal and BMP inhibition directs Baf60c-dependent cardiomyocyte commitment. Genes Dev. 2013, 27, 2332–2344. [Google Scholar] [CrossRef] [Green Version]
- Taha, M.F.; Valojerdi, M.R. Effect of bone morphogenetic protein-4 on cardiac differentiation from mouse embryonic stem cells in serum-free and low-serum media. Int. J. Cardiol. 2008, 127, 78–87. [Google Scholar] [CrossRef]
- Tirosh-Finkel, L.; Zeisel, A.; Brodt-Ivenshitz, M.; Shamai, A.; Yao, Z.; Seger, R.; Domany, E.; Tzahor, E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development 2010, 137, 2989–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; McColl, J.; Camp, E.; Kennerley, N.; Mok, G.F.; McCormick, D.; Grocott, T.; Wheeler, G.N.; Münsterberg, A.E. Smad1 transcription factor integrates BMP2 and Wnt3a signals in migrating cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 7337–7342. [Google Scholar] [CrossRef]
- Luo, W.; Zhao, X.; Jin, H.; Tao, L.; Zhu, J.; Wang, H.; Hemmings, B.A.; Yang, Z. Akt1 signaling coordinates BMP signaling and β-catenin activity to regulate second heart field progenitor development. Development 2015, 142, 732–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Ohta, H.; Nakayama, Y.; Konishi, M. Roles of FGF Signals in Heart Development, Health, and Disease. Front. Cell Dev. Biol. 2016, 4, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, F.; Payan, S.M.; Rochais, F. FGF10 Signaling in Heart Development, Homeostasis, Disease and Repair. Front. Genet. 2018, 9, 599. [Google Scholar] [CrossRef] [PubMed]
- Floriddia, E.M.; Lourenço, T.; Zhang, S.; van Bruggen, D.; Hilscher, M.M.; Kukanja, P.; Gonçalves dos Santos, J.P.; Altınkök, M.; Yokota, C.; Llorens-Bobadilla, E.; et al. Distinct oligodendrocyte populations have spatial preference and different responses to spinal cord injury. Nat. Commun. 2020, 11, 5860. [Google Scholar] [CrossRef] [PubMed]
- Engels, M.C.; Rajarajan, K.; Feistritzer, R.; Sharma, A.; Nielsen, U.B.; Schalij, M.J.; de Vries, A.A.F.; Pijnappels, D.A.; Wu, S.M. Insulin-Like Growth Factor Promotes Cardiac Lineage Induction In Vitro by Selective Expansion of Early Mesoderm. Stem Cells 2014, 32, 1493–1502. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Cavallero, S.; Gu, Y.; Chen, T.H.; Hughes, J.; Hassan, A.B.; Brüning, J.C.; Pashmforoush, M.; Sucov, H.M. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011, 138, 1795–1805. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xie, Y.; Guan, L.; Elkin, K.; Xiao, J. Targets identified from exercised heart: Killing multiple birds with one stone. NPJ Regen. Med. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Weeks, K.L.; Tham, Y.K.; Yildiz, S.G.; Alexander, Y.; Donner, D.G.; Kiriazis, H.; Harmawan, C.A.; Hsu, A.; Bernardo, B.C.; Matsumoto, A.; et al. FoxO1 is required for physiological cardiac hypertrophy induced by exercise but not by constitutively active PI3K. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H1470–H1485. [Google Scholar] [CrossRef] [PubMed]
- Bass-Stringer, S.; Ooi, J.Y.Y.; McMullen, J.R. Clusterin is regulated by IGF1–PI3K signaling in the heart: Implications for biomarker and drug target discovery, and cardiotoxicity. Arch. Toxicol. 2020, 94, 1763–1768. [Google Scholar] [CrossRef]
- Wederell, E.D.; Bilenky, M.; Cullum, R.; Thiessen, N.; Dagpinar, M.; Delaney, A.; Varhol, R.; Zhao, Y.; Zeng, T.; Bernier, B.; et al. Global analysis of in vivo Foxa2-binding sites in mouse adult liver using massively parallel sequencing. Nucl. Acids Res. 2008, 36, 4549–4564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Grunewald, M.; Dor, Y.; Keshet, E. VEGF regulates relative allocation of Isl1+ cardiac progenitors to myocardial and endocardial lineages. Mech. Dev. 2016, 142, 40–49. [Google Scholar] [CrossRef]
- Dor, Y.; Camenisch, T.D.; Itin, A.; Fishman, G.I.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-H.; Gehmert, S.; Sadat, S.; Pinkernell, K.; Bai, X.; Matthias, N.; Alt, E. VEGF is critical for spontaneous differentiation of stem cells into cardiomyocytes. Biochem. Biophys. Res. Commun. 2007, 354, 999–1003. [Google Scholar] [CrossRef]
- Lania, G.; Ferrentino, R.; Baldini, A. TBX1 Represses Vegfr2 Gene Expression and Enhances the Cardiac Fate of VEGFR2+ Cells. PLoS ONE 2015, 10, e0138525. [Google Scholar] [CrossRef] [Green Version]
- Sridurongrit, S.; Larsson, J.; Schwartz, R.; Ruiz-Lozano, P.; Kaartinen, V. Signaling via the Tgf-[beta] type I receptor Alk5 in heart development. Dev. Biol. 2008, 322, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Duelen, R.; Gilbert, G.; Patel, A.; de Schaetzen, N.; De Waele, L.; Roderick, L.; Sipido, K.R.; Verfaillie, C.M.; Buyse, G.M.; Thorrez, L.; et al. Activin A Modulates CRIPTO-1/HNF4α+ Cells to Guide Cardiac Differentiation from Human Embryonic Stem Cells. Stem Cells Int. 2017, 2017, 4651238. [Google Scholar] [CrossRef] [Green Version]
- Deshwar, A.R.; Chng, S.C.; Ho, L.; Reversade, B.; Scott, I.C. The Apelin receptor enhances Nodal/TGFβ signaling to ensure proper cardiac development. eLife 2016, 5, e13758. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Haase, A.; Franke, A.; Bolesani, E.; Schwanke, K.; Robles-Diaz, D.; Coffee, M.; Göhring, G.; Dräger, G.; et al. Bulk cell density and Wnt/TGFbeta signalling regulate mesendodermal patterning of human pluripotent stem cells. Nat. Commun. 2016, 7, 13602. [Google Scholar] [CrossRef] [Green Version]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Cohen, E.D.; Morrisey, E.E. The Importance of Wnt Signaling in Cardiovascular Development. Pediatr. Cardiol. 2010, 31, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, A.G.; Resende, T.P.; Pinto-do-Ó, P. Building and Repairing the Heart: What Can We Learn from Embryonic Development? BioMed Res. Int. 2014, 2014, 679168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onizuka, T.; Yuasa, S.; Kusumoto, D.; Shimoji, K.; Egashira, T.; Ohno, Y.; Kageyama, T.; Tanaka, T.; Hattori, F.; Fujita, J.; et al. Wnt2 accelerates cardiac myocyte differentiation from ES-cell derived mesodermal cells via non-canonical pathway. J. Mol. Cell. Cardiol. 2012, 52, 650–659. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Li, Y.; Qian, H.; Shan, X.; Yan, Y.; Mao, F.; Wu, X.; Xu, W.R. Mesenchymal stem cell-secreted soluble signaling molecules potentiate tumor growth. Cell Cycle 2011, 10, 3198–3207. [Google Scholar] [CrossRef]
- Gnecchi, M.; Zhang, Z.; Ni, A.; Dzau, V.J. Paracrine Mechanisms in Adult Stem Cell Signaling and Therapy. Circ. Res. 2008, 103, 1204–1219. [Google Scholar] [CrossRef] [PubMed]
- Rafatian, G.; Davis, D.R. Concise Review: Heart-Derived Cell Therapy 2.0: Paracrine Strategies to Increase Therapeutic Repair of Injured Myocardium. Stem Cells 2018, 36, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of Angiogenic and Antiapoptotic Factors by Human Adipose Stromal Cells. Circulation 2004, 109, 1292–1298. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.E.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert Rev. Proteom. 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Bayado, N.; He, D.; Li, J.; Chen, H.; Li, L.; Li, J.; Long, X.; Du, T.; Tang, J.; et al. Therapeutic Applications of Extracellular Vesicles for Myocardial Repair. Front. Cardiovasc. Med. 2021, 8, 758050. [Google Scholar] [CrossRef]
- Tian, R.; Colucci, W.S.; Arany, Z.; Bachschmid, M.M.; Ballinger, S.W.; Boudina, S.; Bruce, J.E.; Busija, D.W.; Dikalov, S.; Dorn, G.W.; et al. Unlocking the Secrets of Mitochondria in the Cardiovascular System. Circulation 2019, 140, 1205–1216. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Yellon, D.M.; Sivaraman, V.; Das, D.; Boi-Doku, C.; Arjun, S.; Zheng, Y.; Riquelme, J.A.; Kearney, J.; Sharma, V.; et al. Plasma Exosomes Protect the Myocardium From Ischemia-Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, Y.; Tang, P.; Liu, X.; Chai, X.; Dong, J.; Che, X.; Zhou, Q.; Ni, M.; Jin, F. Maternal serum-derived exosomal lactoferrin as a marker in detecting and predicting ventricular septal defect in fetuses. Exp. Biol. Med. 2022, 247, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Shen, H.; Wu, S.; Ruan, J.; Lyu, G. Construction of the amniotic fluid-derived exosomal ceRNA network associated with ventricular septal defect. Genomics 2021, 113, 4293–4302. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Lionetti, V.; Cervio, E.; Matteucci, M.; Gherghiceanu, M.; Popescu, L.M.; Torre, T.; Siclari, F.; Moccetti, T.; Vassalli, G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc. Res. 2014, 103, 530–541. [Google Scholar] [CrossRef]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.E.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.-h.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Loyer, X.; Vion, A.-C.; Tedgui, A.; Boulanger, C.M. Microvesicles as Cell–Cell Messengers in Cardiovascular Diseases. Circ. Res. 2014, 114, 345–353. [Google Scholar] [CrossRef]
- Wang, X.; Huang, W.; Liu, G.; Cai, W.; Millard, R.W.; Wang, Y.; Chang, J.; Peng, T.; Fan, G.-C. Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J. Mol. Cell. Cardiol. 2014, 74, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Luo, Q. Potential clinical applications of exosomes in the diagnosis, treatment, and prognosis of cardiovascular diseases: A narrative review. Ann. Transl. Med. 2022, 10, 372. [Google Scholar] [CrossRef]
- Kishore, R.; Khan, M. Cardiac cell-derived exosomes: Changing face of regenerative biology. Eur. Heart J. 2016, 38, 212–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafourian, M.; Mahdavi, R.; Akbari Jonoush, Z.; Sadeghi, M.; Ghadiri, N.; Farzaneh, M.; Mousavi Salehi, A. The implications of exosomes in pregnancy: Emerging as new diagnostic markers and therapeutics targets. Cell Commun. Signal. 2022, 20, 51. [Google Scholar] [CrossRef]
- Sarker, S.; Scholz-Romero, K.; Perez, A.; Illanes, S.E.; Mitchell, M.D.; Rice, G.E.; Salomon, C. Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy. J. Transl. Med. 2014, 12, 204. [Google Scholar] [CrossRef] [Green Version]
- Paktinat, S.; Hashemi, S.M.; Ghaffari Novin, M.; Mohammadi-Yeganeh, S.; Salehpour, S.; Karamian, A.; Nazarian, H. Seminal exosomes induce interleukin-6 and interleukin-8 secretion by human endometrial stromal cells. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 235, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Gurung, S.; Greening, D.W.; Catt, S.; Salamonsen, L.; Evans, J. Exosomes and soluble secretome from hormone-treated endometrial epithelial cells direct embryo implantation. Mol. Hum. Reprod. 2020, 26, 510–520. [Google Scholar] [CrossRef]
- Shi, R.; Zhao, L.; Cai, W.; Wei, M.; Zhou, X.; Yang, G.; Yuan, L. Maternal exosomes in diabetes contribute to the cardiac development deficiency. Biochem. Biophys. Res. Commun. 2017, 483, 602–608. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wang, C.; Shi, R.; Zhou, X.; Li, Z.; Sun, W.; Zhao, L.; Yuan, L. Maternal obesity increases the risk of fetal cardiac dysfunction via visceral adipose tissue derived exosomes. Placenta 2021, 105, 85–93. [Google Scholar] [CrossRef]
- Salomon, C.; Guanzon, D.; Scholz-Romero, K.; Longo, S.; Correa, P.; Illanes, S.E.; Rice, G.E. Placental Exosomes as Early Biomarker of Preeclampsia: Potential Role of Exosomal MicroRNAs Across Gestation. J. Clin. Endocrinol. Metab. 2017, 102, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Burkova, E.E.; Sedykh, S.E.; Nevinsky, G.A. Human Placenta Exosomes: Biogenesis, Isolation, Composition, and Prospects for Use in Diagnostics. Int. J. Mol. Sci. 2021, 22, 2158. [Google Scholar] [CrossRef]
- Jin, J.; Menon, R. Placental exosomes: A proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 2018, 79, e12788. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Barnes, R.M.; Black, B.L. Nodal Signaling and Congenital Heart Defects. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer Japan: Tokyo, Japan, 2016; pp. 183–192. [Google Scholar] [CrossRef]
- Pauklin, S.; Vallier, L. Activin/Nodal signalling in stem cells. Development 2015, 142, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef]
- Liu, L.; Nemashkalo, A.; Rezende, L.; Jung, J.Y.; Chhabra, S.; Guerra, M.C.; Heemskerk, I.; Warmflash, A. Nodal is a short-range morphogen with activity that spreads through a relay mechanism in human gastruloids. Nat. Commun. 2022, 13, 497. [Google Scholar] [CrossRef]
- Webber, D.M.; Li, M.; MacLeod, S.L.; Tang, X.; Levy, J.W.; Karim, M.A.; Erickson, S.W.; Hobbs, C.A.; The National Birth Defects Prevention Study. Gene-Folic Acid Interactions and Risk of Conotruncal Heart Defects: Results from the National Birth Defects Prevention Study. Genes 2023, 14, 180. [Google Scholar] [CrossRef]
- Wondemagegn, A.T.; Afework, M. The association between folic acid supplementation and congenital heart defects: Systematic review and meta-analysis. SAGE Open Med. 2022, 10, 20503121221081069. [Google Scholar] [CrossRef]
- Kumar, S.D.; Vijaya, M.; Samy, R.P.; Dheen, S.T.; Ren, M.; Watt, F.; Kang, Y.J.; Bay, B.-H.; Tay, S.S.W. Zinc supplementation prevents cardiomyocyte apoptosis and congenital heart defects in embryos of diabetic mice. Free. Radic. Biol. Med. 2012, 53, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Vermot, J.; Messaddeq, N.; Niederreither, K.; Dierich, A.; Dollé, P. Rescue of morphogenetic defects and of retinoic acid signaling in retinaldehyde dehydrogenase 2 (Raldh2) mouse mutants by chimerism with wild-type cells. Differentiation 2006, 74, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Chiş, A.R.; Moise, A.R. Role of carotenoids and retinoids during heart development. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158636. [Google Scholar] [CrossRef] [PubMed]
- Agnew, E.J.; Ivy, J.R.; Stock, S.J.; Chapman, K.E. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J. Mol. Endocrinol. 2018, 61, R61–R73. [Google Scholar] [CrossRef] [Green Version]
- Breur, J.M.; Visser, G.H.; Kruize, A.A.; Stoutenbeek, P.; Meijboom, E.J. Treatment of fetal heart block with maternal steroid therapy: Case report and review of the literature. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 2004, 24, 467–472. [Google Scholar] [CrossRef]
- Eliasson, H.; Sonesson, S.E.; Sharland, G.; Granath, F.; Simpson, J.M.; Carvalho, J.S.; Jicinska, H.; Tomek, V.; Dangel, J.; Zielinsky, P.; et al. Isolated atrioventricular block in the fetus: A retrospective, multinational, multicenter study of 175 patients. Circulation 2011, 124, 1919–1926. [Google Scholar] [CrossRef] [Green Version]
- Gaber, N.; Gagliardi, M.; Patel, P.; Kinnear, C.; Zhang, C.; Chitayat, D.; Shannon, P.; Jaeggi, E.; Tabori, U.; Keller, G.; et al. Fetal Reprogramming and Senescence in Hypoplastic Left Heart Syndrome and in Human Pluripotent Stem Cells during Cardiac Differentiation. Am. J. Pathol. 2013, 183, 720–734. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.; Bauer, M.; Kimble, R. Antenatal therapy for intrauterine growth retardation. Acta Paediatr. Suppl. 1997, 86, 196–200. [Google Scholar] [CrossRef]
- Laviola, L.; Perrini, S.; Belsanti, G.; Natalicchio, A.; Montrone, C.; Leonardini, A.; Vimercati, A.; Scioscia, M.; Selvaggi, L.; Giorgino, R.; et al. Intrauterine Growth Restriction in Humans Is Associated with Abnormalities in Placental Insulin-Like Growth Factor Signaling. Endocrinology 2005, 146, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Marbán, E. A mechanistic roadmap for the clinical application of cardiac cell therapies. Nat. Biomed. Eng. 2018, 2, 353–361. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Lässer, C.; Eldh, M.; Lötvall, J. Isolation and Characterization of RNA-Containing Exosomes. JoVE 2012, 59, e3037. [Google Scholar] [CrossRef]
- Rosso, G.; Cauda, V. Biomimicking Extracellular Vesicles with Fully Artificial Ones: A Rational Design of EV-BIOMIMETICS toward Effective Theranostic Tools in Nanomedicine. ACS Biomater. Sci. Eng. 2022. [Google Scholar] [CrossRef]
- Cao, S.; Wu, Y.; Albert Reece, E.; Xu, C.; Shen, W.-B.; Kaushal, S.; Yang, P. Functional cargos of exosomes derived from Flk-1+ vascular progenitors enable neurulation and ameliorate embryonic anomalies in diabetic pregnancy. Commun. Biol. 2022, 5, 648. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zhao, Q.; Beck, A.; Vitale, J.M.; Schneider, J.S.; Terzic, A.; Fraidenraich, D. Rescue of Developmental Defects by Blastocyst Stem Cell Injection: Towards Elucidation of Neomorphic Corrective Pathways. J. Cardiovasc. Transl. Res. 2010, 3, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Fraidenraich, D.; Stillwell, E.; Romero, E.; Wilkes, D.; Manova, K.; Basson, C.; Benezra, R. Rescue of cardiac defects in id knockout embryos by injection of embryonic stem cells. Science 2004, 306, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.A.; Mendes-Silva, L.; Afonso, V.; Martins, G.; Machado, R.S.R.; Lopes, J.A.; Cancela, L.; Futschik, M.E.; Sachinidis, A.; Gavaia, P.; et al. Exogenous Wnt5a and Wnt11 proteins rescue Cited2 dysfunction in mouse embryonic stem cells and zebrafish morphants. Cell Death Dis. 2019, 10, 582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragança, J.; Mendes-Silva, L.; Lopes, J.; SM, C. CITED proteins in the heart of pluripotent cells and in heart’s full potential. Regen Med. Front. 2019, 1, e190005. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Leyva, I.; Matias, A.C.; Oliveira, D.V.; Santos, J.M.A.; Nascimento, R.; Guerreiro, E.; Michell, A.C.; van De Vrugt, A.M.; Machado-Oliveira, G.; Ferreira, G.; et al. CITED2 Cooperates with ISL1 and Promotes Cardiac Differentiation of Mouse Embryonic Stem Cells. Stem Cell Rep. 2016, 7, 1037–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, S.T.; Bamforth, S.D.; Bragança, J.; Chen, C.-M.; Broadbent, C.; Schneider, J.E.; Schwartz, R.J.; Bhattacharya, S. A cell-autonomous role of Cited2 in controlling myocardial and coronary vascular development. Eur. Heart J. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-m.; Bentham, J.; Cosgrove, C.; Braganca, J.; Cuenda, A.; Bamforth, S.D.; Schneider, J.E.; Watkins, H.; Keavney, B.; Davies, B.; et al. Functional Significance of SRJ Domain Mutations in CITED2. PLoS ONE 2012, 7, e46256. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, S.T.; Bamforth, S.D.; Chen, C.-M.; Farthing, C.R.; Franklyn, A.; Broadbent, C.; Schneider, J.E.; Saga, Y.; Lewandoski, M.; Bhattacharya, S. Epiblastic Cited2 deficiency results in cardiac phenotypic heterogeneity and provides a mechanism for haploinsufficiency. Cardiovasc. Res. 2008, 79, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Bamforth, S.D.; Bragança, J.; Farthing, C.R.; Schneider, J.E.; Broadbent, C.; Michell, A.C.; Clarke, K.; Neubauer, S.; Norris, D.; Brown, N.A.; et al. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 2004, 36, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamforth, S.D.; Bragança, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.R.; Kranc, K.R.; Farza, H.; Henderson, D.J.; Hurst, H.C.; Bhattacharya, S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef]
- Xu, B.; Doughman, Y.; Turakhia, M.; Jiang, W.; Landsettle, C.E.; Agani, F.H.; Semenza, G.L.; Watanabe, M.; Yang, Y.-C. Partial rescue of defects in Cited2-deficient embryos by HIF-1[alpha] heterozygosity. Dev. Biol. 2007, 301, 130. [Google Scholar] [CrossRef] [Green Version]
- Kranc, K.R.; Oliveira, D.V.; Armesilla-Diaz, A.; Pacheco-Leyva, I.; Matias, A.C.; Escapa, A.L.; Subramani, C.; Wheadon, H.; Trindade, M.; Nichols, J.; et al. Acute loss of Cited2 impairs Nanog expression and decreases self-renewal of mouse embryonic stem cells. Stem Cells 2015, 33, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Webber, S.A.; Wargowski, D.S.; Chitayat, D.; Sandor, G.G.S. Congenital heart disease and Robinow syndrome: Coincidence or an additional component of the syndrome? Am. J. Med. Genet. 1990, 37, 519–521. [Google Scholar] [CrossRef]
- Li, P.; Li, H.; Zheng, Y.; Qiao, B.; Duan, W.; Huang, L.; Liu, W.; Wang, H. Variants in the Regulatory Region of WNT5A Reduced Risk of Cardiac Conotruncal Malformations in the Chinese Population. Sci. Rep. 2015, 5, 13120. [Google Scholar] [CrossRef] [Green Version]
- Škorić-Milosavljević, D.; Tadros, R.; Bosada, F.M.; Tessadori, F.; Weerd, J.H.v.; Woudstra, O.I.; Tjong, F.V.Y.; Lahrouchi, N.; Bajolle, F.; Cordell, H.J.; et al. Common Genetic Variants Contribute to Risk of Transposition of the Great Arteries. Circ. Res. 2022, 130, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Sinha, T.; Li, D.; Théveniau-Ruissy, M.; Hutson, M.R.; Kelly, R.G.; Wang, J. Loss of Wnt5a disrupts second heart field cell deployment and may contribute to OFT malformations in DiGeorge syndrome. Hum. Mol. Genet. 2015, 24, 1704–1716. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Ye, X.; Guo, N.; Nathans, J. Frizzled 2 and frizzled 7 function redundantly in convergent extension and closure of the ventricular septum and palate: Evidence for a network of interacting genes. Development 2012, 139, 4383–4394. [Google Scholar] [CrossRef] [Green Version]
- Schleiffarth, J.R.; Person, A.D.; Martinsen, B.J.; Sukovich, D.J.; Neumann, A.; Baker, C.V.H.; Lohr, J.L.; Cornfield, D.N.; Ekker, S.C.; Petryk, A. Wnt5a Is Required for Cardiac Outflow Tract Septation in Mice. Pediatr. Res. 2007, 61, 386. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Kang, X.; Gao, F.; Guzman, A.; Lau, R.P.; Biniwale, R.; Wadehra, M.; Reemtsen, B.; Garg, M.; Halnon, N.; et al. Gene-environment regulatory circuits of right ventricular pathology in tetralogy of fallot. J. Mol. Med. 2019, 97, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Erikson, D.W.; Tilford, S.A.; Bany, B.M.; Maclean, I.I.J.A.; Rucker, I.I.I.E.B.; Johnson, G.A.; Spencer, T.E. Wnt Genes in the Mouse Uterus: Potential Regulation of Implantation. Biol. Reprod. 2009, 80, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinhardt, G.; Saleh, L.; Otti, G.R.; Haider, S.; Velicky, P.; Fiala, C.; Pollheimer, J.r.; Knöfler, M. Wingless ligand 5a is a critical regulator of placental growth and survival. Sci. Rep. 2016, 6, 28127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, P.; Song, H.; Kim, W.; Kispert, A.; Yang, Y. Wnt5a and Wnt11 regulate mammalian anterior-posterior axis elongation. Development 2015, 142, 1516–1527. [Google Scholar] [CrossRef] [Green Version]
- Sinha, T.; Lin, L.; Li, D.; Davis, J.; Evans, S.; Wynshaw-Boris, A.; Wang, J. Mapping the dynamic expression of Wnt11 and the lineage contribution of Wnt11-expressing cells during early mouse development. Dev. Biol. 2015, 398, 177–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisson, J.A.; Mills, B.; Paul Helt, J.-C.; Zwaka, T.P.; Cohen, E.D. Wnt5a and Wnt11 inhibit the canonical Wnt pathway and promote cardiac progenitor development via the Caspase-dependent degradation of AKT. Dev. Biol. 2015, 398, 80–96. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.D.; Miller, M.F.; Wang, Z.; Moon, R.T.; Morrisey, E.E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development 2012, 139, 1931–1940. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Qian, C.; Bi, L.-L.; Zhao, F.; Zhang, G.-Y.; Wang, Z.-Q.; Gan, X.-D.; Wang, Y.-G. Enrichment of cardiac differentiation by a large starting number of embryonic stem cells in embryoid bodies is mediated by the Wnt11-JNK pathway. Biotechnol. Lett. 2015, 37, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.M.; Garriock, R.J.; Yatskievych, T.A.; D’Agostino, S.L.; Antin, P.B.; Krieg, P.A. Non-canonical Wnt signaling through Wnt5a/b and a novel Wnt11 gene, Wnt11b, regulates cell migration during avian gastrulation. Dev. Biol. 2008, 320, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Vijayaragavan, K.; Szabo, E.; Bossé, M.; Ramos-Mejia, V.; Moon, R.T.; Bhatia, M. Noncanonical Wnt Signaling Orchestrates Early Developmental Events toward Hematopoietic Cell Fate from Human Embryonic Stem Cells. Cell Stem Cell 2009, 4, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastelaro de Rezende, M.; Zenker Justo, G.; Julian Paredes-Gamero, E.; Gosens, R. Wnt-5A/B Signaling in Hematopoiesis throughout Life. Cells 2020, 9, 1801. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, A.; Choi, S.Y.; Kan, Q.; Zhou, W.; Chacon-Heszele, M.F.; Ryu, Y.K.; McKenna, S.; Zuo, X.; Kuruvilla, R.; et al. Wnt5a Is Necessary for Normal Kidney Development in Zebrafish and Mice. Nephron Exp. Nephrol. 2014, 128, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Ryu, Y.K.; Collins, S.E.; Ho, H.-Y.H.; Zhao, H.; Kuruvilla, R. An autocrine Wnt5a-Ror signaling loop mediates sympathetic target innervation. Dev. Biol. 2013, 377, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Goedel, A.; Kang, Y.; Si, C.; Chu, C.; Zheng, Y.; Chen, Z.; Gruber, P.J.; Xiao, Y.; Zhou, C.; et al. Amnion signals are essential for mesoderm formation in primates. Nat. Commun. 2021, 12, 5126. [Google Scholar] [CrossRef]
- Kimbrel, E.A.; Lanza, R. Next-generation stem cells—Ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov. 2020, 19, 463–479. [Google Scholar] [CrossRef]
- Jeensuk, S.; Ortega, M.S.; Saleem, M.; Hawryluk, B.; Scheffler, T.L.; Hansen, P.J. Actions of WNT family member 5A to regulate characteristics of development of the bovine preimplantation embryo. Biol. Reprod. 2022, 107, 928–944. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Bartos, A.; Park, C.; Sun, X.; Li, Y.; Cha, S.-W.; Ajima, R.; Ho, H.-Y.H.; Yamaguchi, T.P.; Dey, S.K. Appropriate crypt formation in the uterus for embryo homing and implantation requires Wnt5a-ROR signaling. Cell Rep. 2014, 8, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhang, Z.; Chen, C.; Zhang, Y.; Zhang, C. Dysfunction of WNT4/WNT5A in deciduas: Possible relevance to the pathogenesis of preeclampsia. J. Hypertens. 2016, 34, 719–727. [Google Scholar] [CrossRef]
- Dietrich, B.; Haider, S.; Meinhardt, G.; Pollheimer, J.; Knöfler, M. WNT and NOTCH signaling in human trophoblast development and differentiation. Cell. Mol. Life Sci. 2022, 79, 292. [Google Scholar] [CrossRef] [PubMed]
- Rider, V.; Talbott, A.; Bhusri, A.; Krumsick, Z.; Foster, S.; Wormington, J.; Kimler, B.F. WINGLESS (WNT) signaling is a progesterone target for rat uterine stromal cell proliferation. J. Endocrinol. 2016, 229, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Cha, S.-W.; Tadjuidje, E.; White, J.; Wells, J.; Mayhew, C.; Wylie, C.; Heasman, J. Wnt11/5a Complex Formation Caused by Tyrosine Sulfation Increases Canonical Signaling Activity. Curr. Biol. 2009, 19, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Laflamme, M.A.; Gold, J.; Xu, C.; Hassanipour, M.; Rosler, E.; Police, S.; Muskheli, V.; Murry, C.E. Formation of Human Myocardium in the Rat Heart from Human Embryonic Stem Cells. Am. J. Pathol. 2005, 167, 663–671. [Google Scholar] [CrossRef] [Green Version]
- van Laake, L.W.; Passier, R.; Monshouwer-Kloots, J.; Verkleij, A.J.; Lips, D.J.; Freund, C.; den Ouden, K.; Ward-van Oostwaard, D.; Korving, J.; Tertoolen, L.G.; et al. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007, 1, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Smart, N.; Risebro, C.A.; Melville, A.A.D.; Moses, K.; Schwartz, R.J.; Chien, K.R.; Riley, P.R. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 2007, 445, 177–182. [Google Scholar] [CrossRef]
- Van den Eynde, J.; Manlhiot, C.; Van De Bruaene, A.; Diller, G.P.; Frangi, A.F.; Budts, W.; Kutty, S. Medicine-Based Evidence in Congenital Heart Disease: How Artificial Intelligence Can Guide Treatment Decisions for Individual Patients. Front. Cardiovasc. Med. 2021, 8, 798215. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, Y.; Sun, L.; Sun, X.; Zhao, X.; Sun, X.; Qian, H.; Xu, W.; Zhu, W. Exosomes Derived from Akt-Modified Human Umbilical Cord Mesenchymal Stem Cells Improve Cardiac Regeneration and Promote Angiogenesis via Activating Platelet-Derived Growth Factor D. Stem Cells Transl. Med. 2017, 6, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menasché, P. Cardiac cell therapy: Current status, challenges and perspectives. Arch. Cardiovasc. Dis. 2020, 113, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Balbi, C.; Vassalli, G. Exosomes: Beyond stem cells for cardiac protection and repair. Stem Cells 2020, 38, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Stastna, M.; Abraham, M.R.; Van Eyk, J.E. Cardiac stem/progenitor cells, secreted proteins, and proteomics. FEBS Lett. 2009, 583, 1800–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, L.; Knoepfler, P. Selling Stem Cells in the USA: Assessing the Direct-to-Consumer Industry. Cell Stem Cell 2016, 19, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Aly, R.M. Current state of stem cell-based therapies: An overview. Stem Cell Investig. 2020, 7, 8. [Google Scholar] [CrossRef]
- Zarzeczny, A.; Atkins, H.; Illes, J.; Kimmelman, J.; Master, Z.; Robillard, J.M.; Snyder, J.; Turner, L.; Zettler, P.J.; Caulfield, T. The stem cell market and policy options: A call for clarity. J. Law Biosci. 2018, 5, 743–758. [Google Scholar] [CrossRef] [Green Version]

| Gene | Main Function in Cardiac Development | Cardiac Phenotype |
|---|---|---|
| Genes Encoding for Transcriptional and Epigenetic/Chromatin Remodeling Factors | ||
| ANKRD1 | Regulation of cardiac gene expression, muscle growth, and heart tissue maturation. | TAPVR |
| CBP * | Transcriptional co-activator regulating the expression of genes critical for heart development, histone acetyltransferase and chromatin remodeling factor, essential for cardiac differentiation processes. | ASD, CoA, HSLS, MVD, PFO, PLSVC, vascular ring, VSD |
| CITED2 | Regulation of cardiac gene expression, cardiac chamber formation, establishment of left–right asymmetry, heart and outflow tract septation. | AS, ASD, AVSD, PDA, PS, RAA, TGA, TOF, VSD |
| FOG2/ZFPM2 | Stimulation of cardiac chamber formation and proper cardiomyocyte differentiation. | DORV, TOF |
| FOXH1 | Forkhead activin signal transducer regulating NODAL signaling to specify the left–right axis and promote proper cardiac morphogenesis and septation. | TGA, TOF |
| GATA4 | Regulation of cardiac gene expression, cardiac cell differentiation, and chamber formation. | ASD, AVSD, PAPVR, PS, TOF, VSD |
| GATA5 | Cardiac cell fate determination and differentiation, and regulation of chamber-specific gene expression. | Bicuspid aortic valve, VSD |
| GATA6 | Regulation of cardiac cell differentiation, heart morphogenesis, and septation. | ASD, AVSD, OFT defects, PDA, PS, TOF, VSD |
| NKX2.5 | Regulation of cardiomyocyte differentiation, cardiac chamber formation, and establishment of the electrical conduction system. | ASD, CoA, DORV, HSLH, IAA, OFT defects, TGA, TOF, VSD |
| P300 * | Transcriptional co-activator regulating the expression of genes critical for heart development, histone acetyltransferase and chromatin remodeling factor, essential for cardiac differentiation processes. | AVD, congenital aortic aneurysm, MVD, PDA, PS, TOF, VDS |
| TBX1 | Regulation of cardiac progenitor cells, conduction system formation, and outflow tract morphogenesis. | TOF, 22q11 deletion syndrome |
| TBX5 | Control of cardiac cell fate, chamber formation. | ASD, AVSD, VSD, Holt–Oram syndrome |
| TBX20 | Control of cardiac cell differentiation, chamber formation, and cardiac gene expression patterns. | ASD, MVD, VSD |
| TFAP2B | Cardiac neural crest migration, outflow tract septation, and aortic arch patterning. | PDA, Char syndrome |
| Cell signaling and adhesion proteins | ||
| ACVR1/ALK2 | Receptor for BMPs (TGF-β signaling pathway family) controlling cardiomyocyte differentiation, valve formation, and heart morphogenesis. | AVSD |
| ACVR2B | Receptor for various ligands of the TGF-beta family, such as activins, myostatin, and growth and differentiation factors (GDFs), modulating signaling pathways involved in cardiomyocyte differentiation, cardiac morphogenesis, and chamber formation. | Dextrocardia, DORV, PS, TGA, TOF |
| CFC1 | Co-receptor for NODAL contributing to left–right patterning and cardiomyocyte differentiation. | ASD, AVSD, DORV, IAA, TGA, TOF, VSD |
| GJA1 | Promotion of the electrical coupling between cardiomyocytes, and contribution to proper cardiac conduction and rhythm establishment. | ASD, HLHS, TAPVR |
| JAG1 | NOTCH ligand important to regulate cardiomyocyte differentiation, cardiac chamber formation, and valve morphogenesis. | PAS, TOF, Alagille syndrome |
| LEFTY2 | NODAL inhibitor playing a role in left–right patterning, cardiac morphogenesis, septation, and chamber formation. | AVSD, CoA, IAA, IVC defects, Left–Right axis defects, TGA |
| NODAL | Control of left–right axis determination and promotion of cardiac looping, chamber formation, and valvulogenesis. | AVSD, Dextrocardia, DORV, IVC defects, PA, TAPVR, TGA, TOF |
| NOTCH1 | Regulation of cardiomyocyte differentiation, cardiac valve formation, cardiac cell fate, and cardiac cell maturation. | AS, BAV, CoA, HLHS |
| PDGFRA | Participation in cardiac neural crest migration, cardiomyocyte proliferation, and proper outflow tract formation. | TAPVR |
| VEGF | Promoting angiogenesis and vascularization, ensuring proper blood supply to developing cardiac tissues. | CoA, OFT defects |
| Structural sarcomere proteins | ||
| ACTC1 | Encoding the major protein component of cardiac muscle, contributing to the formation and contraction of cardiac tissue. | ASD |
| MYH6 | Encoding a major contractile protein in cardiac muscle fibers, contributing to proper heart contraction and function. | AS, ASD, HLHS, PFO, TGA |
| MYH7 | Encoding a major contractile protein in cardiac muscle, playing a key role in cardiac contraction and function. | ASD, Ebstein Anomaly, NVM |
| Cell Source | Active miRNA | Effect on Cardiovascular System | Disease/Experimental Model |
|---|---|---|---|
| Human bone marrow MSC | miR-22 | Cardioprotective during ischemia. | LAD ligation, in vitro |
| miR-19a | Cardiomyocyte survival and preservation of mitochondrial membrane potential. | LAD ligation, in vitro | |
| miR-144 | Reduced apoptosis in embryonic rat cardiomyocytes. | Cell line, in vitro | |
| Human ESC-derived MSC | miR-21 | Reduced infarct size in mouse model. | Myocardial ischemia-reperfusion injury |
| Human heart stromal cells derived from healthy individuals | miR-21 | Prevention of the left ventricular ejection fraction decreased over time. | Mouse model of acute myocardial infarction |
| Human CDC | miR-210 | Mitigation of adverse remodeling and improvement of angiogenesis in pig models with myocardium infarct. | In vivo |
| miR-146a | Inhibition of apoptosis and stimulation of the proliferation of cardiomyocytes and angiogenesis in vitro. Improvement of heart function in mouse myocardial infarction model. | In vitro, using human umbilical-derived cells, and in vivo mouse model | |
| Human CPC | miR-132 | Inhibition of cardiomyocyte apoptosis and improvement of cardiac function after myocardial infarction. | In vitro and in vivo, using rat models |
| Human ADSC | miR-126 | Increased angiogenesis of endothelial cells. Exosomes from ADSC of obese subjects present a reduced load of miR-126 and a low pro-angiogenic capacity. | In vitro, using human umbilical-derived endothelial cells |
| miR-31 | Increase the migration and tube formation of human umbilical vein endothelial cells. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bragança, J.; Pinto, R.; Silva, B.; Marques, N.; Leitão, H.S.; Fernandes, M.T. Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects. J. Pers. Med. 2023, 13, 1263. https://doi.org/10.3390/jpm13081263
Bragança J, Pinto R, Silva B, Marques N, Leitão HS, Fernandes MT. Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects. Journal of Personalized Medicine. 2023; 13(8):1263. https://doi.org/10.3390/jpm13081263
Chicago/Turabian StyleBragança, José, Rute Pinto, Bárbara Silva, Nuno Marques, Helena S. Leitão, and Mónica T. Fernandes. 2023. "Charting the Path: Navigating Embryonic Development to Potentially Safeguard against Congenital Heart Defects" Journal of Personalized Medicine 13, no. 8: 1263. https://doi.org/10.3390/jpm13081263