
Neurocutaneous Diseases: Diagnosis, Management, and Treatment

 , ,
, ,
Abstract
:1. Introduction
2. Neurofibromatosis 1 & 2

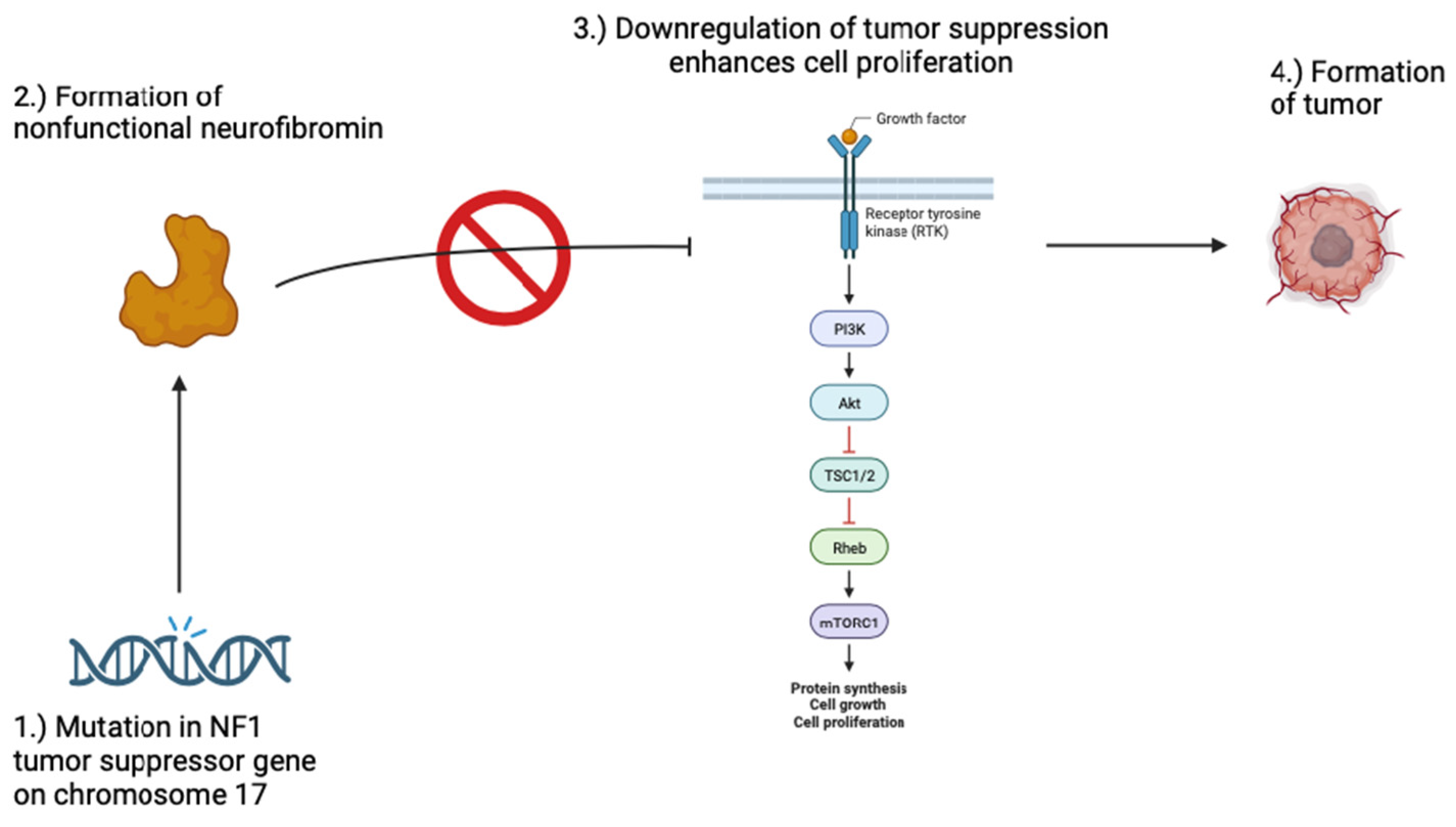{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnostic Criteria of NF1 |
|---|
|
3. Tuberous Sclerosis
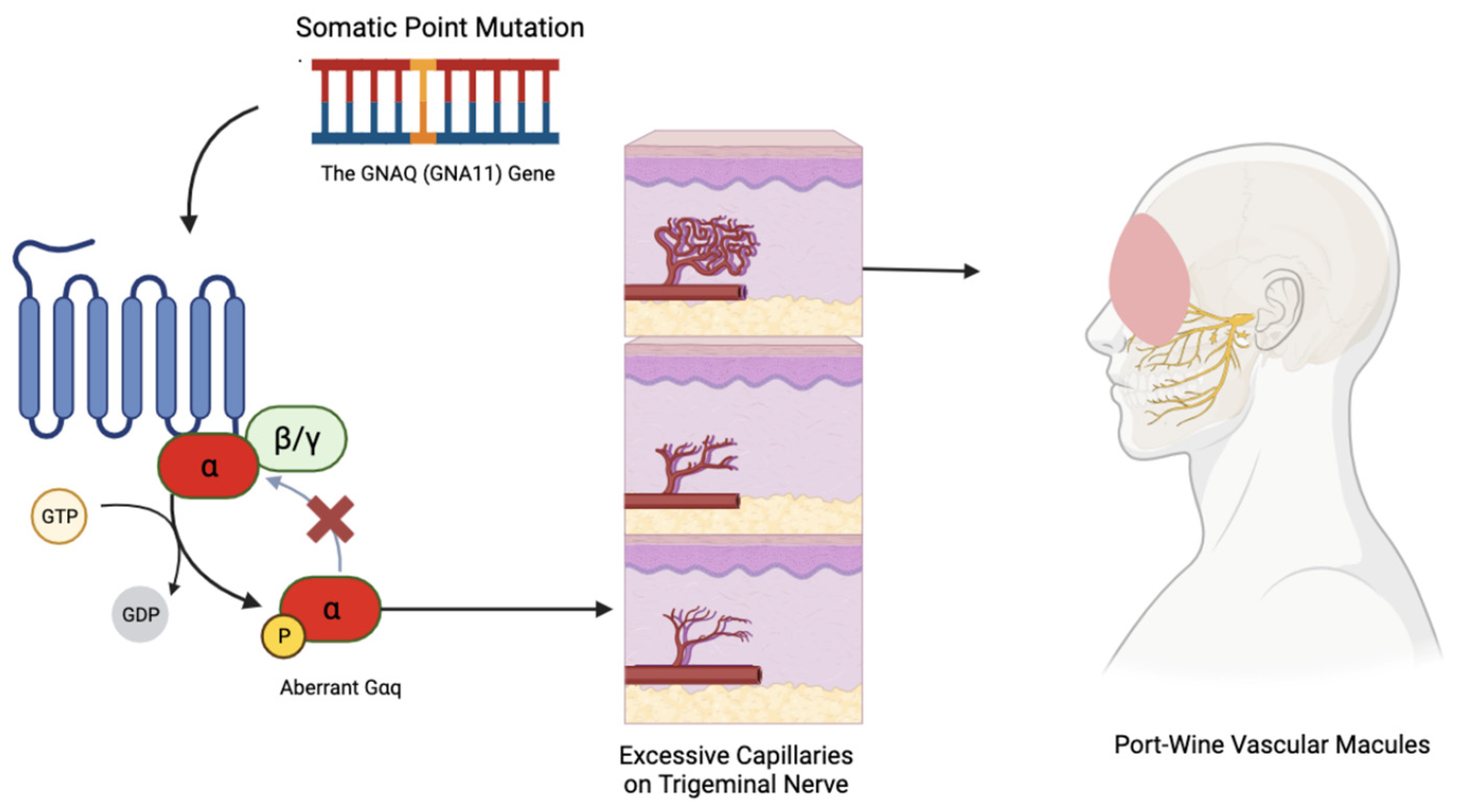
4. Sturge–Weber Syndrome
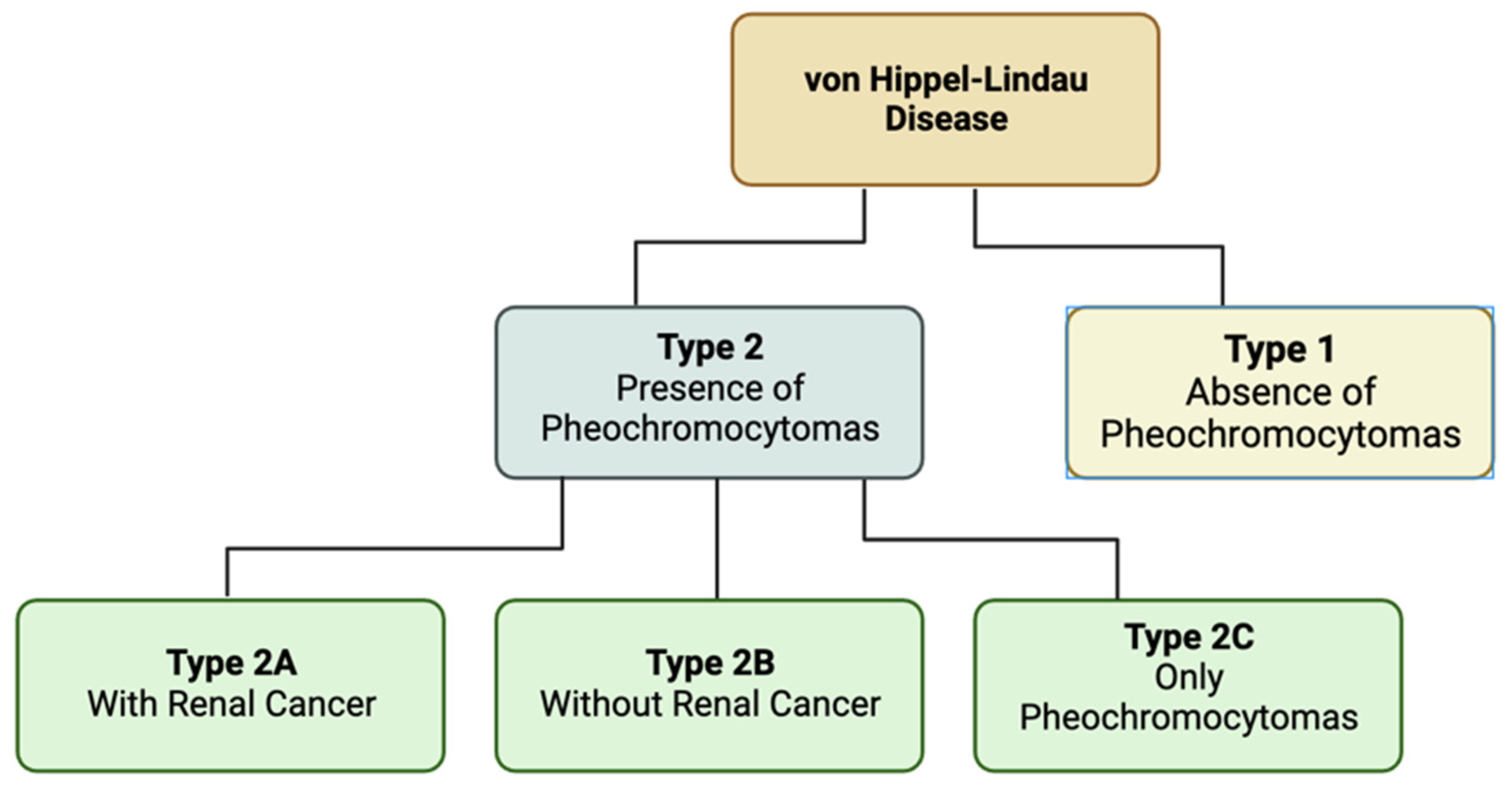
5. Von Hippel-Lindau Disease
6. Ataxia–Telangiectasia
7. Osler-Weber-Rendu Syndrome
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klar, N.; Cohen, B.; Lin, D.D.M. Neurocutaneous syndromes. Handb. Clin. Neurol. 2016, 135, 565–589. [Google Scholar]
- Le, C.; Bedocs, P.M. Neurofibromatosis. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459329/ (accessed on 10 December 2023).
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum. Genom. 2011, 5, 623. [Google Scholar] [CrossRef]
- Wilson, B.N.; John, A.M.; Handler, M.Z.; Schwartz, R.A. Neurofibromatosis type 1: New developments in genetics and treatment. J. Am. Acad. Dermatol. 2020, 84, 1667–1676. [Google Scholar] [CrossRef]
- Giraud, J.-S.; Bièche, I.; Pasmant, É.; Tlemsani, C. NF1 alterations in cancers: Therapeutic implications in precision medicine. Expert. Opin. Investig. Drugs 2023, 32, 941–957. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Legius, E.; Brems, H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism. Child’s Nerv. Syst. 2020, 36, 2285–2295. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Moye, S.L.; McKay, R.M.; Le, L.Q. Neurofibromin and suppression of tumorigenesis: Beyond the GAP. Oncogene 2022, 41, 1235–1251. [Google Scholar] [CrossRef]
- Bikowska-Opalach, B.; Jackowska, T. Neurofibromatosis type 1—Description of clinical features and molecular mechanism of the disease. Med. Wieku Rozwoj. 2013, 17, 334–340. [Google Scholar] [PubMed]
- Nasi, L.; Alexopoulos, A.; Kokkinou, E.; Roka, K.; Tzetis, M.; Tsipi, M.; Kakourou, T.; Kanaka-Gantenbein, C.; Chrousos, G.; Kattamis, A.; et al. Characteristics of Café-au-lait Macules and their Association with the Neurofibromatosis type I Genotype in a Cohort of Greek Children. Acta Derm.-Venereol. 2023, 103, adv5758. [Google Scholar] [CrossRef] [PubMed]
- Drouet, A.; Wolkenstein, P.; Lefaucheur, J.-P.; Pinson, S.; Combemale, P.; Gherardi, R.K.; Brugières, P.; Salama, J.; Ehre, P.; Decq, P.; et al. Neurofibromatosis 1-associated neuropathies: A reappraisal. Brain 2004, 127, 1993–2009. [Google Scholar] [CrossRef]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: A critical review of clinical and genetic features. Clin. Genet. 2014, 87, 401–410. [Google Scholar] [CrossRef]
- Yoshida, Y. Neurofibromatosis 1 (von Recklinghausen Disease). Keio J. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Idogawa, M.; Okura, M.; Sugita, S.; Sugawara, T.; Sasaki, Y.; Uhara, H. Genetic analyses of mosaic neurofibromatosis type 1 with giant café-au-lait macule, plexiform neurofibroma and multiple melanocytic nevi. J. Dermatol. 2020, 47, 658–662. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Bäzner, U.; Krämer, J.; Lewerenz, J.; Pfeiffer, C. The NF1 microdeletion syndrome: Early genetic diagnosis facilitates the management of a clinically defined disease. J. Dtsch. Dermatol. Ges. 2022, 20, 273–277. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Plotkin, S.R. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Magwood, G.S.; Ellis, C.; Buie, J.N.J.; Slan, S.; Bonilha, L.; Adams, R.J. High tech and high touch: Recruitment strategies for enrolling African American stroke survivors in Community Based Intervention under Nurse Guidance after stroke (CINGS) trial. Contemp. Clin. Trials Commun. 2021, 24, 100844. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Santo, V.E.; Passos, J.; Nzwalo, H.; Carvalho, I.; Santos, F.; Martins, C.; Salgado, L.; e Silva, C.; Vinhais, S.; Vilares, M.; et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: A single-institution experience. J. Neuro-Oncol. 2020, 147, 459–463. [Google Scholar] [CrossRef]
- Armstrong, A.E.; Belzberg, A.J.; Crawford, J.R.; Hirbe, A.C.; Wang, Z.J. Treatment decisions and the use of MEK inhibitors for children with neurofibromatosis type 1-related plexiform neurofibromas. BMC Cancer 2023, 23, 553. [Google Scholar] [CrossRef]
- Zheng, T.; Zhu, B.; Wang, Z.; Li, Q. Gene therapy strategies and prospects for neurofibromatosis type 1. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2024, 38, 1–8. [Google Scholar] [PubMed]
- Ardern-Holmes, S.; Fisher, G.; North, K. Neurofibromatosis Type 2. J. Child. Neurol. 2017, 32, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Singh, A.K. Neurofibromatosis Type 2. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470350/ (accessed on 8 March 2024).
- Evans, D.G.; Freeman, S.; Gokhale, C.; Wallace, A.; Lloyd, S.K.; Axon, P.; Ramsden, R.T. Bilateral vestibular schwannomas in older patients: NF2 or chance? J. Med. Genet. 2015, 52, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Halliday, D.; Emmanouil, B.; Pretorius, P.; MacKeith, S.; Painter, S.; Tomkins, H.; Evans, D.G.; Parry, A. Genetic Severity Score predicts clinical phenotype in NF2. J. Med. Genet. 2017, 54, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.E.; Jones, A.P.; Haley, M.J.; Hoyle, C.; Zeef, L.A.H.; Lin, I.-H.; Coope, D.J.; King, A.T.; Evans, D.G.; Paszek, P.; et al. The comparable tumour microenvironment in sporadic and NF2-related schwannomatosis vestibular schwannoma. Brain Commun. 2023, 5, fcad197. [Google Scholar] [CrossRef]
- Lloyd, S.K.W.; Evans, D.G.R. Neurofibromatosis type 2 (NF2): Diagnosis and management. Handb. Clin. Neurol. 2013, 115, 957–967. [Google Scholar]
- Plotkin, S.R.; Messiaen, L.; Legius, E.; Pancza, P.; Avery, R.A.; Blakeley, J.O.; Evans, D.G. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet. Med. 2022, 24, 1967–1977. [Google Scholar] [CrossRef]
- Cumpston, E.C.; Rhodes, S.D.; Yates, C.W. Advances in Targeted Therapy for Neurofibromatosis Type 2 (NF2)-Associated Vestibular Schwannomas. Curr. Oncol. Rep. 2023, 25, 531–537. [Google Scholar] [CrossRef]
- Di Maio, S.; Akagami, R. Prospective comparison of quality of life before and after observation, radiation, or surgery for vestibular schwannomas. J. Neurosurg. 2009, 111, 855–862. [Google Scholar] [CrossRef]
- Lu, V.M.; Ravindran, K.; Graffeo, C.S.; Perry, A.; Van Gompel, J.J.; Daniels, D.J.; Link, M.J. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: A systematic review and meta-analysis of treatment outcomes. J. Neurooncol. 2019, 144, 239–248. [Google Scholar] [CrossRef]
- Ren, Y.; Chari, D.A.; Vasilijic, S.; Welling, D.B.; Stankovic, K.M. New developments in neurofibromatosis type 2 and vestibular schwannoma. Neurooncol. Adv. 2021, 3, vdaa153. [Google Scholar] [CrossRef]
- Hiruta, R.; Saito, K.; Bakhit, M.; Fujii, M. Current progress in genomics and targeted therapies for neurofibromatosis type 2. Fukushima J. Med. Sci. 2023, 69, 95–103. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D.A.; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef]
- Roach, E.S. Applying the Lessons of Tuberous Sclerosis: The 2015 Hower Award Lecture. Pediatr. Neurol. 2016, 63, 6–22. [Google Scholar] [CrossRef]
- Northrup, H.; Koenig, M.K.; Pearson, D.A.; Au, K.S. Tuberous Sclerosis Complex. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1220/ (accessed on 8 March 2024).
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive mutation analysis of TSC1 and TSC2—And phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef]
- Au, K.S.; Williams, A.T.; Roach, E.S.; Batchelor, L.; Sparagana, S.P.; Delgado, M.R.; Wheless, J.W.; Baumgartner, J.E.; Roa, B.B.; Wilson, C.M.; et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Anesth. Analg. 2007, 9, 88–100. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; Ouweland, A.V.D.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Caban, C.; Khan, N.; Hasbani, D.M.; Crino, P.B. Genetics of tuberous sclerosis complex: Implications for clinical practice. Appl. Clin. Genet. 2017, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rosset, C.; Netto, C.B.O.; Ashton-Prolla, P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: A review. Genet. Mol. Biol. 2017, 40, 69–79. [Google Scholar] [CrossRef]
- Feliciano, D.M. The Neurodevelopmental Pathogenesis of Tuberous Sclerosis Complex (TSC). Front. Neuroanat. 2020, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.M.C.; Cowen, E.W.; Wataya-Kaneda, M.; Gosnell, E.S.; Witman, P.M.; Hebert, A.A.; Mlynarczyk, G.; Soltani, K.; Darling, T.N. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014, 150, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Jacks, S.K.; Witman, P.M. Tuberous Sclerosis Complex: An Update for Dermatologists. Pediatr. Dermatol. 2015, 32, 563–570. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Kanish, B.; Bhatia, A. Tuberous sclerosis. Indian Dermatol. Online J. 2015, 6, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Cardis, M.A.; DeKlotz, C.M.C. Cutaneous manifestations of tuberous sclerosis complex and the paediatrician’s role. Arch. Dis. Child. 2017, 102, 858–863. [Google Scholar] [CrossRef]
- Dao, D.-P.D.; Pixley, J.N.; Akkurt, Z.M.; Feldman, S.R. A Review of Topical Sirolimus for the Treatment of Facial Angiofibromas in Tuberous Sclerosis Complex. Ann. Pharmacother. 2023. [Google Scholar] [CrossRef] [PubMed]
- Borzęcki, A.; Chyl-Surdacka, K.; Turska, M. Spectacular Effect of Massive Facial Angiofibromas Removal with a Carbon Dioxide Laser as a Manifestation of a Tuberous Sclerosis Complex. J. Lasers Med. Sci. 2021, 12, e24. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; Pevsner, J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef]
- Happle, R. Phacomatosis pigmentovascularis revisited and reclassified. Arch. Dermatol. 2005, 141, 385–388. [Google Scholar] [CrossRef]
- Fois, A.; Calistri, L.; Balestri, P.; Vivarelli, R.; Bartalini, G.; Mancini, L.; Berardi, A.; Vanni, M. Relationship between café-au-lait spots as the only symptom and peripheral neurofibromatosis (NF1): A follow-up study. Eur. J. Pediatr. 1993, 152, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Espino, L.F.; Ivars, M.; Antoñanzas, J.; Baselga, E. Sturge-Weber Syndrome: A Review of Pathophysiology, Genetics, Clinical Features, and Current Management Approache. Appl. Clin. Genet. 2023, 16, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Poliner, A.; Faith, E.F.; Blieden, L.; Kelly, K.M.; Metry, D. Port-wine Birthmarks: Update on Diagnosis, Risk Assessment for Sturge-Weber Syndrome, and Management. Pediatr. Rev. 2022, 43, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Wetzel-Strong, S.E.; Galeffi, F.; Benavides, C.; Patrucco, M.; Bullock, J.L.; Gallione, C.J.; Lee, H.K.; Marchuk, D.A. Developmental expression of the Sturge–Weber syndrome-associated genetic mutation in Gnaq: A formal test of Happle’s paradominant inheritance hypothesis. Genetics 2023, 224, iyad077. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.; Frelin, L.P.; McCann, M.; Pardo, C.A.; Cohen, B.A.; Comi, A.M.; Pevsner, J. Identification of a Mosaic Activating Mutation in GNA11 in Atypical Sturge-Weber Syndrome. J. Investig. Dermatol. 2020, 141, 685–688. [Google Scholar] [CrossRef]
- Thomas, A.C.; Zeng, Z.; Rivière, J.B.; O’Shaughnessy, R.; Al-Olabi, L.; St-Onge, J.; Kinsler, V.A. Mosaic Activating Mutations in GNA11 and GNAQ Are Associated with Phakomatosis Pigmentovascularis and Extensive Dermal Melanocytosis. J. Investig. Dermatol. 2016, 136, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Waelchli, R.; Aylett, S.; Robinson, K.; Chong, W.; Martinez, A.; Kinsler, V. New vascular classification of port-wine stains: Improving prediction of Sturge–Weber risk. Br. J. Dermatol. 2014, 171, 861–867. [Google Scholar] [CrossRef]
- Dompmartin, A.; van der Vleuten, C.J.M.; Dekeuleneer, V.; Duprez, T.; Revencu, N.; Désir, J.; Loo, D.M.W.M.T.; Flucke, U.; Eijkelenboom, A.; Kool, L.S.; et al. GNA11-mutated Sturge–Weber syndrome has distinct neurological and dermatological features. Eur. J. Neurol. 2022, 29, 3061–3070. [Google Scholar] [CrossRef]
- Sanghvi, J.; Mehta, S.; Mulye, S. Paroxysmal vascular events in Sturge–Weber syndrome: Role of aspirin. J. Pediatr. Neurosci. 2014, 9, 39–41. [Google Scholar] [CrossRef]
- Yeom, S.; Comi, A.M. Updates on Sturge-Weber Syndrome. Stroke 2022, 53, 3769–3779. [Google Scholar] [CrossRef]
- Comi, A.M. Sturge–Weber syndrome and epilepsy: An argument for aggressive seizure management in these patients. Expert. Rev. Neurother. 2007, 7, 951–956. [Google Scholar] [CrossRef]
- Day, A.M.; McCulloch, C.E.; Hammill, A.M.; Juhász, C.; Lo, W.D.; Pinto, A.L.; Miles, D.K.; Fisher, B.J.; Ball, K.L.; Wilfong, A.A.; et al. Physical and Family History Variables Associated with Neurological and Cognitive Development in Sturge-Weber Syndrome. Pediatr. Neurol. 2019, 96, 30–36. [Google Scholar] [CrossRef]
- Koenraads, Y.; van Egmond-Ebbeling, M.B.; de Boer, J.H.; Imhof, S.M.; Braun, K.P.J.; Porro, G.L.; the SWS study group. Visual outcome in Sturge-Weber syndrome: A systematic review and Dutch multicentre cohort. Acta Ophthalmol. 2016, 94, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Sujansky, E.; Conradi, S. Sturge-Weber syndrome: Age of onset of seizures and glaucoma and the prognosis for affected children. J. Child. Neurol. 1995, 10, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Gittins, S.; Steel, D.; Brunklaus, A.; Newsom-Davis, I.; Hawkins, C.; Aylett, S.E. Autism spectrum disorder, social communication difficulties, and developmental comorbidities in Sturge-Weber syndrome. Epilepsy Behav. 2018, 88, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chapieski, L.; Friedman, A.; Lachar, D. Psychological functioning in children and adolescents with Sturge-Weber syndrome. J. Child. Neurol. 2000, 15, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Léauté-Labréze, C.; Boralevi, F.; Pedespan, J.-M.; Meymat, Y.; Taïeb, A. Pulsed dye laser for Sturge-Weber syndrome. Arch. Dis. Child. 2002, 87, 434–435. [Google Scholar] [CrossRef] [PubMed]
- Oakes, W.J. The natural history of patients with the Sturge-Weber syndrome. Pediatr. Neurosurg. 1992, 18, 287–290. [Google Scholar] [CrossRef]
- Parsa, C.F. Sturge-weber syndrome: A unified pathophysiologic mechanism. Curr. Treat. Options Neurol. 2008, 10, 47–54. [Google Scholar] [CrossRef]
- Nakashima, M.; Miyajima, M.; Sugano, H.; Iimura, Y.; Kato, M.; Tsurusaki, Y.; Matsumoto, N. The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef]
- Sabeti, S.; Ball, K.L.; Bhattacharya, S.K.; Bitrian, E.; Blieden, L.S.; Brandt, J.D.; Burkhart, C.; Chugani, H.T.; Falchek, S.J.; Jain, B.G.; et al. Consensus Statement for the Management and Treatment of Sturge-Weber Syndrome: Neurology, Neuroimaging, and Ophthalmology Recommendations. Pediatr. Neurol. 2021, 121, 59–66. [Google Scholar] [CrossRef]
- Smegal, L.F.; Sebold, A.J.; Hammill, A.M.; Juhász, C.; Lo, W.D.; Miles, D.K.; Wilfong, A.A.; Levin, A.V.; Fisher, B.; Ball, K.L.; et al. Multicenter Research Data of Epilepsy Management in Patients with Sturge-Weber Syndrome. Pediatr. Neurol. 2021, 119, 3–10. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, B.; Yang, J.; Bi, M.; Bi, L.; Fan, W. Efficacy of hemoporfin-mediated photodynamic therapy in treating Sturge–Weber syndrome associated port-wine stains: A retrospective study. Indian J. Dermatol. Venereol. Leprol. 2023, 90, 202–209. [Google Scholar] [CrossRef]
- De la Torre, A.J.; Luat, A.F.; Juhász, C.; Ho, M.L.; Argersinger, D.P.; Cavuoto, K.M.; Enriquez-Algeciras, M.; Tikkanen, S.; North, P.; Burkhart, C.N.; et al. A Multidisciplinary Consensus for Clinical Care and Research Needs for Sturge-Weber Syndrome. Pediatr. Neurol. 2018, 84, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, M.I.; Singh, A.K. Von Hippel-Lindau Syndrome. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459242/ (accessed on 10 December 2023).
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Klingler, J.-H.; Gläsker, S.; Bausch, B.; Urbach, H.; Krauss, T.; Jilg, C.A.; Steiert, C.; Puzik, A.; Neumann-Haefelin, E.; Kotsis, F.; et al. Hemangioblastoma and von Hippel-Lindau disease: Genetic background, spectrum of disease, and neurosurgical treatment. Child’s Nerv. Syst. 2020, 36, 2537–2552. [Google Scholar] [CrossRef]
- Reisch, N.; Peczkowska, M.; Januszewicz, A.; Neumann, H.P.H. Pheochromocytoma: Presentation, diagnosis and treatment. J. Hypertens. 2006, 24, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Gläsker, S.; Neumann, H.P.H.; Koch, C.A.; Vortmeyer, A. Von Hippel-Lindau Disease. In Endotext [Internet]; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK279124/ (accessed on 10 December 2023).
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [PubMed]
- Sgambati, M.T.; Stolle, C.; Choyke, P.L.; Walther, M.M.; Zbar, B.; Linehan, W.M.; Glenn, G. Mosaicism in von Hippel–Lindau disease: Lessons from kindreds with germline mutations identified in offspring with mosaic parents. Am. J. Hum. Genet. 2000, 66, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Chappell, J.C.; Payne, L.B.; Rathmell, W.K. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J. Clin. Investig. 2019, 129, 442–451. [Google Scholar] [CrossRef]
- Ferzli, P.G.; Millett, C.R.; Newman, M.D.; Heymann, W.R. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis 2008, 81, 41–48. [Google Scholar]
- Singh, A.D.; Shields, C.L.; Shields, J.A. von Hippel-Lindau disease. Surv. Ophthalmol. 2001, 46, 117–142. [Google Scholar] [CrossRef]
- Ahmad, W.A.W.; Khanom, M.; Yaakob, Z.H. Heart failure in pregnancy: An overview. Int. J. Clin. Pract. 2011, 65, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.E.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.K.; Mendez, M.D. Cafe Au Lait Macules. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK557492/ (accessed on 10 December 2023).
- Van Leeuwaarde, R.S.; Ahmad, S.; van Nesselrooij, B.; Zandee, W.; Giles, R.H. Von Hippel-Lindau Syndrome. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1463/ (accessed on 10 December 2023).
- Wolters, W.P.G.; Dreijerink, K.M.A.; Giles, R.H.; van der Horst-Schrivers, A.N.A.; van Nesselrooij, B.; Zandee, W.T.; Timmers, H.J.L.M.; Seute, T.; de Herder, W.W.; Stuart, A.A.V.; et al. Multidisciplinary integrated care pathway for von Hippel–Lindau disease. Cancer 2022, 128, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kakutani, S.; Sato, Y.; Hanashi, A.; Kinoshita, Y.; Ishikawa, A. Drug review: Pazopanib. Jpn. J. Clin. Oncol. 2018, 48, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel–Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Chan, V.W.S.; Lenton, J.; Smith, J.; Jagdev, S.; Ralph, C.; Vasudev, N.; Bhattarai, S.; Lewington, A.; Kimuli, M.; Cartledge, J.; et al. Multimodal image-guided ablation on management of renal cancer in Von-Hippel-Lindau syndrome patients from 2004 to 2021 at a specialist centre: A longitudinal observational study. Eur. J. Surg. Oncol. 2022, 48, 672–679. [Google Scholar] [CrossRef]
- Gläsker, S.; Krüger, M.T.; Klingler, J.-H.; Wlodarski, M.; Klompen, J.; Schatlo, B.; Hippchen, B.; Neumann, H.P.; van Velthoven, V. Hemangioblastomas and neurogenic polyglobulia. Neurosurgery 2013, 72, 930–935, discussion 935. [Google Scholar] [CrossRef]
- Krüger, M.T.; Steiert, C.; Gläsker, S.; Klingler, J.-H. Minimally invasive resection of spinal hemangioblastoma: Feasibility and clinical results in a series of 18 patients. J. Neurosurg. Spine 2019, 31, 880–889. [Google Scholar] [CrossRef]
- Greenberger, S.; Berkun, Y.; Ben-Zeev, B.; Levi, Y.B.; Barziliai, A.; Nissenkorn, A. Dermatologic manifestations of ataxia-telangiectasia syndrome. J. Am. Acad. Dermatol. 2013, 68, 932–936. [Google Scholar] [CrossRef]
- Tang, S.Y.; Shaikh, A.G. Past and Present of Eye Movement Abnormalities in Ataxia-Telangiectasia. Cerebellum 2019, 18, 556–564. [Google Scholar] [CrossRef]
- Van Os, N.J.H.; van Deuren, M.; Weemaes, C.M.R.; van Gaalen, J.; Hijdra, H.; Taylor, A.M.R.; van de Warrenburg, B.P.C.; Willemsen, M.A.A.P. Classic ataxia-telangiectasia: The phenotype of long-term survivors. J. Neurol. 2020, 267, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Sirajwala, A.A.; Khan, S.; Rathod, V.M.; Gevariya, V.C.; Jansari, J.R.; Patel, Y.M.; Sirajwala, A.A. Ataxia-Telangiectasia: A Case Report and a Brief Review. Cureus 2023, 15, e39346. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, G.M.; Samanta, D.; Frucht, S. Ataxia Telangiectasia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK519542/ (accessed on 10 December 2023).
- Jiang, Y.; Chen, H.-C.; Su, X.; Thompson, P.A.; Liu, X.; Do, K.-A.; Wierda, W.; Keating, M.J.; Plunkett, W. ATM function and its relationship with ATM gene mutations in chronic lymphocytic leukemia with the recurrent deletion (11q22.3–23.2). Blood Cancer J. 2016, 6, e465. [Google Scholar] [CrossRef]
- Amirifar, P.; Ranjouri, M.R.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-telangiectasia: A review of clinical features and molecular pathology. Pediatr. Allergy Immunol. 2019, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, A.; Kaya, Y.; Tanriverdi, O. Effect of the Interaction Between Selenium and Zinc on DNA Repair in Association with Cancer Prevention. J. Cancer Prev. 2019, 24, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef]
- Cavone, F.; Cappelli, S.; Bonuccelli, A.; D’Elios, S.; Costagliola, G.; Peroni, D.; Orsini, A.; Consolini, R. Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis. J. Clin. Med. 2023, 12, 6041. [Google Scholar] [CrossRef]
- Kumar, N.; Aggarwal, P.; Dev, N.; Kumar, G. Ataxia telangiectasia: Learning from previous mistakes. BMJ Case Rep. 2012, 2012, bcr2012007246. [Google Scholar] [CrossRef] [PubMed]
- Asadollahi, R.; Britschgi, C.; Joset, P.; Oneda, B.; Schindler, D.; Meier, U.R.; Rauch, A. Severe reaction to radiotherapy provoked by hypomorphic germline mutations in ATM (ataxia–telangiectasia mutated gene). Mol. Genet. Genom. Med. 2020, 8, e1409. [Google Scholar] [CrossRef] [PubMed]
- Shad, T.M.; Yazdani, R.; Amirifar, P.; Delavari, S.; Arani, M.H.; Mahdaviani, S.A.; Sadeghi-Shabestari, M.; Aghamohammadi, A.; Rezaei, N.; Abolhassani, H. Atypical Ataxia Presentation in Variant Ataxia Telangiectasia: Iranian Case-Series and Review of the Literature. Front. Immunol. 2021, 12, 779502. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, S.; van Os, N.; Weemaes, C.; Kamsteeg, E.J.; Willemsen, M. Ataxia-Telangiectasia. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK26468/ (accessed on 10 December 2023).
- Perlman, S.; Becker-Catania, S.; Gatti, R.A. Ataxia-telangiectasia: Diagnosis and treatment. Semin. Pediatr. Neurol. 2003, 10, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.J.; Lange, K.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Paucar, M.; Taylor, A.M.; Hadjivassiliou, M.; Fogel, B.L.; Svenningsson, P. Progressive Ataxia with Elevated Alpha-Fetoprotein: Diagnostic Issues and Review of the Literature. Tremor Other Hyperkinetic Mov. 2019, 9. [Google Scholar] [CrossRef]
- Chopra, C.; Davies, G.; Taylor, M.; Anderson, M.; Bainbridge, S.; Tighe, P.; McDermott, E.M. Immune deficiency in Ataxia-Telangiectasia: A longitudinal study of 44 patients. Clin. Exp. Immunol. 2014, 176, 275–282. [Google Scholar] [CrossRef]
- Long, S.S. Parents can accurately observe the severity of respiratory illness in their infants born term and preterm. J. Pediatr. 2019, 214, 1–3. [Google Scholar] [CrossRef]
- Giardino, G.; Fusco, A.; Romano, R.; Gallo, V.; Maio, F.; Esposito, T.; Palamaro, L.; Parenti, G.; Salerno, M.C.; Vajro, P.; et al. Betamethasone therapy in ataxia telangiectasia: Unraveling the rationale of this serendipitous observation on the basis of the pathogenesis. Eur. J. Neurol. 2013, 20, 740–747. [Google Scholar] [CrossRef]
- Hirch, T.; Brander, N.; Schenk, F.; Pöllmann, S.J.; Reichenbach, J.; Schubert, R.; Modlich, U. Expression of a large coding sequence: Gene therapy vectors for Ataxia Telangiectasia. Sci. Rep. 2023, 13, 19386. [Google Scholar] [CrossRef]
- McGrath-Morrow, S.A.; Rothblum-Oviatt, C.C.; Wright, J.; Schlechter, H.; Lefton-Greif, M.A.; Natale, V.A.; Crawford, T.O.; Lederman, H.M. Multidisciplinary Management of Ataxia Telangiectasia: Current Perspectives. J. Multidiscip. Healthc. 2021, 14, 1637–1643. [Google Scholar] [CrossRef]
- Unes, S.; Tuncdemir, M.; Eroglu-Ertugrul, N.G.; Gunel, M.K. Effectiveness of Physical Therapy on Ataxia-Telangiectasia: A Case Report. Pediatr. Phys. Ther. 2021, 33, E103–E107. [Google Scholar] [CrossRef] [PubMed]
- Zannolli, R.; Buoni, S.; Betti, G.; Salvucci, S.; Plebani, A.; Soresina, A.; Pietrogrande, M.C.; Martino, S.; Leuzzi, V.; Finocchi, A.; et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Mov. Disord. 2012, 27, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Kwei, K.T.; Kuo, S.-H. An Overview of the Current State and the Future of Ataxia Treatments. Neurol. Clin. 2020, 38, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Sabino Pinho de Oliveira, B.; Putti, S.; Naro, F.; Pellegrini, M. Bone Marrow Transplantation as Therapy for Ataxia-Telangiectasia: A Systematic Review. Cancers 2020, 12, 3207. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, S.; de Gusmao, C.M.; Zhao, B.; Chin, D.H.; DiDonato, R.L.; Nguyen, M.A.; Nakayama, T.; Hu, C.A.; Soucy, A.; et al. A framework for individualized splice-switching oligonucleotide therapy. Nature 2023, 619, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Macri, A.; Wilson, A.M.; Shafaat, O.; Sharma, S. Osler-Weber-Rendu Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK482361/ (accessed on 10 December 2023).
- Hammill, A.M.; Wusik, K.; Kasthuri, R.S. Hereditary hemorrhagic telangiectasia (HHT): A practical guide to management. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.A.; Turcios, N.L. Pulmonary Manifestations of Skin Disorders in Children. Pediatr. Clin. N. Am. 2020, 68, 261–276. [Google Scholar] [CrossRef]
- Hyldahl, S.J.; El-Jaji, M.Q.; Schuster, A.; Kjeldsen, A.D. Skin and mucosal telangiectatic lesions in hereditary hemorrhagic telangiectasia patients. Int. J. Dermatol. 2022, 61, 1497–1505. [Google Scholar] [CrossRef]
- Mylavarapu, C.; Lu, A.J.; Burns, E.A.; Samorajski, J.; Gotur, D.; Baker, K. Diffuse Cerebral Edema and Impending Herniation Complicating Hepatic Encephalopathy in Hereditary Hemorrhagic Telangiectasia. Case Rep. Med. 2022, 2022, 2612544. [Google Scholar] [CrossRef] [PubMed]
- Alkhalid, Y.; Darji, Z.; Shenkar, R.; Clancy, M.; Dyamenahalli, U.; Awad, I.A.; the multidisciplinary faculty of the HHT Center of Excellence at University of Chicago Medicine. Multidisciplinary coordinated care of hereditary hemorrhagic telangiectasia (Osler–Weber–Rendu disease). Vasc. Med. 2023, 28, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H. Hereditary hemorrhagic telangiectasia: Systemic therapies, guidelines, and an evolving standard of care. Blood 2021, 137, 888–895. [Google Scholar] [CrossRef] [PubMed]






| Tuberous Sclerosis Diagnostic Criteria | |
|---|---|
| Major | Minor |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kioutchoukova, I.; Foster, D.; Thakkar, R.; Ciesla, C.; Cabassa, J.S.; Strouse, J.; Kurz, H.; Lucke-Wold, B. Neurocutaneous Diseases: Diagnosis, Management, and Treatment. J. Clin. Med. 2024, 13, 1648. https://doi.org/10.3390/jcm13061648
Kioutchoukova I, Foster D, Thakkar R, Ciesla C, Cabassa JS, Strouse J, Kurz H, Lucke-Wold B. Neurocutaneous Diseases: Diagnosis, Management, and Treatment. Journal of Clinical Medicine. 2024; 13(6):1648. https://doi.org/10.3390/jcm13061648
Chicago/Turabian StyleKioutchoukova, Ivelina, Devon Foster, Rajvi Thakkar, Christopher Ciesla, Jake Salvatore Cabassa, Jacob Strouse, Hayley Kurz, and Brandon Lucke-Wold. 2024. "Neurocutaneous Diseases: Diagnosis, Management, and Treatment" Journal of Clinical Medicine 13, no. 6: 1648. https://doi.org/10.3390/jcm13061648