

Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis
 , , and
, , and
Abstract
:1. Introduction
2. Case Report
3. Discussion
The Differential Diagnosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Bredemeyer, A.L.; Sharma, G.G.; Huang, C.-Y.; Helmink, B.A.; Walker, L.M.; Khor, K.C.; Nuskey, B.; Sullivan, K.E.; Pandita, T.K.; Bassing, C.H.; et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006, 442, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Alyasin, S.; Esmaeilzadeh, H.; Ebrahimi, N.; Nabavizadeh, S.H.; Nemati, H. Clinical Presentation of Ataxia-Telangiectasia. Arch. Iran. Med. 2019, 22, 682–686. [Google Scholar] [PubMed]
- van Os, N.J.; Chessa, L.; Weemaes, C.M.; van Deuren, M.; Fiévet, A.; van Gaalen, J.; Mahlaoui, N.; Roeleveld, N.; Schrader, C.; Schindler, D.; et al. Genotype-phenotype correlations in ataxia telangiectasia patients with ATM c.3576G>A and c.8147T>C mutations. J. Med. Genet. 2019, 56, 308–316. [Google Scholar] [CrossRef]
- Sahama, I.; Sinclair, K.; Pannek, K.; Lavin, M.; Rose, S. Radiological imaging in ataxia telangiectasia: A review. Cerebellum 2014, 13, 521–530. [Google Scholar] [CrossRef]
- Cabana, M.D.; Crawford, T.O.; Winkelstein, J.A.; Christensen, J.R.; Lederman, H.M. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics 1998, 102, 98–100. [Google Scholar] [CrossRef]
- Bredemeyer, A.L.; Helmink, B.A.; Innes, C.L.; Calderon, B.; McGinnis, L.M.; Mahowald, G.K.; Gapud, E.J.; Walker, L.M.; Collins, J.B.; Weaver, B.K.; et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 2008, 456, 819–823. [Google Scholar] [CrossRef]
- Nowak-Wegrzyn, A.; Crawford, T.O.; Winkelstein, J.A.; Carson, K.A.; Lederman, H.M. Immunodeficiency and infections in ataxia-telangiectasia. J. Pediatr. 2004, 144, 505–511. [Google Scholar] [CrossRef]
- Boyarchuk, O.; Makukh, H.; Kostyuchenko, L.; Yarema, N.; Haiboniuk, I.; Kravets, V.; Shulhai, O.; Tretyak, B. TREC/KREC levels in children with ataxia-telangiectasia. Immunol. Res. 2021, 69, 436–444. [Google Scholar] [CrossRef]
- McGrath-Morrow, S.A.; Collaco, J.M.; Detrick, B.; Lederman, H.M. Serum Interleukin-6 Levels and Pulmonary Function in Ataxia-Telangiectasia. J. Pediatr. 2016, 171, 256–261.e1. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Massey, R.B.; Curtis, M.A.; Pelachyk, J.M.; Dombrowski, H.C.; Leickly, F.E.; Swift, M. Cutaneous granulomatous lesions in patients with ataxia-telangiectasia. J. Pediatr. 1991, 119, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Reiman, A.; Srinivasan, V.; Barone, G.; Last, J.I.; Wootton, L.L.; Davies, E.G.; Verhagen, M.M.; Willemsen, M.A.; Weemaes, C.M.; Byrd, P.J.; et al. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. Br. J. Cancer 2011, 105, 586–591. [Google Scholar] [CrossRef] [PubMed]
- van Os, N.J.; Haaxma, C.A.; van der Flier, M.; Merkus, P.J.; van Deuren, M.; de Groot, I.J.; Loeffen, J.; van de Warrenburg, B.P.; Willemsen, M.A.; A-T Study Group. Ataxia-telangiectasia: Recommendations for multidisciplinary treatment. Dev. Med. Child. Neurol. 2017, 59, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Gueven, N.; Bottle, S.; Gatti, R.A. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br. Med. Bull. 2007, 81–82, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Biagiotti, S.; Bianchi, M.; Rossi, L.; Chessa, L.; Magnani, M. Activation of NRF2 by dexamethasone in ataxia telangiectasia cells involves KEAP1 inhibition but not the inhibition of p38. PLoS ONE 2019, 14, e0216668. [Google Scholar] [CrossRef]
- Buoni, S.; Zannolli, R.; Sorrentino, L.; Fois, A. Betamethasone and improvement symptoms in ataxia-telangiectasia. Arch. Neurol. 2006, 63, 1479–1482. [Google Scholar] [CrossRef]
- Martire, B.; Azzari, C.; Badolato, R.; Canessa, C.; Cirillo, E.; Gallo, V.; Graziani, S.; Lorenzini, T.; Milito, C.; Panza, R.; et al. Vaccination in immunocompromised host: Recommendations of Italian Primary Immunodeficiency Network Centers (IPINET). Vaccine 2018, 36, 3541–3554. [Google Scholar] [CrossRef]
- Herskind, A.; Greisen, G.; Nielsen, J.B. Early identification and intervention in cerebral palsy. Dev. Med. Child. Neurol. 2014, 57, 29–36. [Google Scholar] [CrossRef]
- Salman, M.S.; Ikeda, K.M. The syndrome of infantile-onset saccade initiation delay. Can. J. Neurol. Sci./J. Can. des Sci. Neurol. 2013, 40, 235–240. [Google Scholar] [CrossRef]
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef]
- ESID Registry—Working Definitions for Clinical Diagnosis of PID 2019. Available online: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (accessed on 22 January 2019).
- Dehkordy, S.F.; Aghamohammadi, A.; Ochs, H.D.; Rezaei, N. Primary immunodeficiency diseases associated with neurologic manifestations. J. Clin. Immunol. 2011, 32, 1–24. [Google Scholar] [CrossRef]
- Taylor, A.M.; Groom, A.; Byrd, P.J. Ataxia-telangiectasia-like disorder (ATLD)—Its clinical presentation and molecular basis. DNA Repair. 2004, 3, 1219–1225. [Google Scholar] [CrossRef]
- Sauer, A.V.; Aiuti, A. New insights into the pathogenesis of adenosine deaminase-severe combined immunodeficiency and progress in gene therapy. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 496–502. [Google Scholar] [CrossRef]
- Nofech-Mozes, Y.; Blaser, S.I.; Kobayashi, J.; Grunebaum, E.; Roifman, C.M. Neurologic abnormalities in patients with adenosine deaminase deficiency. Pediatr. Neurol. 2007, 37, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Grunebaum, E.; Arpaia, E.; Roifman, C.M. Immunodeficiency caused by purine nucleotide phosphorylase deficiency. Immunol. Allergy Clin. N. Am. 2000, 20, 143–159. [Google Scholar] [CrossRef]
- Dalal, I.; Grunebaum, E.; Cohen, A.; Roifman, C. Two novel mutations in a purine nucleoside phosphorylase (PNP)-deficient patient. Clin. Genet. 2001, 59, 430–437. [Google Scholar] [CrossRef]
- Tabarki, B.; Yacoub, M.; Tlili, K.; Trabelsi, A.; Dogui, M.; Essoussi, A.S. Familial spastic paraplegia as the presenting manifestation in patients with purine nucleoside phosphorylase deficiency. J. Child. Neurol. 2003, 18, 140–141. [Google Scholar] [CrossRef] [PubMed]
- Digweed, M.; Sperling, K. Nijmegen breakage syndrome: Clinical manifestation of defective response to DNA double-strand breaks. DNA Repair. 2004, 3, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, A.P. Ligase IV syndrome. Eur. J. Med. Genet. 2009, 52, 373–378. [Google Scholar] [CrossRef]
- Enders, A.; Fisch, P.; Schwarz, K.; Duffner, U.; Pannicke, U.; Nikolopoulos, E.; Peters, A.; Orlowska-Volk, M.; Schindler, D.; Friedrich, W.; et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J. Immunol. 2006, 176, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondanèche, M.-C.; Sanal, O.; Plebani, A.; Stéphan, J.-L.; Hufnagel, M.; le Deist, F.; et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 2006, 124, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.R.; Wei, Q.; Jiang, M.; Zhang, X.B.; Zhang, Z.X.; Nong, G.M. Hoyeraal-Hreidarsson syndrome with combined immunodeficiency and enterocolitis caused by a DCK1 gene variant. Chin. J. Pediatr. 2022, 60, 248–249. [Google Scholar]
- Hagleitner, M.M.; Lankester, A.; Maraschio, P.; Hulten, M.; Fryns, J.P.; Schuetz, C.; Gimelli, G.; Davies, E.G.; Gennery, A.; Belohradsky, B.H.; et al. Clinical spectrum of immunodeficiency, centromeric instability and facial dysmorphism (ICF syndrome). J. Med. Genet. 2007, 45, 93–99. [Google Scholar] [CrossRef]
- Ehrlich, M. The ICF syndrome, a DNA methyltransferase 3B deficiency and immunodeficiency disease. Clin. Immunol. 2003, 109, 17–28. [Google Scholar] [CrossRef]
- Nguyen, J.T.U.; Green, A.; Wilson, M.R.; DeRisi, J.L.; Gundling, K. Neurologic Complications of Common Variable Immunodeficiency. J. Clin. Immunol. 2016, 36, 793–800. [Google Scholar] [CrossRef]
- Sassi, A.; Lazaroski, S.; Wu, G.; Haslam, S.M.; Fliegauf, M.; Mellouli, F.; Patiroglu, T.; Unal, E.; Ozdemir, M.A.; Jouhadi, Z.; et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J. Allergy Clin. Immunol. 2014, 133, 1410–1419. [Google Scholar] [CrossRef]
- Dasovic, B.; Borys, E.; Schneck, M.J. Granulomatous Diseases of the Central Nervous System. Curr. Neurol. Neurosci. Rep. 2022, 22, 33–45. [Google Scholar] [CrossRef]



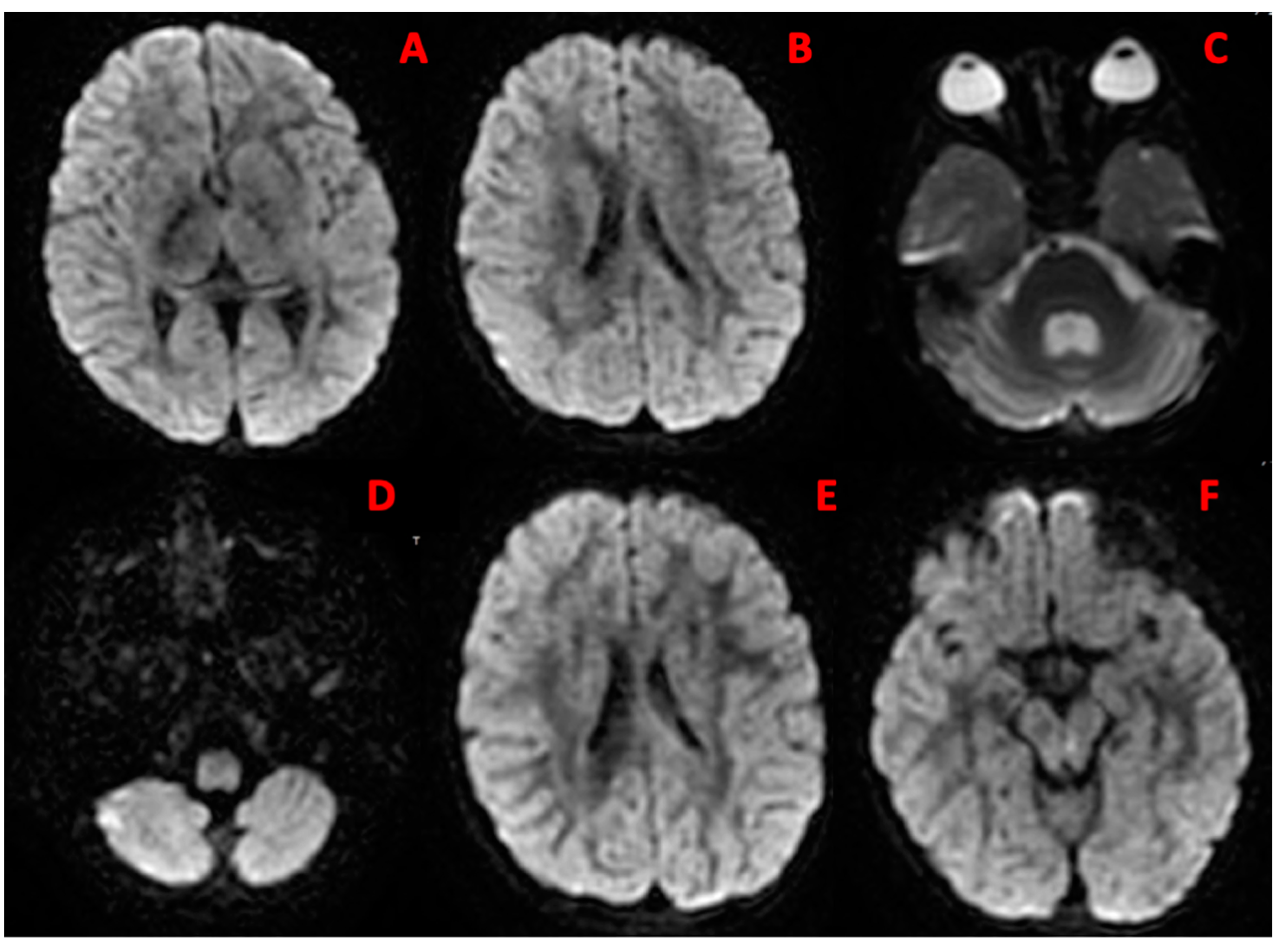{kind=link}
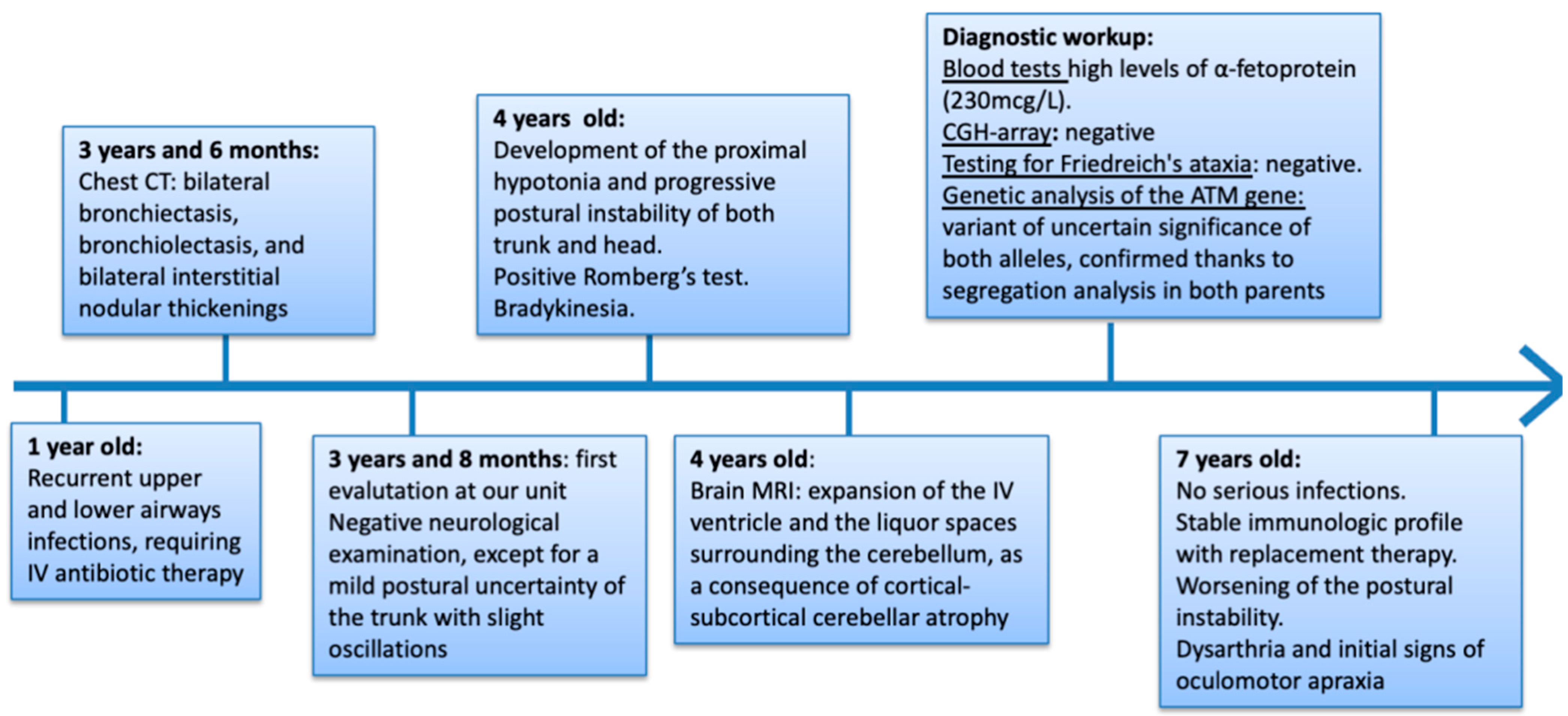{kind=link}
| Similarities in Clinical and Laboratory Features with AT | Differentiating Features Not Commonly Observed in AT | |
|---|---|---|
| Ataxia-Telangiectasia-like Disorders (ATDL) | -Ataxia -Involuntary movements (tremor, chorea, dystonia) -Central/peripheral neuropathy | -Late onset -Less severe phenotype, slow progression -Telangiectasias, immunodeficiency, AFP increase rarely present |
| Adenosine Deaminase (ADA) Deficiency | -Ataxia -Motor dysfunction -Hypotonia -Combined immunodeficiency, recurrent severe infections | -Neurocognitive delay, epilepsy -Sensorineural deafness -Skeletal abnormalities -Normal AFP level -Brain MRI: leukoencephalopathy, dilatation of ventricles and subarachnoid spaces |
| Purine Nucleoside Phosphorylase (PNP) Deficiency | -Ataxia -Involuntary movements (tremor, chorea, dystonia) -Hyper/hypotonia -T cell lymphopenia, recurrent, unusual infections -Autoimmune disorders (one third of patients) | -Delay in neurocognitive development -Sensorineural deafness -Normal level of AFP -Brain MRI: absence of cerebellar atrophy |
| Nijmegen Breakage Syndrome (NBS) | -Severe combined immunodeficiency, severe infections -Radiosensitivity -Increased susceptibility to lymphoid malignancies | -Intrauterine growth retardation -Microcephaly -Facial dysmorphism (mid-facial prominence accentuated by the obliquity of the forehead and the receding jaw) -Neurocognitive delay, behavioral disorders -Skeletal abnormalities (clinodactyly of the fifth finger and partial syndactyly of the second and third toes) |
| DNA Ligase IV Deficiency (LIG4) | -Ataxia -Growth delay | -Microcephaly, facial dysmorphisms, bone deformations -Cognitive delay, learning difficulties -Skin abnormalities (psoriasis, eczema, erythroderma) |
| Cerunnos/XLF Deficiency | -Recurrent infections -Lymphopenia, hypogammaglobulinemia | -Microcephaly -Dysmorphic features, including “bird-like” facial dysmorphism |
| Riddle Syndrome | -Mild motor control -Ataxia -Conjunctival teleangectasias -Radiosensitivity and cancer susceptibility -Combined immunodeficiency -Recurrent infections -Increase in AFP | -Psychomotor retardation -Atopy, serum IgE elevation -Sensorineural hearing loss |
| Hoyeraal–Hreidarsson Syndrome (HHS) | -Ataxia -Hypotonia -Progressive combined immunodeficiency | -Prenatal growth retardation -Microcephaly -Neurocognitive delay -Epilepsy -Pancytopenia -Hyperpigmentation, nail dystrophy -Premalignant leukoplakia oral and gastrointestinal |
| Immunodeficiency, Centromeric Region Instability and Facial Anomalies Syndrome (ICF) | -Ataxia -Hypotonia -Hypo/agammaglobulinemia, decrease in T cell count (half of cases) | -Psychomotor delay -Facial abnormalities: hypertelorism and epicant folds, micrognathia, low ear implantation, and macroglossia -Macrocephaly |
| Common Variable Immunodeficiency Disease (CVID) | -Humoral immunodeficiency, recurrent infections -Ataxia, dysarthria, tremor -Paraesthesia -Myoclonic dystonia | -Entheropathy -Vitamin E deficiency -Guillain Barré Syndrome |
| Phosphoglucomutase 3 (PGM3) Deficiency | -Ataxia, hypotonia, dysarthria -Thymic dysfunction, recurrent infections -Cancer susceptibility | -Retinitis pigmentosa |
| Chronic Granulomatous Disease (CGD) | -CNS granulomatous disease -Cutaneous granulomas, chronic inflammation -Recurrent severe infections | -Defective bactericidal function -Hypergammaglobulinemia -Normal level of AFP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavone, F.; Cappelli, S.; Bonuccelli, A.; D’Elios, S.; Costagliola, G.; Peroni, D.; Orsini, A.; Consolini, R. Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis. J. Clin. Med. 2023, 12, 6041. https://doi.org/10.3390/jcm12186041
Cavone F, Cappelli S, Bonuccelli A, D’Elios S, Costagliola G, Peroni D, Orsini A, Consolini R. Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis. Journal of Clinical Medicine. 2023; 12(18):6041. https://doi.org/10.3390/jcm12186041
Chicago/Turabian StyleCavone, Federica, Susanna Cappelli, Alice Bonuccelli, Sofia D’Elios, Giorgio Costagliola, Diego Peroni, Alessandro Orsini, and Rita Consolini. 2023. "Ataxia Telangiectasia Arising as Immunodeficiency: The Intriguing Differential Diagnosis" Journal of Clinical Medicine 12, no. 18: 6041. https://doi.org/10.3390/jcm12186041