
The Fundamental Role of Oxime and Oxime Ether Moieties in Improving the Physicochemical and Anticancer Properties of Structurally Diverse Scaffolds
 ,
,
Abstract
:1. Introduction
2. Classes of Anticancer Oximes
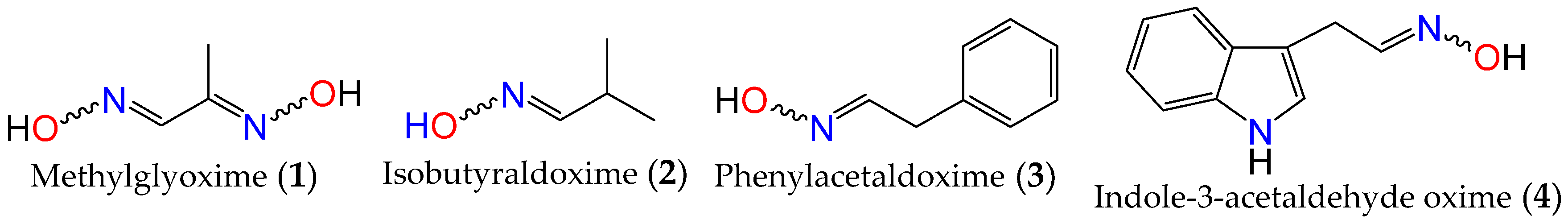
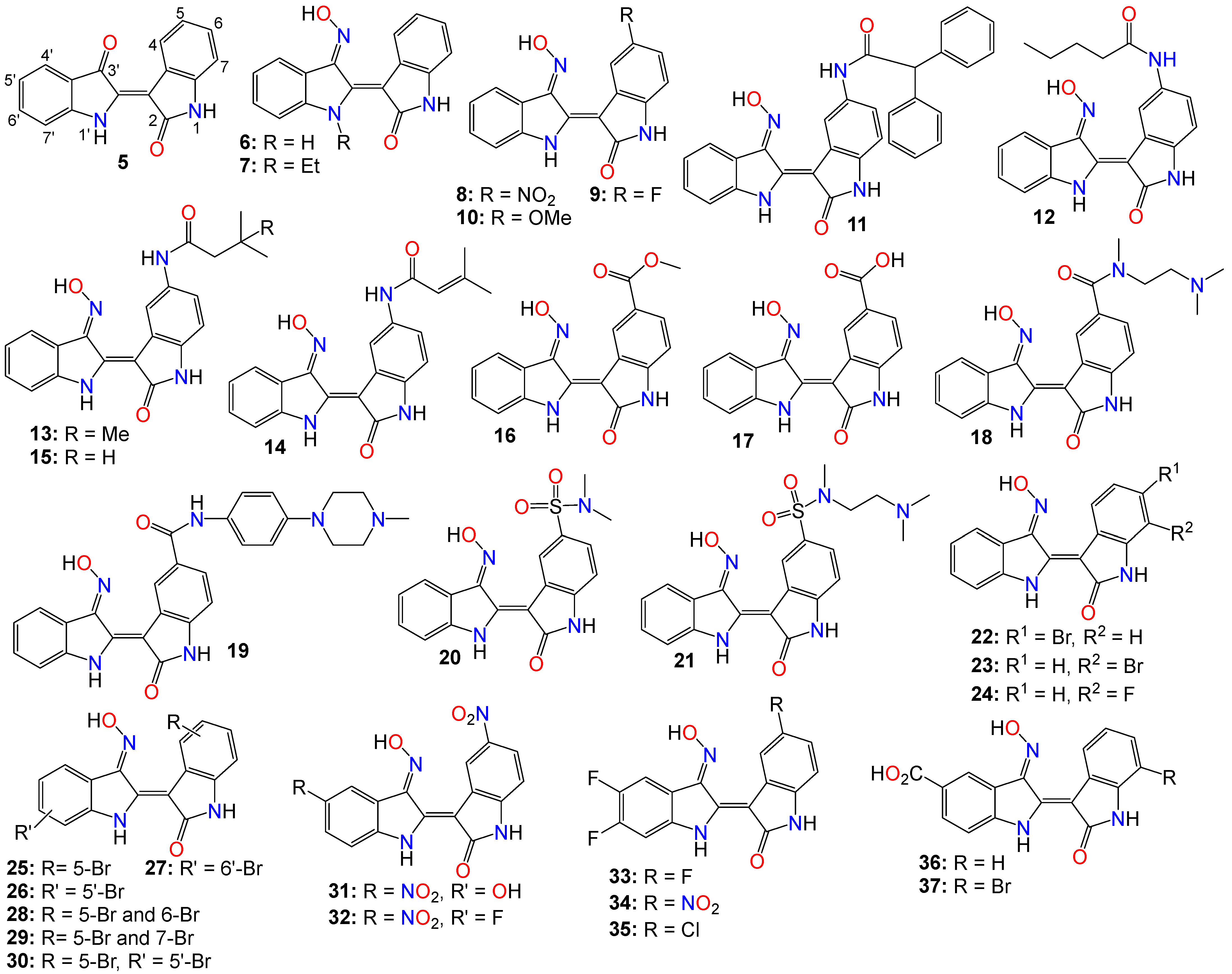
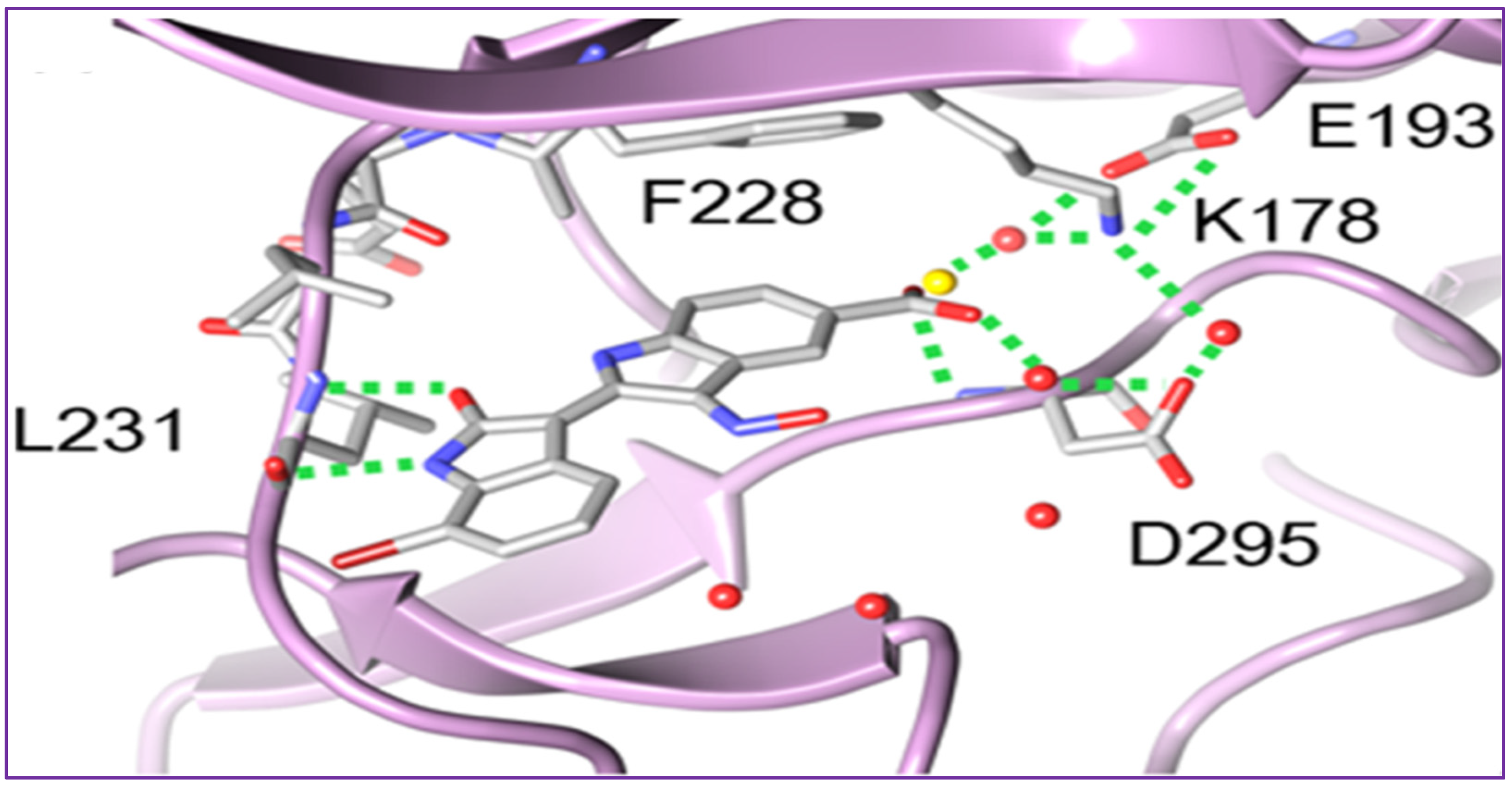
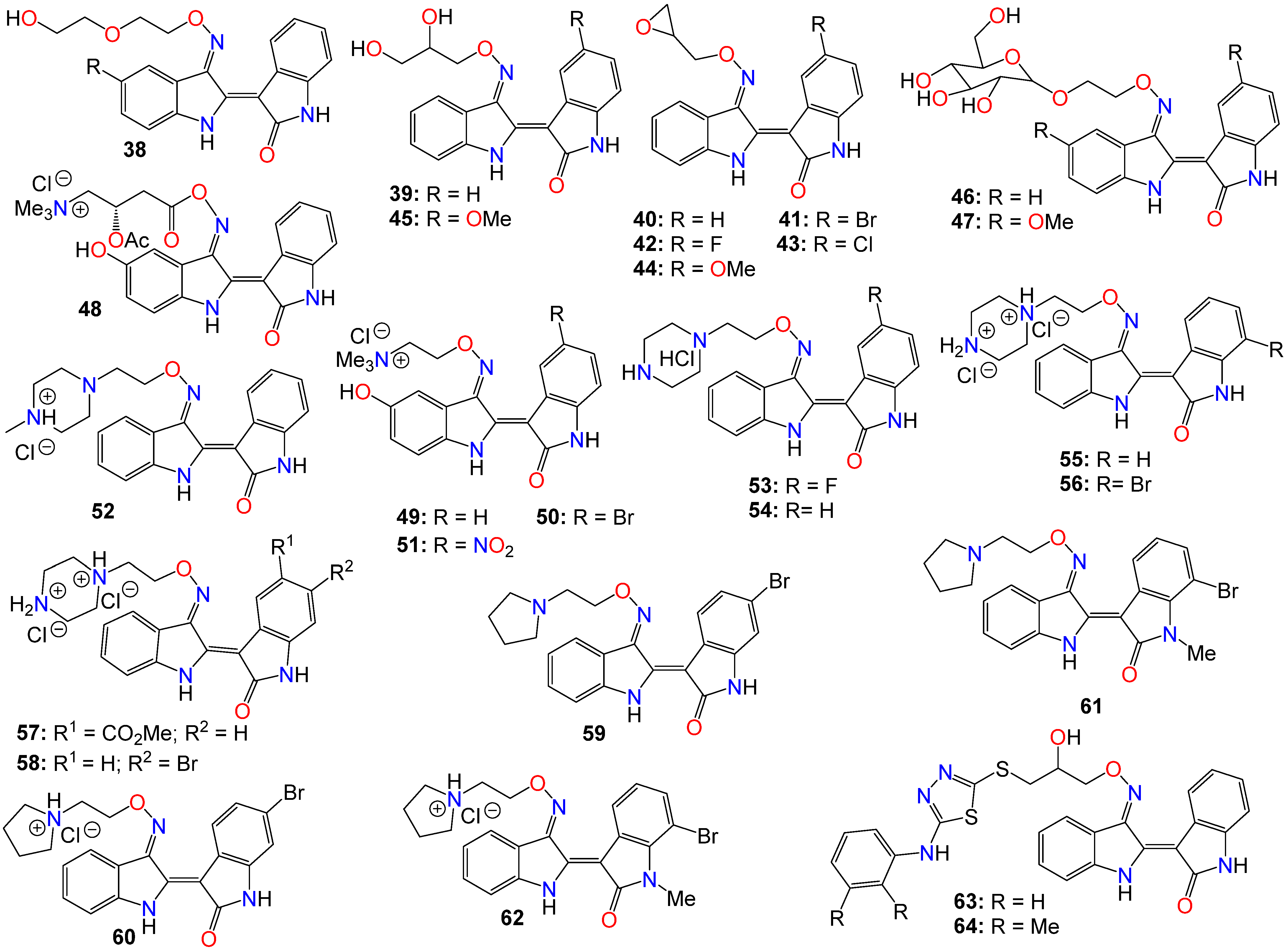
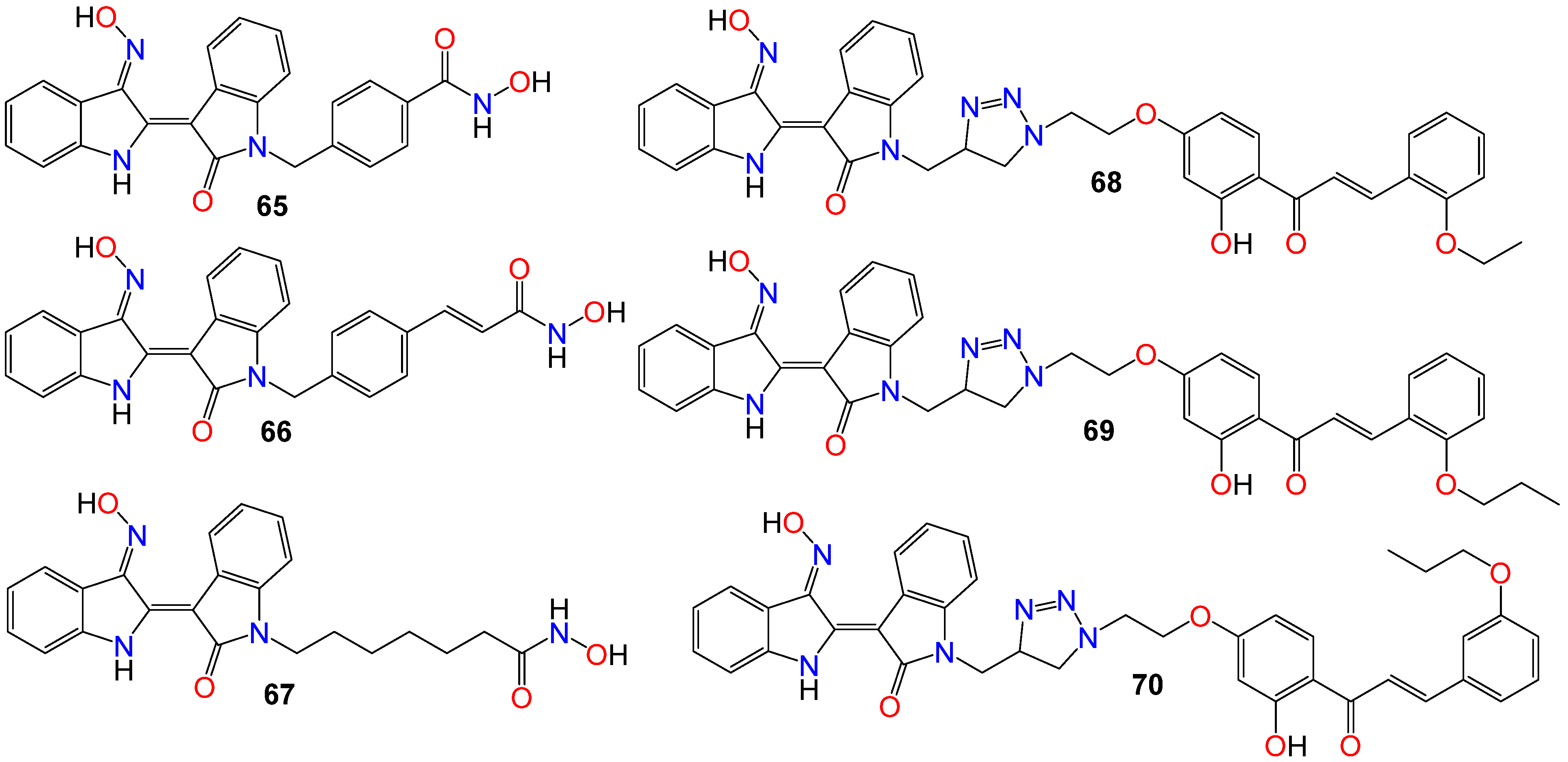
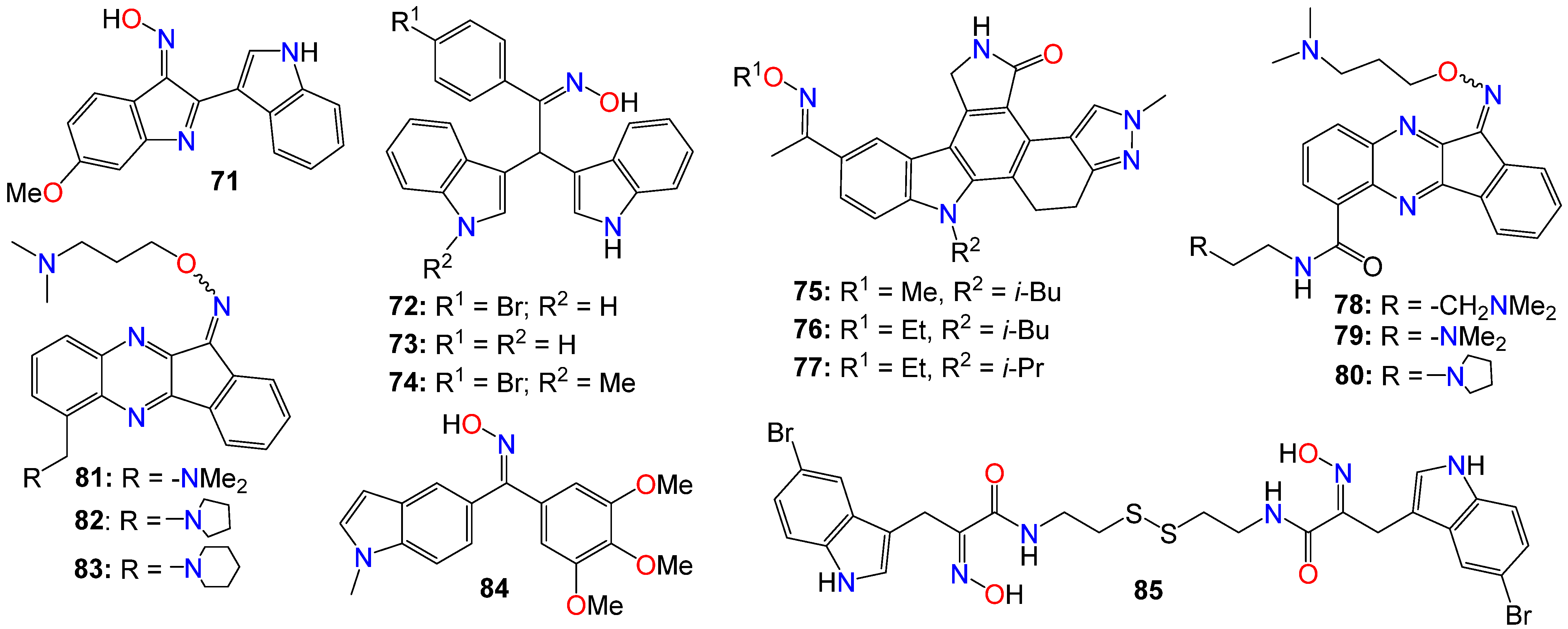
2.1. Anticancer Activity of Indole Based-Oximes and Oxime Ethers
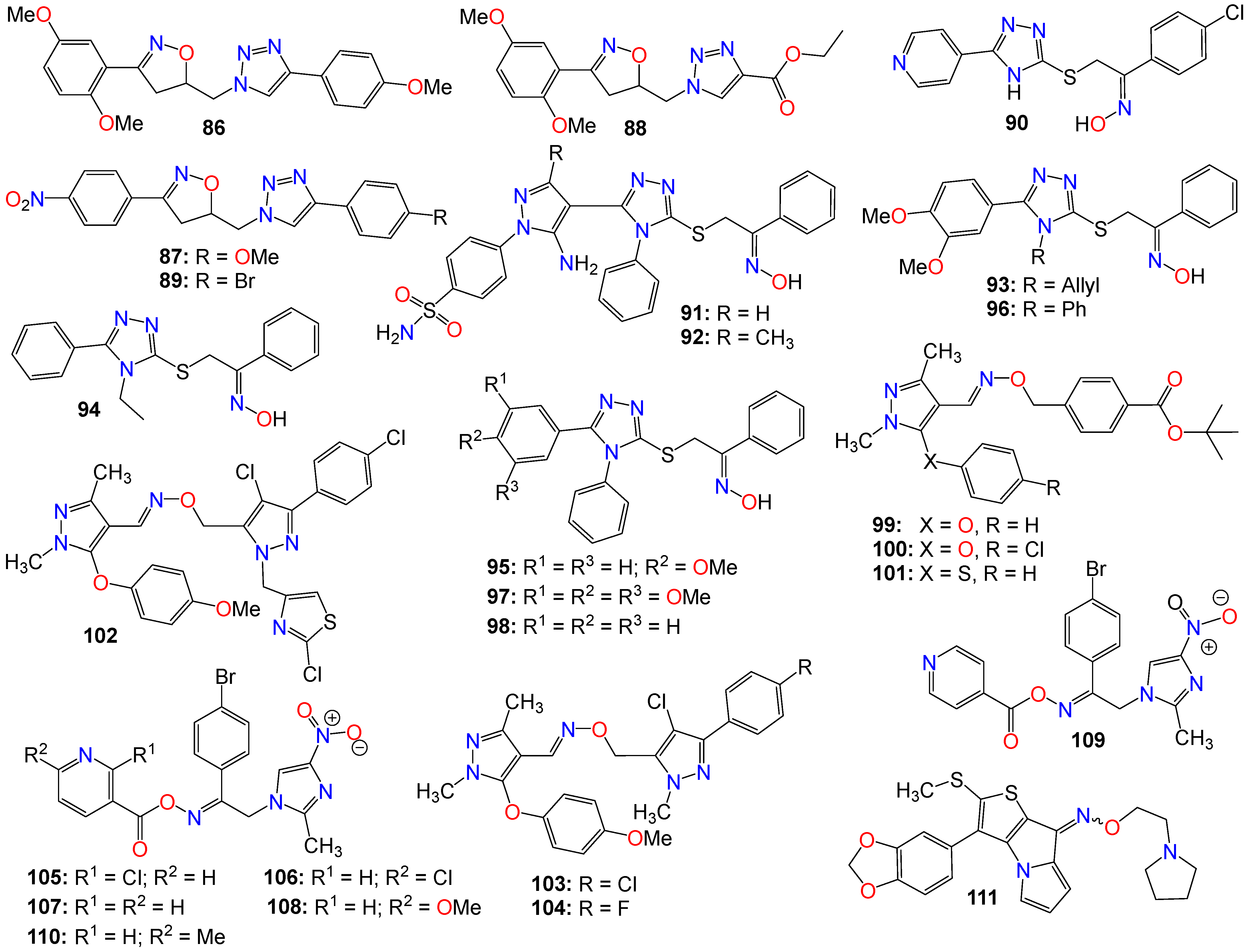
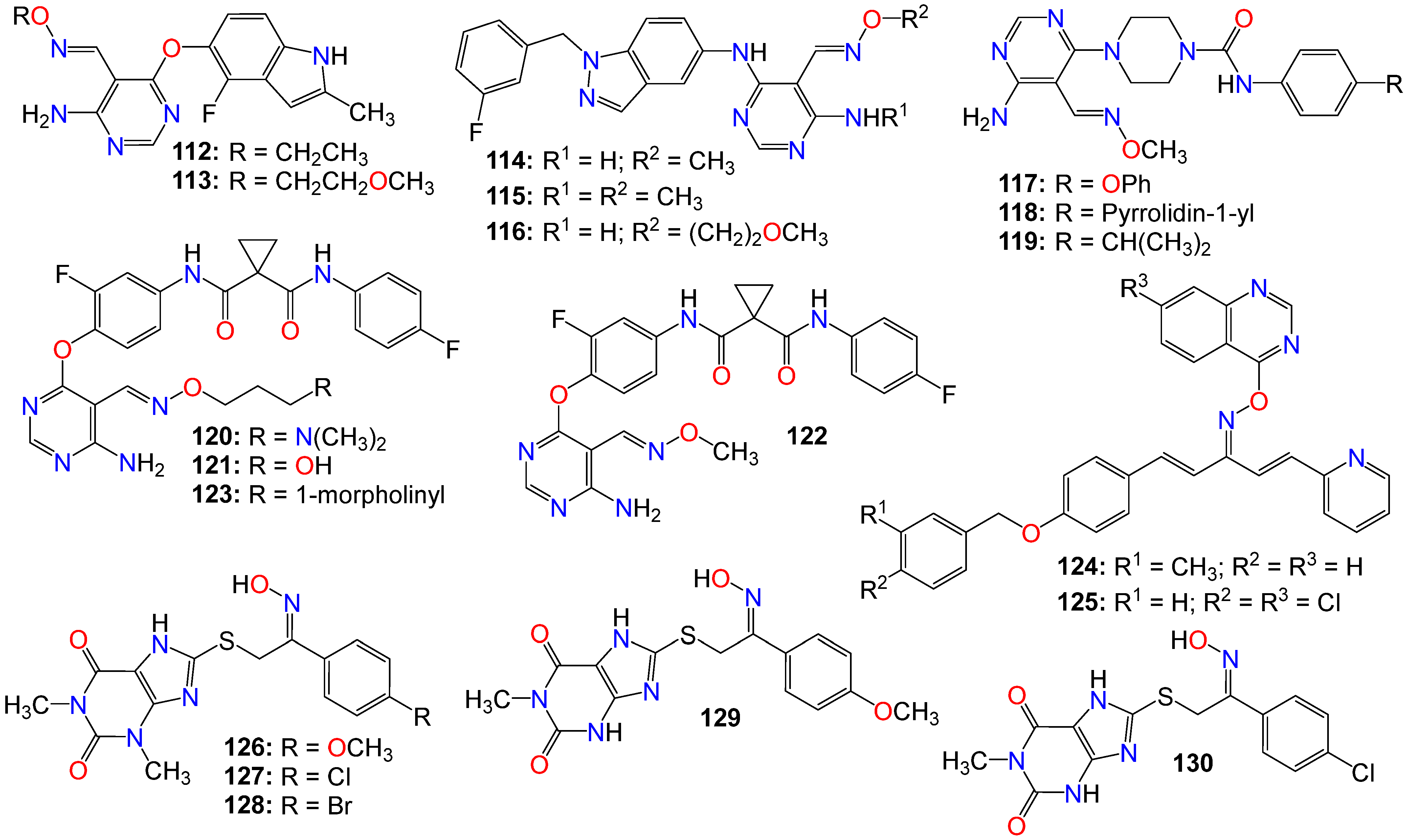
2.2. Anticancer Activity of Small Nitrogen-Containing Heterocyclic-Based Oximes
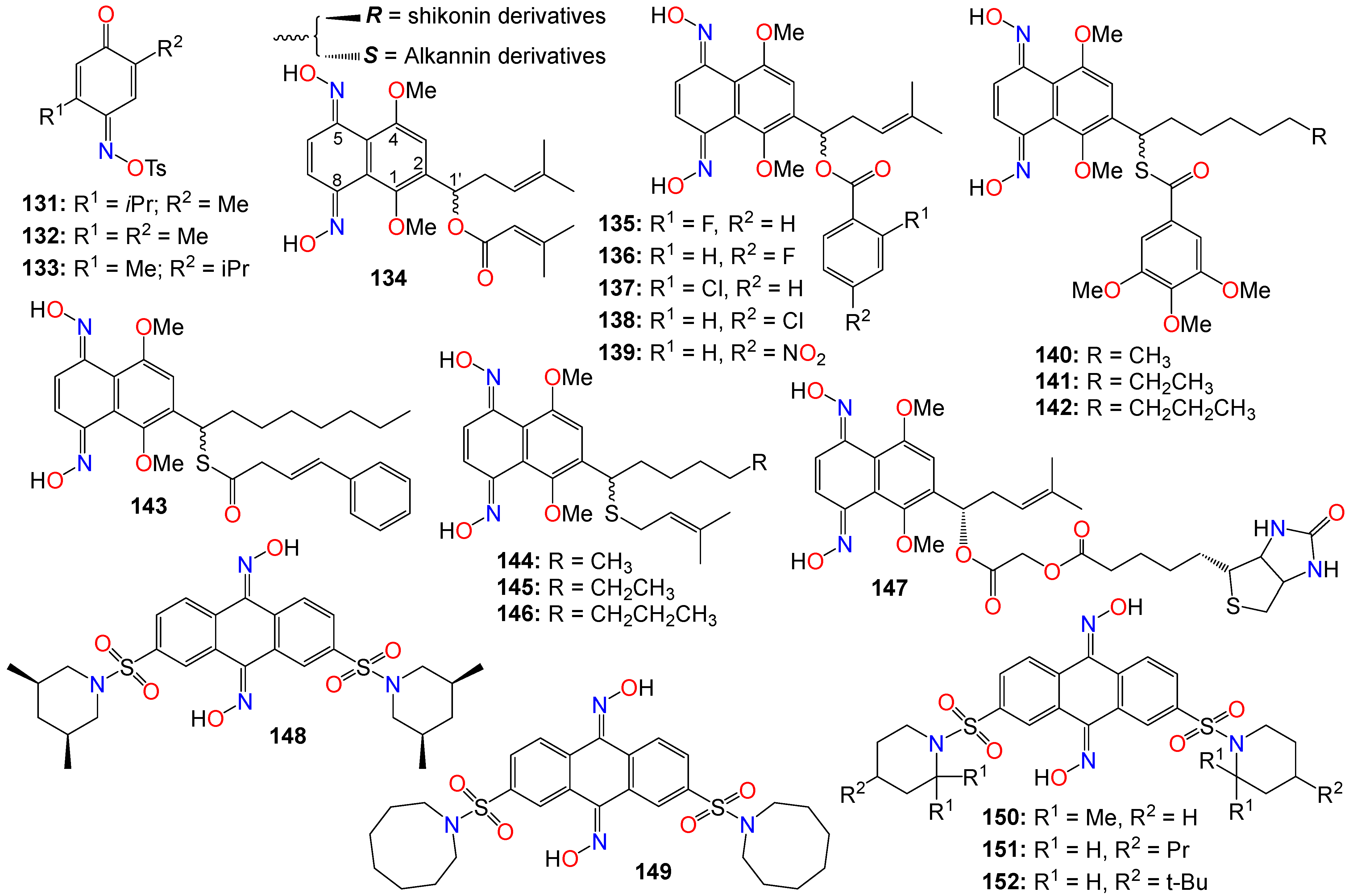
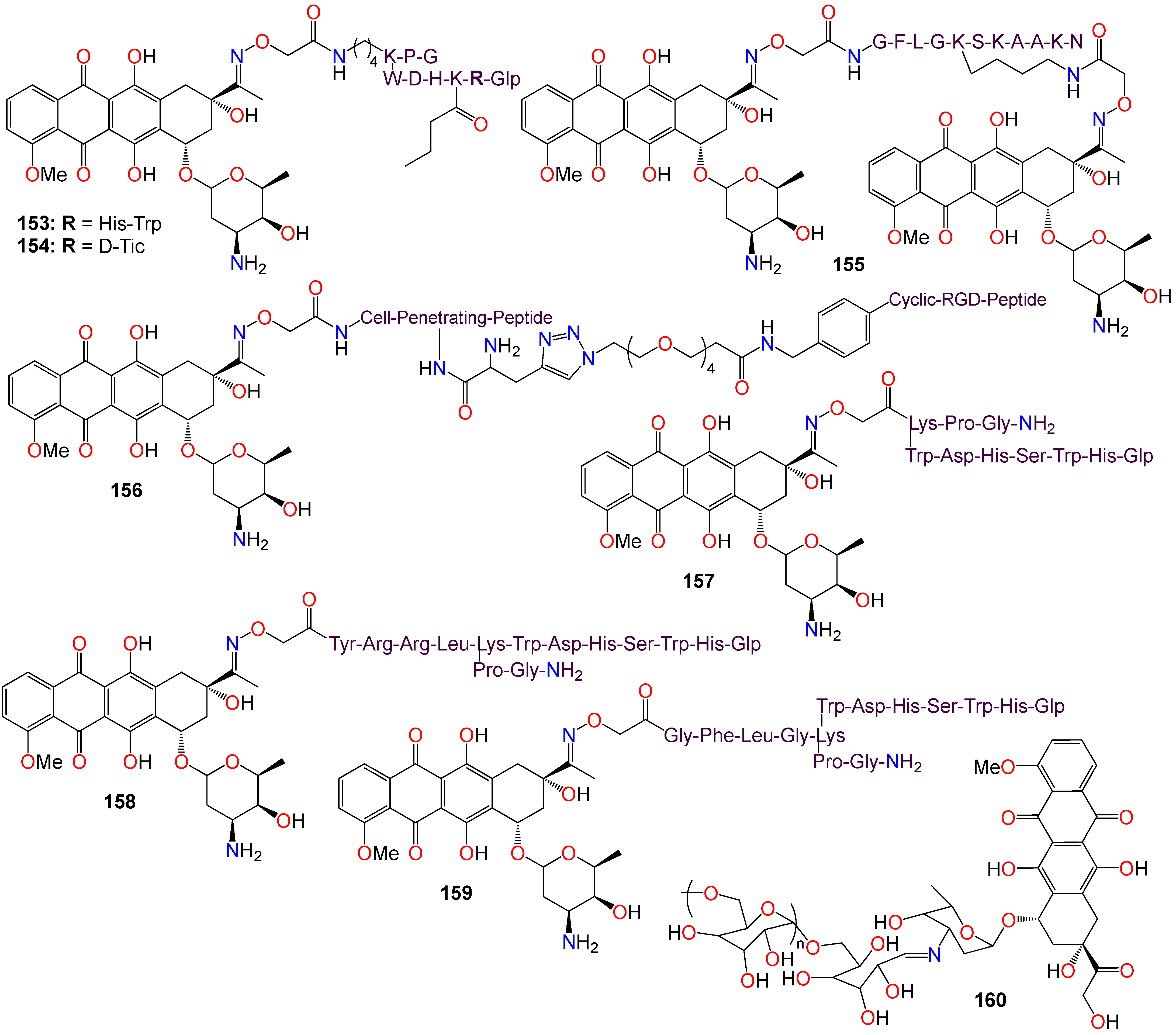
2.3. Anticancer Activity of Quinone-Based Oximes and Oxime Ethers
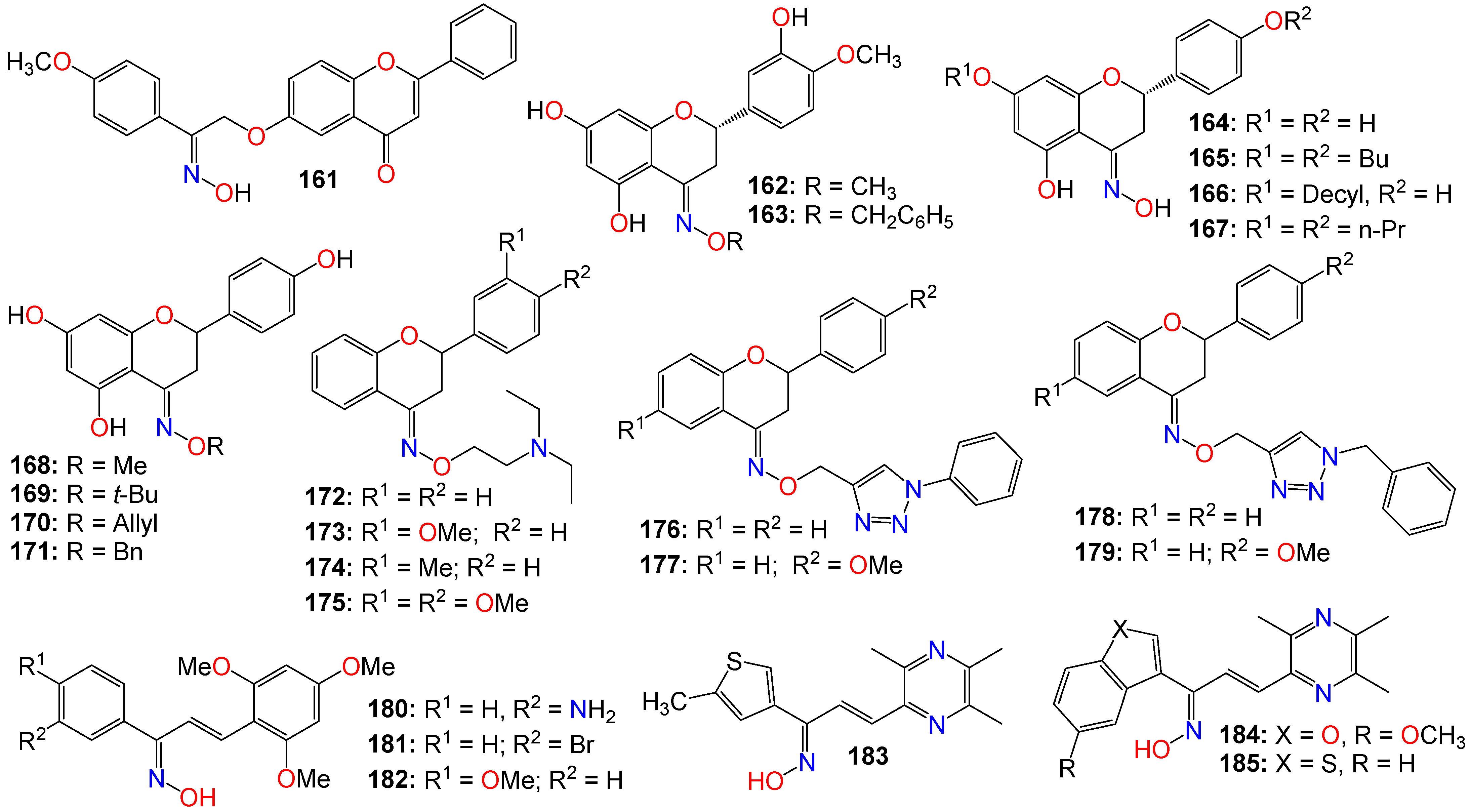
2.4. Anticancer Activity of Flavonoid-Based Oximes and Oxime Ethers
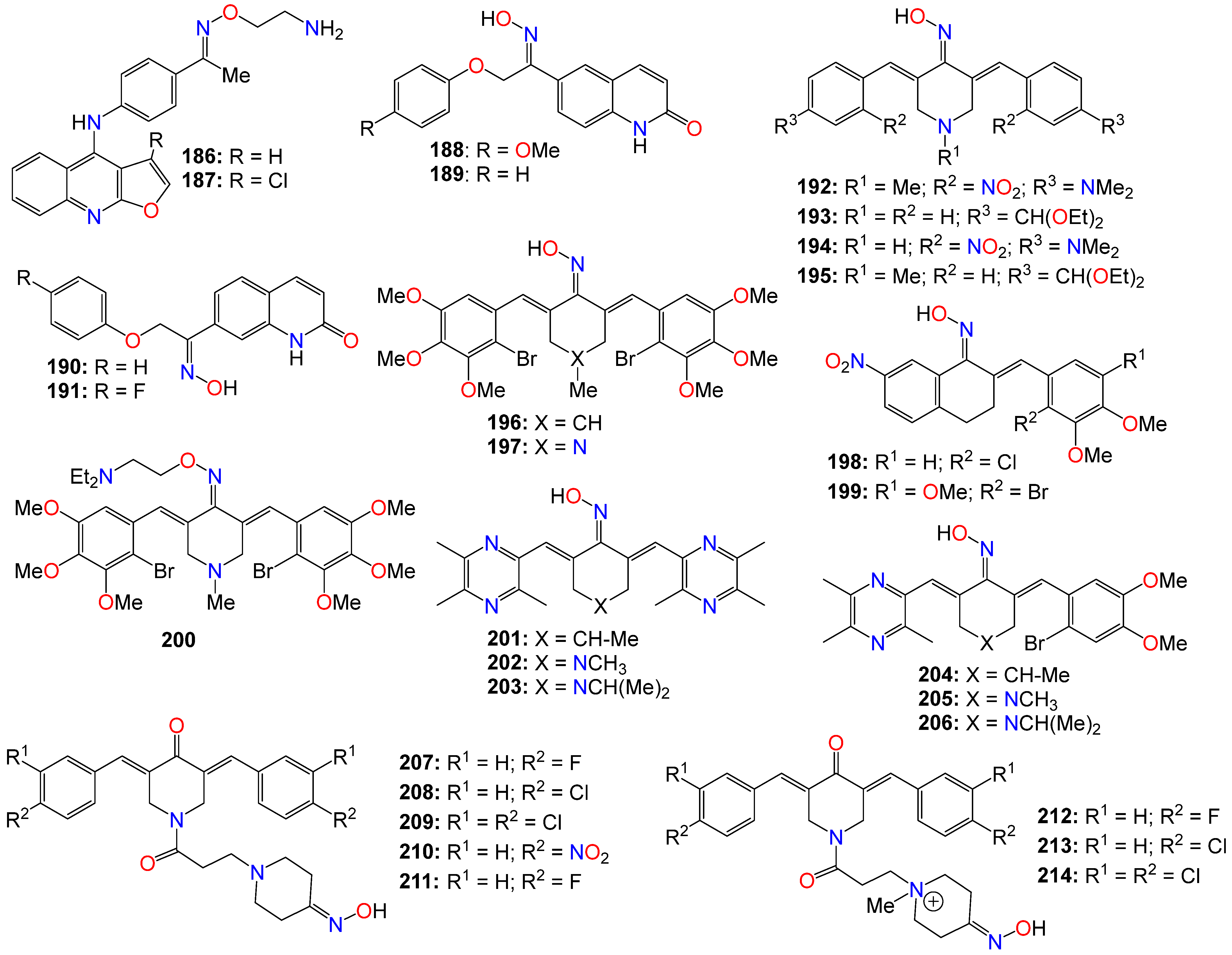
2.5. Anticancer Activity of Miscellaneous Aromatic- and Heterocyclic-Based Oximes and Oxime Ethers
2.6. Anticancer Activity of Small Aromatic and Phenolic-Based Oximes and Oxime Ethers
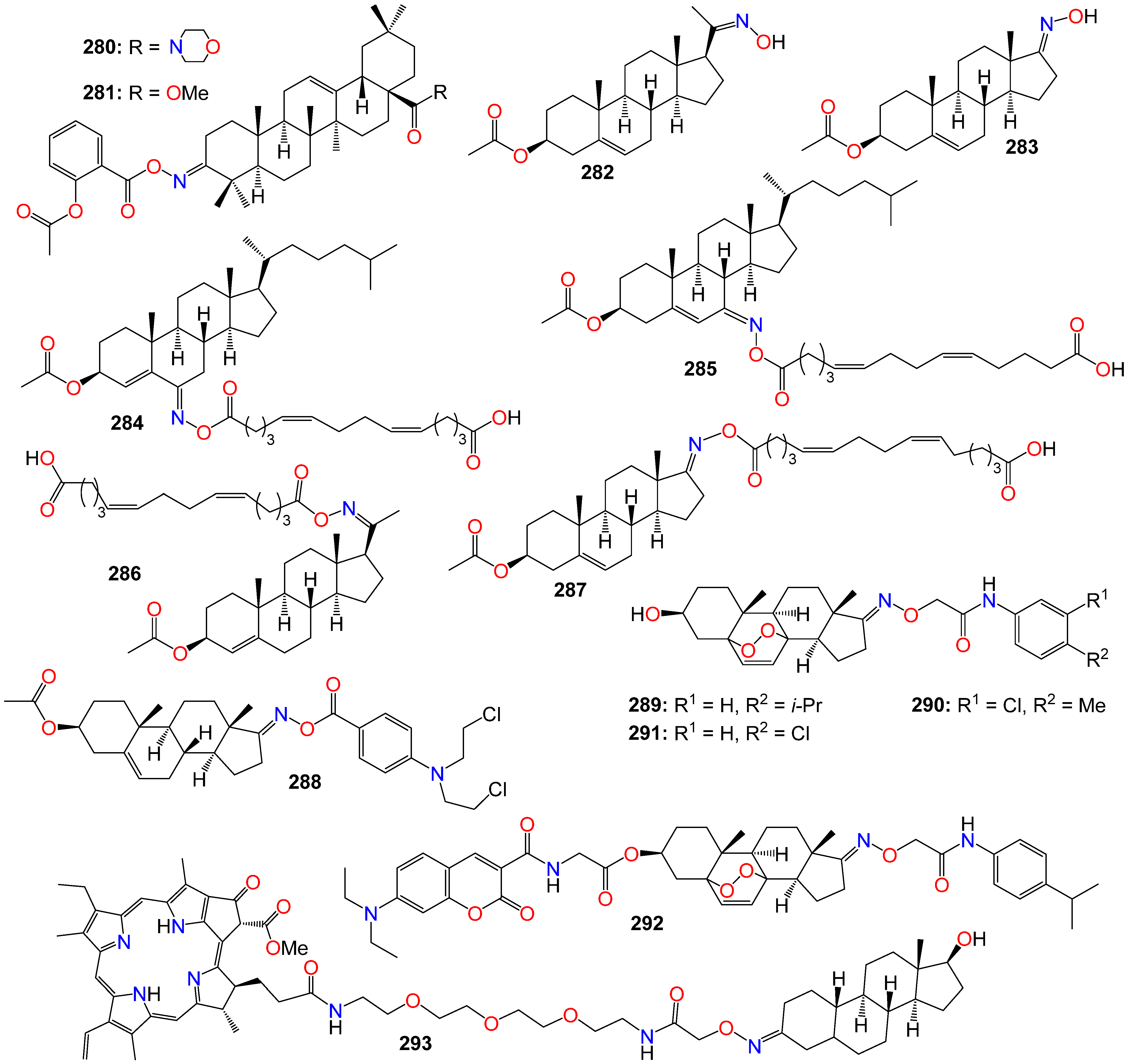
2.7. Anticancer Activity of Steroidal Oximes and Oxime Ethers
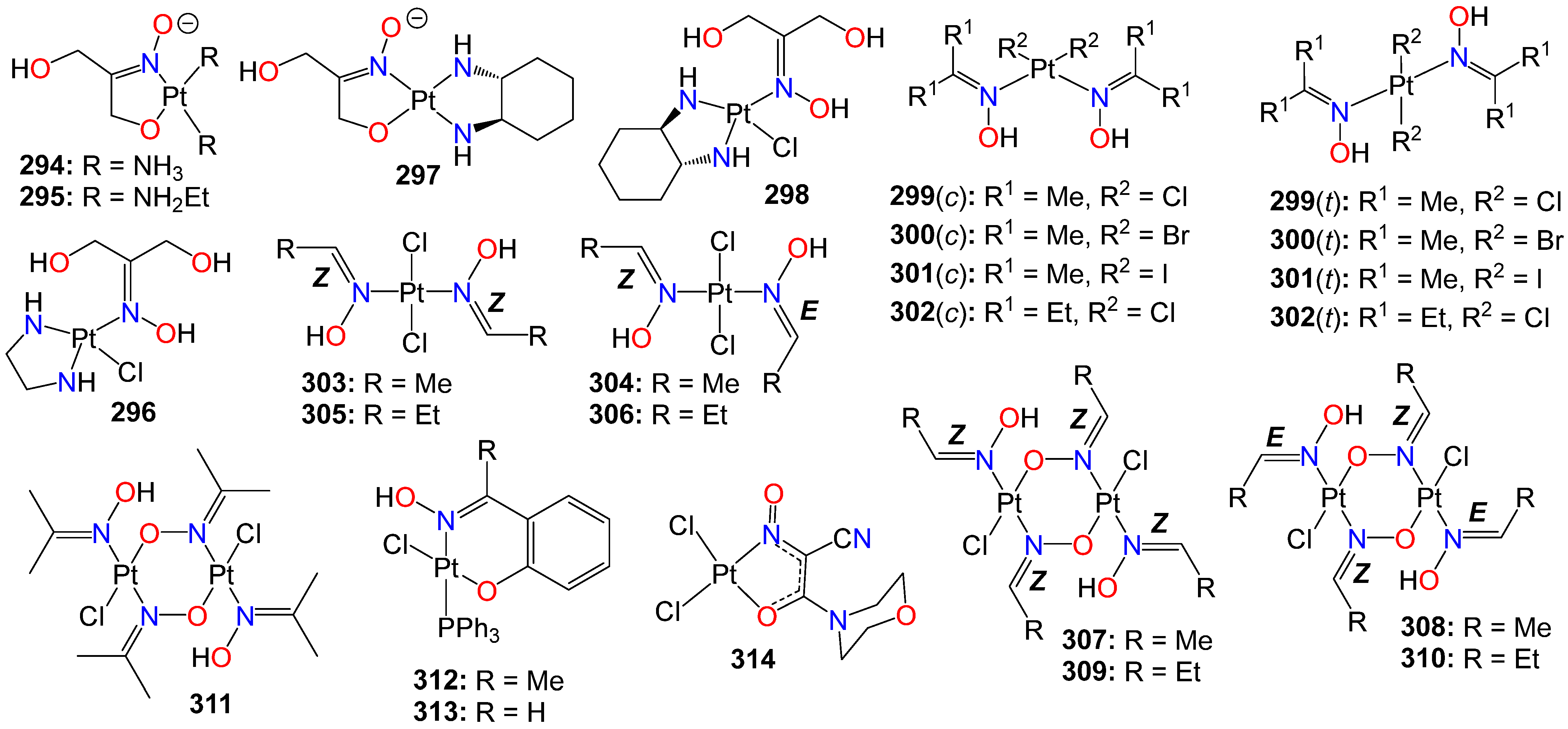
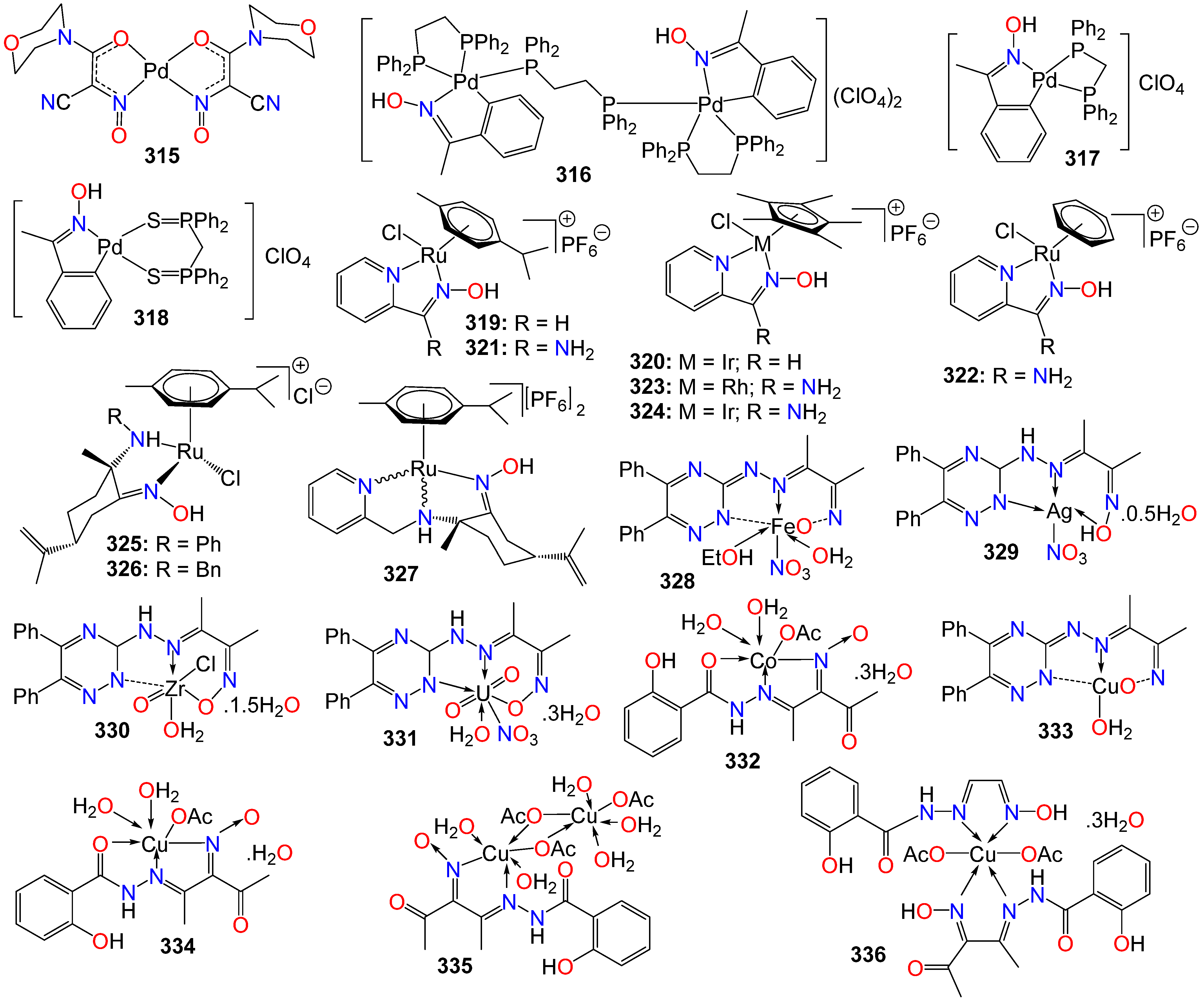
2.8. Anticancer Activity of Oxime Liganded Metals and Cyanoximates Metallic Complexes
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, Y.-B.; Chen, S.-Y.; Liu, R.-B. The new insights into autophagy in thyroid cancer progression. J. Transl. Med. 2023, 21, 413. [Google Scholar] [CrossRef]
- Cao, J.; Wan, S.; Chen, S.; Yang, L. ANXA6: A key molecular player in cancer progression and drug resistance. Discov. Oncol. 2023, 14, 53. [Google Scholar] [CrossRef]
- Zhou, M.; Zheng, M.; Zhou, X.; Tian, S.; Yang, X.; Ning, Y.; Li, Y.; Zhang, S. The roles of connexins and gap junctions in the progression of cancer. Cell Commun. Signal. 2023, 21, 8. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Deo, S.V.S.; Sharma, J.; Kumar, S. GLOBOCAN 2020 report on global cancer burden: Challenges and opportunities for surgical oncologists. Ann. Surg. Oncol. 2022, 29, 6497–6500. [Google Scholar] [CrossRef]
- La Vecchia, C.; Negri, E.; Carioli, G. Progress in cancer epidemiology: Avoided deaths in Europe over the last three decades. Eur. J. Cancer Prev. 2022, 31, 388–392. [Google Scholar] [CrossRef]
- Mazidimoradi, A.; Momenimovahed, Z.; Allahqoli, L.; Tiznobaik, A.; Hajinasab, N.; Salehiniya, H.; Alkatout, I. The global, regional and national epidemiology, incidence, mortality, and burden of ovarian cancer. Health Sci. Rep. 2022, 5, e936. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global cancer observatory: Cancer Today. In Lyon: International Agency for Research on Cancer; Organization WH: Geneva, Switzerland, 2020; Available online: https://gco.iarc.fr/today2020 (accessed on 12 May 2023).
- Li, C.; He, W.-Q. Global prediction of primary liver cancer incidences and mortality in 2040. J. Hepatol. 2023, 78, e144–e146. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global burden of colorectal cancer in 2020 and 2040: Incidence and mortality estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Yu, N.; Hwang, M.; Lee, Y.; Song, B.R.; Kang, E.H.; Sim, H.; Ahn, B.-C.; Hwang, K.H.; Kim, J.; Hong, S.; et al. Patient-derived cell-based pharmacogenomic assessment to unveil underlying resistance mechanisms and novel therapeutics for advanced lung cancer. J. Exp. Clin. Cancer Res. 2023, 42, 37. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Amero, P.; Noh, K.; Mangala, L.S.; Wen, Y.; Bayraktar, E.; Umamaheswaran, S.; Stur, E.; Dasari, S.K.; Ivan, C.; et al. Overcoming adaptive resistance to anti-VEGF therapy by targeting CD5L. Nat. Commun. 2023, 14, 2407. [Google Scholar] [CrossRef]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Amaresan, R.; Gopal, U. Cell surface GRP78: A potential mechanism of therapeutic resistant tumors. Cancer Cell Int. 2023, 23, 100. [Google Scholar] [CrossRef]
- Zhuang, H.; Yu, B.; Tao, D.; Xu, X.; Xu, Y.; Wang, J.; Jiao, Y.; Wang, L. The role of m6A methylation in therapy resistance in cancer. Mol. Cancer 2023, 22, 91. [Google Scholar] [CrossRef]
- Naldini, M.M.; Casirati, G.; Barcella, M.; Rancoita, P.M.V.; Cosentino, A.; Caserta, C.; Pavesi, F.; Zonari, E.; Desantis, G.; Gilioli, D.; et al. Longitudinal single-cell profiling of chemotherapy response in acute myeloid leukemia. Nat. Commun. 2023, 14, 1285. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, Y.; Li, W.; Gao, C.; Huang, F.; Cheng, L.; Jin, M.; Xu, X.; Huang, J. Single cell profiling of female breast fibroadenoma reveals distinct epithelial cell compositions and therapeutic targets. Nat. Commun. 2023, 14, 3469. [Google Scholar] [CrossRef]
- Cortés-Selva, D.; Dasgupta, B.; Singh, S.; Grewal, I.S. Innate and innate-like cells: The future of chimeric antigen receptor (CAR) cell therapy. Trends Pharmacol. Sci. 2021, 42, 45–59. [Google Scholar] [CrossRef]
- Bohineust, A.; Tourret, M.; Derivry, L.; Caillat-Zucman, S. Mucosal-associated invariant T (MAIT) cells, a new source of universal immune cells for chimeric antigen receptor (CAR)-cell therapy. Bull. Cancer 2021, 108, S92–S95. [Google Scholar] [CrossRef]
- Gao, Z.; Bai, Y.; Lin, A.; Jiang, A.; Zhou, C.; Cheng, Q.; Liu, Z.; Chen, X.; Zhang, J.; Luo, P. Gamma delta T-cell-based immune checkpoint therapy: Attractive candidate for antitumor treatment. Mol. Cancer 2023, 22, 31. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Aminu, M.; Li, S.; Lu, X.; Petranovic, M.; Saad, M.B.; Chen, P.; Qin, K.; Varghese, S.; Rinsurongkawong, W.; et al. Efficacy and clinicogenomic correlates of response to immune checkpoint inhibitors alone or with chemotherapy in non-small cell lung cancer. Nat. Commun. 2023, 14, 695. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Gong, M.; Deng, Y.; Wang, H.; Ye, D. T cell effects and mechanisms in immunotherapy of head and neck tumors. Cell Commun. Signal. 2023, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-González, R.V.; González-Díaz, S.; Vidal-Gutiérrez, O.; Cruz, C.d.l.C.-D.l.; Pérez-Ibave, D.C.; Garza-Rodríguez, M.L. Hypersensitivity reactions to anticancer chemotherapy and monoclonal antibodies: Safety and efficacy of desensitization. J. Oncol. Pharm. Pract. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Suzuki, H.; Asano, T.; Ohishi, T.; Yoshikawa, T.; Suzuki, H.; Mizuno, T.; Tanaka, T.; Kawada, M.; Kaneko, M.K.; Kato, Y. Antitumor activities in mouse xenograft models of canine fibroblastic tumor by defucosylated mouse-dog chimeric anti-HER2 monoclonal antibody (H77Bf). Monoclon. Antibodies Immunodiagn. Immunother. 2023, 42, 34–40. [Google Scholar] [CrossRef]
- Tanaka, M.; Ito, A.; Shiozawa, S.; Hara-Chikuma, M. Anti-tumor effect of aquaporin 3 monoclonal antibody on syngeneic mouse tumor model. Transl. Oncol. 2022, 24, 101498. [Google Scholar] [CrossRef]
- Yong, T.; Wei, Z.; Gan, L.; Yang, X. Extracellular-vesicle-based drug delivery systems for enhanced antitumor therapies through modulating the cancer-immunity cycle. Adv. Mater. 2022, 34, e2201054. [Google Scholar] [CrossRef]
- Moslehi, M.; Moazamiyanfar, R.; Dakkali, M.S.; Rezaei, S.; Rastegar-Pouyani, N.; Jafarzadeh, E.; Mouludi, K.; Khodamoradi, E.; Taeb, S.; Najafi, M. Modulation of the immune system by melatonin; implications for cancer therapy. Int. Immunopharmacol. 2022, 108, 108890. [Google Scholar] [CrossRef]
- Castelli, C.; Rivoltini, L.; Rodolfo, M.; Tazzari, M.; Belgiovine, C.; Allavena, P. Modulation of the myeloid compartment of the immune system by angiogenic- and kinase inhibitor-targeted anti-cancer therapies. Cancer Immunol. Immunother. 2015, 64, 83–89. [Google Scholar] [CrossRef]
- Mahadevan, S. Role of oximes in nitrogen metabolism in plants. Annu. Rev. Plant Physiol. 1973, 24, 69–88. [Google Scholar] [CrossRef]
- Sørensen, M.; Neilson, E.H.; Møller, B.L. Oximes: Unrecognized chameleons in general and specialized plant metabolism. Mol. Plant 2018, 11, 95–117. [Google Scholar] [CrossRef]
- Meyer, V.; Janny, A. Nitrogenous acetone derivatives. Ber. Dtsch. Chem. Ges. 1882, 15, 1164–1167. [Google Scholar] [CrossRef]
- Tapper, B.; Conn, E.; Butler, G. Conversion of α-ketoisovaleric acid oxime and isobutyraldoxime to linamarin in flax seedlings. Arch. Biochem. Biophys. 1967, 119, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Underhill, E.W. Biosynthesis of mustard oil glucosides. Conversion of phenylacetaldehyde oxime and 3-phenylpropionaldehyde oxime to glucotropaeolin and gluconasturtiin. Eur. J. Biochem. 1967, 2, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Kindl, H. Oxidases and oxygenases in higher plants. I. Occurrence of indole-3-acetaldehyde oxime and its formation from L-tryptophan. Hoppe-Seyler’s Z Physiol Chem. 1968, 349, 519. [Google Scholar]
- A Halkier, B.; E Olsen, C.; Møller, B.L. The biosynthesis of cyanogenic glucosides in higher plants. The (E)- and (Z)-isomers of p-hydroxyphenylacetaldehyde oxime as intermediates in the biosynthesis of dhurrin in Sorghum bicolor (L.) Moench. J. Biol. Chem. 1989, 264, 19487–19494. [Google Scholar]
- Andrawes, N.R.; Bagley, W.P.; Herrett, R.A. Metabolism of 2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime (Temik aldicarb pesticide) in potato plants. J. Agric. Food Chem. 1971, 19, 731–737. [Google Scholar] [CrossRef]
- Bartley, W.J.; Andrawes, N.R.; Chancey, E.L.; Bagley, W.P.; Spurr, H.W. Metabolism of Temik aldicarb pesticide [2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime] in the cotton plant. J. Agric. Food Chem. 1970, 18, 446–453. [Google Scholar] [CrossRef]
- Ridgway, R.L.; Jones, S.L.; Coppedge, J.R.; Lindquist, D.A. Systemic activity of 2-methyl-2-(methylthio)propionaldehyde O-(methylcarbamoyl)oxime (UC-21149) in the cotton plant with special reference to the boll weevil. J. Econ. Entomol. 1968, 61, 1705–1712. [Google Scholar] [CrossRef]
- Massolini, G.; Carmellino, M.L.; Baruffini, A. Activity of some benzophenone oximes on plant-pathogenic fungi. Farm. Ed Sci. 1987, 42, 117. [Google Scholar]
- Kula, K.; Nagatsky, R.; Sadowski, M.; Siumka, Y.; Demchuk, O.M. Arylcyanomethylenequinone oximes: An overview of synthesis, chemical transformations, and biological activity. Molecules 2023, 28, 5229. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Pires, A.S.; Roleira, F.M.F.; Tavares-Da-Silva, E.J. The structural diversity and biological activity of steroid oximes. Molecules 2023, 28, 1690. [Google Scholar] [CrossRef]
- Omidi, S.; Kakanejadifard, A. A review on biological activities of Schiff base, hydrazone, and oxime derivatives of curcumin. RSC Adv. 2020, 10, 30186–30202. [Google Scholar] [CrossRef] [PubMed]
- Abele, E. Synthesis, reactions and biological activity of derivatives of oximes of three-membered heterocycles. Heterocycl. Lett. 2015, 46, 229. [Google Scholar] [CrossRef]
- Abele, E.; Abele, R. Oximes of nucleosides and related compounds: Synthesis, reactions and biological activity. Curr. Org. Synth. 2018, 15, 650–665. [Google Scholar] [CrossRef]
- Dai, H.; Li, G.; Chen, J.; Shi, Y.; Ge, S.; Fan, C.; He, H. Synthesis and biological activities of novel 1,3,4-thiadiazole-containing pyrazole oxime derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 3818–3821. [Google Scholar] [CrossRef]
- Dhuguru, J.; Zviagin, E.; Skouta, R. FDA-Approved oximes and their significance in medicinal chemistry. Pharmaceuticals 2022, 15, 66. [Google Scholar] [CrossRef]
- Rosenthal, G.A. l-Canaline: A potent antimetabolite and anti-cancer agent from leguminous plants. Life Sci. 1997, 60, 1635–1641. [Google Scholar] [CrossRef]
- Fylaktakidou, K.; Hadjipavlou-Litina, D.; Litinas, K.; Varella, E.; Nicolaides, D. Recent developments in the chemistry and in the biological applications of amidoximes. Curr. Pharm. Des. 2008, 14, 1001–1047. [Google Scholar] [CrossRef]
- Canario, C.; Silvestre, S.; Falcao, A.; Alves, G. Steroidal oximes: Useful compounds with antitumor activities. Curr. Med. Chem. 2018, 25, 660–686. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Plotnikov, M.B.; Khlebnikov, A.I.; Plotnikova, T.M.; Quinn, M.T. Oximes: Novel therapeutics with anticancer and anti-inflammatory potential. Biomolecules 2021, 11, 777. [Google Scholar] [CrossRef]
- Dhuguru, J.; Skouta, R. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615. [Google Scholar] [CrossRef]
- Ma, M.Z.; Yao, B.Y. Progress in indirubin treatment of chronic myelocytic leukemia. J. Tradit. Chin. Med. 1983, 3, 245–248. [Google Scholar]
- Wang, H.; Wang, Z.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L.; Chen, Y. Anticancer potential of indirubins in medicinal chemistry: Biological activity, structural modification, and structure-activity relationship. Eur. J. Med. Chem. 2021, 223, 113652. [Google Scholar] [CrossRef]
- Xiao, Z.; Hao, Y.; Liu, B.; Qian, L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leuk. Lymphoma 2002, 43, 1763–1768. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, G.-B.; Liu, P.; Song, J.-H.; Liang, Y.; Yan, X.-J.; Xu, F.; Wang, B.-S.; Mao, J.-H.; Shen, Z.-X.; et al. Dissection of mechanisms of Chinese medicinal formula Realgar- Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 4826–4831. [Google Scholar] [CrossRef]
- Fogaça, M.V.; Cândido-Bacani, P.D.M.; Benicio, L.M.; Zapata, L.M.; Cardoso, P.D.F.; De Oliveira, M.T.; Calvo, T.R.; Varanda, E.A.; Vilegas, W.; Cólus, I.M.D.S. Effects of indirubin and isatin on cell viability, mutagenicity, genotoxicity and BAX/ERCC1 gene expression. Pharm. Biol. 2017, 55, 2005–2014. [Google Scholar] [CrossRef]
- Alex, D.; Lam, I.K.; Lin, Z.; Lee, S.M.Y. Indirubin shows anti-angiogenic activity in an in vivo zebrafish model and an in vitro HUVEC model. J. Ethnopharmacol. 2010, 131, 242–247. [Google Scholar] [CrossRef]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Knockaert, M.; Greengard, P.; Meijer, L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol. Sci. 2002, 23, 417–425. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.-Z.; Mandelkow, E.-M.; et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Aratsu, Y.; Sugawara, R.; Sasaki, T.; Miyairi, S.; Nagata, K. Indirubin, a component of Ban-Lan-Gen, activates CYP3A4 gene transcription through the human pregnane X receptor. Drug Metab. Pharmacokinet. 2016, 31, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Eisenbrand, G.; Hippe, F.; Jakobs, S.; Muehlbeyer, S. Molecular mechanisms of indirubin and its derivatives: Novel anticancer molecules with their origin in traditional Chinese phytomedicine. J. Cancer Res. Clin. Oncol. 2004, 130, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Campa, V.M.; Baltziskueta, E.; Bengoa-Vergniory, N.; Gorroño-Etxebarria, I.; Wesołowski, R.; Waxman, J.; Kypta, R.M. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget 2014, 5, 8173–8187. [Google Scholar] [CrossRef]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.-M.; Boucher, M.-J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef]
- Sutherland, C.; A Leighton, I.; Cohen, P. Inactivation of glycogen synthase kinase-3β by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Hur, E.-M.; Zhou, F.-Q. GSK3 signaling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Skaltsounis, A.-L. Indirubin derivatives: A patent review (2010-present). Expert Opin. Ther. Pat. 2015, 25, 583–593. [Google Scholar] [CrossRef]
- Ribas, J.; Bettayeb, K.; Ferandin, Y.; Knockaert, M.; Garrofé-Ochoa, X.; Totzke, F.; Schächtele, C.; Mester, J.; Polychronopoulos, P.; Magiatis, P.; et al. 7-Bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene 2006, 25, 6304–6318. [Google Scholar] [CrossRef]
- Cheng, X.; Merz, K.-H.; Vatter, S.; Zeller, J.; Muehlbeyer, S.; Thommet, A.; Christ, J.; Wölfl, S.; Eisenbrand, G. Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. J. Med. Chem. 2017, 60, 4949–4962. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Feng, M.; Tan, X.; Cheng, J.; Hua, W.; Yao, Q. Facile synthesis, X-ray structure and anticancer activity of N1-alkylindirubin-3’-oxime. Youji Huaxue 2009, 29, 1606–1610. [Google Scholar]
- Lee, J.-W.; Moon, M.J.; Min, H.-Y.; Chung, H.-J.; Park, E.-J.; Park, H.J.; Hong, J.-Y.; Kim, Y.-C.; Lee, S.K. Induction of apoptosis by a novel indirubin-5-nitro-3′-monoxime, a CDK inhibitor, in human lung cancer cells. Bioorg. Med. Chem. Lett. 2005, 15, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Moon, M.J.; Lee, S.D.; Choi, S.-U.; Han, S.-Y.; Kim, Y.-C. Indirubin derivatives as potent FLT3 inhibitors with anti-proliferative activity of acute myeloid leukemic cells. Bioorg. Med. Chem. Lett. 2010, 20, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ichimaru, Y.; Kurita, M.; Hayashi, E.; Homma, T.; Saito, H.; Masuda, S.; Nemoto, N.; Hemmi, A.; Suzuki, T.; et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 3′-oxime and 5-methoxyindirubin 3′-oxime in vitro and in vivo. Cancer Lett. 2017, 397, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Lee, J.-E.; Cho, K.-M.; Park, S.-H.; Kim, H.-J.; Kim, Y.-C.; Kim, T.S. 5-diphenylacetamido-indirubin-3′-oxime as a novel mitochondria-targeting agent with anti-leukemic activities. Mol. Carcinog. 2016, 55, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, X.; Kuang, L.; Wang, Y. New insights into the characteristics of DRAK2 and its role in apoptosis: From molecular mechanisms to clinically applied potential. Front. Pharmacol. 2022, 13, 1014508. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Byun, B.J.; Kim, H.-M.; Lee, J.Y.; Park, J.-H.; Lee, N.; Son, Y.H.; Choi, S.U.; Yang, K.-M.; Kim, S.-J.; et al. Discovery of indirubin derivatives as new class of DRAK2 inhibitors from high throughput screening. Bioorg. Med. Chem. Lett. 2016, 26, 2719–2723. [Google Scholar] [CrossRef]
- Yoon, K.; Lee, H.J.; Chung, H.J.; Lee, J.; Choi, J.; Heo, J.D.; Kim, Y.; Han, S. Discovery of LDD-1075 as a potent FLT3 inhibitor. Oncol. Lett. 2019, 17, 4735–4741. [Google Scholar] [CrossRef]
- Cheng, X.; Rasqué, P.; Vatter, S.; Merz, K.-H.; Eisenbrand, G. Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg. Med. Chem. 2010, 18, 4509–4515. [Google Scholar] [CrossRef]
- Jautelat, R.; Brumby, T.; Schäfer, M.; Briem, H.; Eisenbrand, G.; Schwahn, S.; Krüger, M.; Lücking, U.; Prien, O.; Siemeister, G. From the insoluble dye indirubin towards highly active, soluble CDK2-inhibitors. ChemBioChem 2005, 6, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.-S.; Zhao, L. Effects of the GSK-3β inhibitor (2Z,3E)-6-bromoindirubin-3′-oxime upon ovarian cancer cells. Tumor Biol. 2016, 37, 4857–4864. [Google Scholar] [CrossRef] [PubMed]
- Ribas, J.; Yuste, V.J.; Garrofé-Ochoa, X.; Meijer, L.; Esquerda, J.E.; Boix, J. 7-Bromoindirubin-3′-oxime uncovers a serine protease-mediated paradigm of necrotic cell death. Biochem. Pharmacol. 2008, 76, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Broecker-Preuss, M.; Becher-Boveleth, N.; Gall, S.; Rehmann, K.; Schenke, S.; Mann, K. Induction of atypical cell death in thyroid carcinoma cells by the indirubin derivative 7-bromoindirubin-3’-oxime (7BIO). Cancer Cell Int. 2015, 15, 97. [Google Scholar] [CrossRef]
- Ichimaru, Y.; Fujii, T.; Saito, H.; Sano, M.; Uchiyama, T.; Miyairi, S. 5-Bromoindirubin 3′-(O-oxiran-2-ylmethyl)oxime: A long-acting anticancer agent and a suicide inhibitor for epoxide hydrolase. Bioorg. Med. Chem. 2017, 25, 4665–4676. [Google Scholar] [CrossRef]
- Ichimaru, Y.; Saito, H.; Uchiyama, T.; Metori, K.; Tabata, K.; Suzuki, T.; Miyairi, S. Indirubin 3′-(O-oxiran-2-ylmethyl)oxime: A novel anticancer agent. Bioorg. Med. Chem. Lett. 2015, 25, 1403–1406. [Google Scholar] [CrossRef]
- Choi, S.-J.; Lee, J.-E.; Jeong, S.-Y.; Im, I.; Lee, S.-D.; Lee, E.-J.; Lee, S.K.; Kwon, S.-M.; Ahn, S.-G.; Yoon, J.-H.; et al. 5,5’-Substituted Indirubin-3’-oxime derivatives as potent cyclin-dependent kinase inhibitors with anticancer activity. J. Med. Chem. 2010, 53, 3696–3706. [Google Scholar] [CrossRef]
- Kim, J.; Shin, E.K.; Kang, Y.; Park, J.H.Y. Indirubin-3’-monoxime, a derivative of a chinese antileukemia medicine, inhibits angiogenesis. J. Cell. Biochem. 2011, 112, 1384–1391. [Google Scholar] [CrossRef]
- Kim, W.-S.; Lee, M.-J.; Kim, D.-H.; Lee, J.-E.; Kim, J.-I.; Kim, Y.-C.; Song, M.-R.; Park, S.-G. 5′-OH-5-nitro-Indirubin oxime (AGM130), an Indirubin derivative, induces apoptosis of Imatinib-resistant chronic myeloid leukemia cells. Leuk. Res. 2013, 37, 427–433. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Kim, T.-H.; Kwon, S.-M.; Yoon, H.-E.; Kim, H.-S.; Kim, J.-I.; Kim, Y.-C.; Kang, K.-W.; Ahn, S.-G.; Yoon, J.-H. 5-Nitro-5′-hydroxy-indirubin-3′-oxime (AGM130), an indirubin-3′-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur. J. Pharm. Sci. 2015, 79, 122–131. [Google Scholar] [CrossRef]
- Yan, L.; Lai, F.; Chen, X.; Xiao, Z. Discovery of novel indirubin-3′-monoxime derivatives as potent inhibitors against CDK2 and CDK9. Bioorg. Med. Chem. Lett. 2015, 25, 2447–2451. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-specificity, tyrosine phosphorylation-regulated kinases (DYRKs) and cdc2-like kinases (CLKs) in human disease, an overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Kritsanida, M.; Gaboriaud-Kolar, N.; Magiatis, P.; Ferandin, Y.; Durieu, E.; Lozach, O.; Cappel, D.; Soundararajan, M.; Filippakopoulos, P.; et al. Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. ACS Med. Chem. Lett. 2013, 4, 22–26. [Google Scholar] [CrossRef]
- Meijer, L.; Skaltsounis, A.-L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-selective inhibitors derived from tyrian purple indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef]
- Nam, S.; Buettner, R.; Turkson, J.; Kim, D.; Cheng, J.Q.; Muehlbeyer, S.; Hippe, F.; Vatter, S.; Merz, K.-H.; Eisenbrand, G.; et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5998–6003. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef]
- Kurita, M.; Hanada, S.; Ichimaru, Y.; Saito, H.; Tabata, K.; Asami, S.; Miyairi, S.; Suzuki, T. Indirubin 3’-epoxide induces caspase-independent cell death in human neuroblastoma. Biol. Pharm. Bull. 2016, 39, 993–999. [Google Scholar] [CrossRef]
- Shin, E.-K.; Kim, J.-K. Indirubin derivative E804 inhibits angiogenesis. BMC Cancer 2012, 12, 164. [Google Scholar] [CrossRef]
- Chan, Y.-K.; Kwok, H.-H.; Chan, L.-S.; Leung, K.S.-Y.; Shi, J.; Mak, N.-K.; Wong, R.N.-S.; Yue, P.Y.-K. An indirubin derivative, E804, exhibits potent angiosuppressive activity. Biochem. Pharmacol. 2012, 83, 598–607. [Google Scholar] [CrossRef]
- Nam, S.; Scuto, A.; Yang, F.; Chen, W.; Park, S.; Yoo, H.-S.; Konig, H.; Bhatia, R.; Cheng, X.; Merz, K.-H.; et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of Stat5 signaling. Mol. Oncol. 2012, 6, 276–283. [Google Scholar] [CrossRef]
- Cheng, X.; Alborzinia, H.; Merz, K.-H.; Steinbeisser, H.; Mrowka, R.; Scholl, C.; Kitanovic, I.; Eisenbrand, G.; Wölfl, S. Indirubin derivatives modulate TGFβ/BMP signaling at different levels and trigger ubiquitin-mediated depletion of nonactivated R-smads. Chem. Biol. 2012, 19, 1423–1436. [Google Scholar] [CrossRef]
- Nam, S.; Wen, W.; Schroeder, A.; Herrmann, A.; Yu, H.; Cheng, X.; Merz, K.-H.; Eisenbrand, G.; Li, H.; Yuan, Y.-C.; et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013, 7, 369–378. [Google Scholar] [CrossRef]
- Heshmati, N.; Wagner, B.; Cheng, X.; Scholz, T.; Kansy, M.; Eisenbrand, G.; Fricker, G. Physicochemical characterization and in vitro permeation of an indirubin derivative. Eur. J. Pharm. Sci. 2013, 50, 467–475. [Google Scholar] [CrossRef]
- Jakobs, S.; Merz, K.; Vatter, S.; Eisenbrand, G. Molecular targets of indirubins. Int. J. Clin. Pharmacol. Ther. 2005, 43, 592–594. [Google Scholar] [CrossRef]
- Ginzinger, W.; Egger, A.; Mühlgassner, G.; Arion, V.B.; Jakupec, M.A.; Galanski, M.S.; Berger, W.; Keppler, B.K. Water-soluble cationic derivatives of indirubin, the active anticancer component from Indigo naturalis. Chem Biodivers. 2012, 9, 2175–2185. [Google Scholar] [CrossRef]
- Kim, W.-H.; Jeong, P.; Kim, S.-W.; Cho, H.; Lee, J.-M.; Seo, S.; Shen, H.; Ahn, Y.; Jung, D.-W.; Kim, Y.-C.; et al. A novel indirubin derivative that increases somatic cell plasticity and inhibits tumorigenicity. Bioorg. Med. Chem. 2019, 27, 2923–2934. [Google Scholar] [CrossRef]
- Jeong, P.; Moon, Y.; Lee, J.-H.; Lee, S.-D.; Park, J.; Lee, J.; Kim, J.; Lee, H.J.; Kim, N.Y.; Choi, J.; et al. Discovery of orally active indirubin-3′-oxime derivatives as potent type 1 FLT3 inhibitors for acute myeloid leukemia. Eur. J. Med. Chem. 2020, 195, 112205. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, P.; Moon, Y.; Choi, J.; Heo, J.D.; Kim, Y.-C.; Han, S.-Y. Characterization of LDD-2633 as a novel RET kinase inhibitor with anti-tumor effects in thyroid cancer. Pharmaceuticals 2021, 14, 38. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.; Jeong, P.; Choi, J.; Baek, J.; Ahn, S.J.; Moon, Y.; Heo, J.D.; Choi, Y.H.; Chin, Y.-W.; et al. Discovery of a FLT3 inhibitor LDD1937 as an anti-leukemic agent for acute myeloid leukemia. Oncotarget 2018, 9, 924–936. [Google Scholar] [CrossRef]
- Gaboriaud-Kolar, N.; Myrianthopoulos, V.; Vougogiannopoulou, K.; Gerolymatos, P.; Horne, D.A.; Jove, R.; Mikros, E.; Nam, S.; Skaltsounis, A.-L. Natural-based indirubins display potent cytotoxicity toward wild-type and T315I-resistant leukemia cell lines. J. Nat. Prod. 2016, 79, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Ferandin, Y.; Bettayeb, K.; Kritsanida, M.; Lozach, O.; Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.-L.; Meijer, L. 3’-Substituted 7-halogenoindirubins, a new class of cell death inducing agents. J. Med. Chem. 2006, 49, 4638–4649. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Kritsanida, M.; Magiatis, P.; Gaboriaud, N.; Wang, Y.; Wu, J.; Buettner, R.; Yang, F.; Nam, S.; Skaltsounis, L.; et al. A novel 7-bromoindirubin with potent anticancer activity suppresses survival of human melanoma cells associated with inhibition of STAT3 and Akt signaling. Cancer Biol. Ther. 2012, 13, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Ndolo, K.M.; An, S.J.; Park, K.R.; Lee, H.J.; Bin Yoon, K.; Kim, Y.-C.; Han, S.-Y. Discovery of an indirubin derivative as a novel c-Met kinase inhibitor with in vitro anti-tumor effects. Biomol. Ther. 2019, 27, 216–221. [Google Scholar] [CrossRef]
- Vougogiannopoulou, K.; Ferandin, Y.; Bettayeb, K.; Myrianthopoulos, V.; Lozach, O.; Fan, Y.; Johnson, C.H.; Magiatis, P.; Skaltsounis, A.-L.; Mikros, E.; et al. Soluble 3’,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J. Med. Chem. 2008, 51, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Truong, G.N.; Van Vuong, T.; Van, T.N.; Manh, C.N.; Dao, C.T.; Thuy, T.D.T.; Van, C.L.; Khac, V.T. Synthesis of new indirubin derivatives and their in vitro anticancer activity. Chem. Pap. 2019, 73, 1083–1092. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Dung, D.T.M.; Dung, P.T.P.; Huong, L.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Tung, T.T.; et al. Design, synthesis and evaluation of novel indirubin-based N-hydroxybenzamides, N-hydroxypropenamides and N-hydroxyheptanamides as histone deacetylase inhibitors and antitumor agents. Bioorg. Med. Chem. Lett. 2020, 30, 127537. [Google Scholar] [CrossRef]
- Dan, N.T.; Quang, H.D.; Van Truong, V.; Nghi, D.H.; Cuong, N.M.; Cuong, T.D.; Toan, T.Q.; Bach, L.G.; Anh, N.H.T.; Mai, N.T.; et al. Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3β of new indirubin-3ʹ-oxime derivatives. Sci. Rep. 2020, 10, 11429. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.-E.; Huang, R.-Z.; Yao, G.-Y.; Li, J.-L.; Ye, M.-Y.; Wang, H.-S.; Liu, L. Synthesis and pharmacological evaluation of novel bisindole derivatives bearing oximes moiety: Identification of novel proapoptotic agents. Eur. J. Med. Chem. 2015, 95, 400–415. [Google Scholar] [CrossRef]
- Grosso, C.; Cardoso, A.L.; Lemos, A.; Varela, J.; Rodrigues, M.J.; Custódio, L.; Barreira, L.; e Melo, T.M.P. Novel approach to bis(indolyl)methanes: De novo synthesis of 1-hydroxyiminomethyl derivatives with anti-cancer properties. Eur. J. Med. Chem. 2015, 93, 9–15. [Google Scholar] [CrossRef]
- Dandu, R.; Zulli, A.L.; Bacon, E.R.; Underiner, T.; Robinson, C.; Chang, H.; Miknyoczki, S.; Grobelny, J.; Ruggeri, B.A.; Yang, S.; et al. Design and synthesis of dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole oximes as potent dual inhibitors of TIE-2 and VEGF-R2 receptor tyrosine kinases. Bioorg. Med. Chem. Lett. 2008, 18, 1916–1921. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Chen, Y.-R.; Tzeng, C.-C.; Liu, W.; Chou, C.-K.; Chiu, C.-C.; Chen, Y.-L. Discovery of indeno[1,2-b]quinoxaline derivatives as potential anticancer agents. Eur. J. Med. Chem. 2016, 108, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, C.; Álvarez, R.; Corchete, P.; López, J.L.; Pérez-Melero, C.; Peláez, R.; Medarde, M. Diarylmethyloxime and hydrazone derivatives with 5-indolyl moieties as potent inhibitors of tubulin polymerization. Bioorg. Med. Chem. 2008, 16, 5952–5961. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Benedetti, R.; Pérez-Rodríguez, S.; Nebbioso, A.; García-Rodríguez, J.; Carafa, V.; Stuhldreier, M.; Conte, M.; Rodríguez-Barrios, F.; Stunnenberg, H.G.; et al. Indole-Derived Psammaplin A analogues as epigenetic modulators with multiple inhibitory activities. J. Med. Chem. 2012, 55, 9467–9491. [Google Scholar] [CrossRef]
- Sharma, A.; Talimarada, D.; Yadav, U.P.; Singh, N.; Reddy, A.S.; Bag, D.; Biswas, K.; Baidya, A.; Borale, A.N.; Shinde, D.; et al. Design and synthesis of new tubulin polymerization inhibitors inspired from combretastatin A-4: An anticancer agent. ChemistrySelect 2020, 5, 11560–11572. [Google Scholar] [CrossRef]
- El-Wahab, H.A.A.A.; Ali, A.M.; Abdel-Rahman, H.M.; Qayed, W.S. Synthesis, biological evaluation, and molecular modeling studies of acetophenones-tethered 1,2,4-triazoles and their oximes as epidermal growth factor receptor inhibitors. Chem. Biol. Drug Des. 2022, 100, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Fadaly, W.A.; Elshaier, Y.A.; Hassanein, E.H.; Abdellatif, K.R. New 1,2,4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies. Bioorg. Chem. 2020, 98, 103752. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.-M.; El-Azab, A.S.; AlSaif, N.A.; Alanazi, M.M.; El-Gendy, M.A.; Obaidullah, A.J.; Alkahtani, H.M.; Almehizia, A.A.; Al-Suwaidan, I.A. Synthesis, anti-inflammatory, cytotoxic, and COX-1/2 inhibitory activities of cyclic imides bearing 3-benzenesulfonamide, oxime, and β-phenylalanine scaffolds: A molecular docking study. J. Enzym. Inhib. Med. Chem. 2020, 35, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, M.; Abuo-Rahma, G.E.-D.A.; Beshr, E.A.; Ali, T.F. New nitric oxide donating 1,2,4-triazole/oxime hybrids: Synthesis, investigation of anti-inflammatory, ulcerogenic liability and antiproliferative activities. Bioorg. Med. Chem. 2013, 21, 3839–3849. [Google Scholar] [CrossRef]
- Abuo-Rahma, G.E.-D.A.; Abdel-Aziz, M.; Beshr, E.A.; Ali, T.F. 1,2,4-Triazole/oxime hybrids as new strategy for nitric oxide donors: Synthesis, anti-inflammatory, ulcerogenicity and antiproliferative activities. Eur. J. Med. Chem. 2014, 71, 185–198. [Google Scholar] [CrossRef]
- Park, H.-J.; Lee, K.; Park, S.-J.; Ahn, B.; Lee, J.-C.; Cho, H.; Lee, K.-I. Identification of antitumor activity of pyrazole oxime ethers. Bioorg. Med. Chem. Lett. 2005, 15, 3307–3312. [Google Scholar] [CrossRef]
- Xiong, B.; Chen, S.; Zhu, P.; Huang, M.; Gao, W.; Zhu, R.; Qian, J.; Peng, Y.; Zhang, Y.; Dai, H.; et al. Design, synthesis, and biological evaluation of novel thiazolyl substituted bis-pyrazole oxime derivatives with potent antitumor activities by selectively inducing apoptosis and ROS in cancer cells. Med. Chem. 2019, 15, 743–754. [Google Scholar] [CrossRef]
- Dai, H.; Ge, S.; Guo, J.; Chen, S.; Huang, M.; Yang, J.; Sun, S.; Ling, Y.; Shi, Y. Development of novel bis-pyrazole derivatives as antitumor agents with potent apoptosis induction effects and DNA damage. Eur. J. Med. Chem. 2018, 143, 1066–1076. [Google Scholar] [CrossRef]
- Sun, J.; Liu, H.-Y.; Xu, R.-F.; Zhu, H.-L. Identification of nitroimidazole-oxime derivatives targeting the polo-box domain of polo-like kinase 1. Bioorg. Med. Chem. 2017, 25, 6581–6588. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-C.; Zhao, Y.-F.; Li, R.-D.; Xie, L.-J.; Yang, Y.-B.; Gong, P. Synthesis and biological evaluation of novel tricyclic oximino derivatives as antitumor agents. Chem. Res. Chin. Univ. 2008, 24, 47–53. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Connolly, P.J.; Xu, G.; Gaul, M.D.; Emanuel, S.L.; LaMontagne, K.R.; Greenberger, L.M. Synthesis and biological study of 4-aminopyrimidine-5-carboxaldehyde oximes as antiproliferative VEGFR-2 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 6063–6066. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Searle, L.L.; Hughes, T.V.; Beck, A.K.; Connolly, P.J.; Abad, M.C.; Neeper, M.P.; Struble, G.T.; Springer, B.A.; Emanuel, S.L.; et al. Discovery of novel 4-amino-6-arylaminopyrimidine-5-carbaldehyde oximes as dual inhibitors of EGFR and ErbB-2 protein tyrosine kinases. Bioorg. Med. Chem. Lett. 2008, 18, 3495–3499. [Google Scholar] [CrossRef] [PubMed]
- Gaul, M.D.; Xu, G.; Kirkpatrick, J.; Ott, H.; Baumann, C.A. 4-Amino-6-piperazin-1-yl-pyrimidine-5-carbaldehyde oximes as potent FLT-3 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 4861–4865. [Google Scholar] [CrossRef]
- Qiang, H.; Gu, W.; Huang, D.; Shi, W.; Qiu, Q.; Dai, Y.; Huang, W.; Qian, H. Design, synthesis and biological evaluation of 4-aminopyrimidine-5-cabaldehyde oximes as dual inhibitors of c-Met and VEGFR-2. Bioorg. Med. Chem. 2016, 24, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Li, Q.; Fu, Y.; Liu, F.; Huai, Z.; Xia, R.; Guo, T.; Chen, Y.; Jiang, S.; Tang, X. Process for preparation of quinazoline-containing 1,4-pentadiene-3-one oxime ether derivative, and application to treatment of human liver cancer. Patent CN108530427 A, 14 September 2018. [Google Scholar]
- Su, S.; Chen, M.; Li, Q.; Wang, Y.; Chen, S.; Sun, N.; Xie, C.; Huai, Z.; Huang, Y.; Xue, W. Novel penta-1,4-diene-3-one derivatives containing quinazoline and oxime ether fragments: Design, synthesis and bioactivity. Bioorg. Med. Chem. 2021, 32, 115999. [Google Scholar] [CrossRef]
- Hisham, M.; Youssif, B.G.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef]
- Huynh, M.T.; Anson, C.W.; Cavell, A.C.; Stahl, S.S.; Hammes-Schiffer, S. Quinone 1 e− and 2 e−/2 H+ reduction potentials: Identification and analysis of deviations from systematic scaling relationships. J. Am. Chem. Soc. 2016, 138, 15903–15910. [Google Scholar] [CrossRef] [PubMed]
- El-Najjar, N.; Gali-Muhtasib, H.; Ketola, R.A.; Vuorela, P.; Urtti, A.; Vuorela, H. The chemical and biological activities of quinones: Overview and implications in analytical detection. Phytochem. Rev. 2011, 10, 353–370. [Google Scholar] [CrossRef]
- O’Brien, P. Molecular mechanisms of quinone cytotoxicity. Chem-Biol. Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Lianeri, M.; Kozubski, W. Molecular effects of L-dopa therapy in Parkinson disease. Curr. Genom. 2014, 15, 11–17. [Google Scholar] [CrossRef]
- Feitosa, C.M.; de Sousa Silva, S.; Militao, G.G.C.; Pergentino de Sousa, D.; Rashed, K.; Lima, L.K.F. Benzoquinone mono oximes derivatives with anticancer activity. Int. J. Pharmacogn. 2020, 7, 369–375. [Google Scholar] [CrossRef]
- Kaur, K.; Sharma, R.; Singh, A.; Attri, S.; Arora, S.; Kaur, S.; Bedi, N. Pharmacological and analytical aspects of alkannin/shikonin and their derivatives: An update from 2008 to 2022. Chin. Herb. Med. 2022, 14, 511–527. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, S.; Li, S. Cancer therapeutic agents targeting hypoxia-inducible factor-1. Curr. Med. Chem. 2011, 18, 3168–3189. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, X.; Song, H.; Zhou, S.; Li, S. Synthesis and evaluation of novel alkannin and shikonin oxime derivatives as potent antitumor agents. Bioorg. Med. Chem. Lett. 2014, 24, 4304–4307. [Google Scholar] [CrossRef]
- Huang, G.; Dong, J.-Y.; Zhang, Q.-J.; Meng, Q.-Q.; Zhao, H.-R.; Zhu, B.-Q.; Li, S.-S. Discovery and synthesis of sulfur-containing 6-substituted 5,8-dimethoxy-1,4-naphthoquinone oxime derivatives as new and potential anti-MDR cancer agents. Eur. J. Med. Chem. 2019, 165, 160–171. [Google Scholar] [CrossRef]
- Huang, G.; Meng, Q.-Q.; Zhou, W.; Zhang, Q.-J.; Dong, J.-Y.; Li, S.-S. Design and synthesis of biotinylated dimethylation of alkannin oxime derivatives. Chin. Chem. Lett. 2017, 28, 453–457. [Google Scholar] [CrossRef]
- Huang, G.; Zhao, H.-R.; Meng, Q.-Q.; Zhang, Q.-J.; Dong, J.-Y.; Zhu, B.-Q.; Li, S.-S. Synthesis and biological evaluation of sulfur-containing shikonin oxime derivatives as potential antineoplastic agents. Eur. J. Med. Chem. 2018, 143, 166–181. [Google Scholar] [CrossRef]
- Huang, G.; Zhao, H.-R.; Zhou, W.; Dong, J.-Y.; Zhang, Q.-J.; Meng, Q.-Q.; Zhu, B.-Q.; Li, S.-S. 6-Substituted 1,4-naphthoquinone oxime derivatives (I): Synthesis and evaluation of their cytotoxic activity. Monatsh. Chem. 2017, 148, 1011–1023. [Google Scholar] [CrossRef]
- Macdonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Klaus, A.; Saga, Y.; Taketo, M.M.; Tzahor, E.; Birchmeier, W. Distinct roles of Wnt/β-catenin and Bmp signaling during early cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18531–18536. [Google Scholar] [CrossRef]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Soldi, R.; Horrigan, S.K.; Cholody, M.W.; Padia, J.; Sorna, V.; Bearss, J.; Gilcrease, G.; Bhalla, K.; Verma, A.; Vankayalapati, H.; et al. Design, synthesis, and biological evaluation of a series of anthracene-9,10-dione dioxime β-catenin pathway inhibitors. J. Med. Chem. 2015, 58, 5854–5862. [Google Scholar] [CrossRef] [PubMed]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody–drug conjugates: A review of approved drugs and their clinical level of evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Niu, X.; He, Q.; Liu, M.; Qiao, S.; Qi, R.-Q. Development, efficacy and side effects of antibody-drug conjugates for cancer therapy (Review). Mol. Clin. Oncol. 2023, 18, 47. [Google Scholar] [CrossRef]
- Samantasinghar, A.; Sunildutt, N.P.; Ahmed, F.; Soomro, A.M.; Salih, A.R.C.; Parihar, P.; Memon, F.H.; Kim, K.H.; Kang, I.S.; Choi, K.H. A comprehensive review of key factors affecting the efficacy of antibody drug conjugate. Biomed. Pharmacother. 2023, 161, 114408. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Zhou, S.; Ota, Y.; Harrington, M.; Miyagi, E.; Takagi, H.; Kuno, T.; Wright, J.D. Toxicity profiles of antibody drug conjugates for anticancer treatment: A systematic review and meta-analysis. JNCI Cancer Spectr. 2023, 7, pkad069. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Ali, S.; Mata, D.G.M.M.; Lohmann, A.E.; Blanchette, P.S. Antibody–drug conjugates in breast cancer: Ascent to destiny and beyond—A 2023 review. Curr. Oncol. 2023, 30, 6447–6461. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide–drug conjugates with different linkers for cancer therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Nakano, I.; Soe, C.Z.; Codd, R. Isolation of doxorubicin from a bacterial culture using immobilised metal ion affinity chromatography. RSC Adv. 2015, 5, 46437–46442. [Google Scholar] [CrossRef]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Momparler, R.L.; Karon, M.; E Siegel, S.; Avila, F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar]
- Fornari, F.A.; Randolph, J.K.; Yalowich, J.C.; Ritke, M.K.; Gewirtz, D.A. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol. Pharmacol. 1994, 45, 649–656. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced in vitro antitumor activity of GnRH-III-daunorubicin bioconjugates influenced by sequence modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Farkas, V.; Buday, L.; Szabó, Z.; Halmos, G.; Mező, G. Synthesis and in vitro biochemical evaluation of oxime bond-linked daunorubicin-GnRH-III conjugates developed for targeted drug delivery. Beilstein J. Org. Chem. 2018, 14, 756–771. [Google Scholar] [CrossRef]
- Orbán, E.; Mező, G.; Schlage, P.; Csík, G.; Kulić, Ž.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In vitro degradation and antitumor activity of oxime bond-linked daunorubicin–GnRH-III bioconjugates and DNA-binding properties of daunorubicin–amino acid metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef]
- Ranđelović, I.; Schuster, S.; Kapuvári, B.; Fossati, G.; Steinkühler, C.; Mező, G.; Tóvári, J. Improved in vivo anti-tumor and anti-metastatic effect of GnRH-III-daunorubicin analogs on colorectal and breast carcinoma bearing mice. Int. J. Mol. Sci. 2019, 20, 4763. [Google Scholar] [CrossRef] [PubMed]
- Dókus, L.E.; Lajkó, E.; Ranđelović, I.; Mező, D.; Schlosser, G.; Kőhidai, L.; Tóvári, J.; Mező, G. Phage display-based homing peptide-daunomycin conjugates for selective drug targeting to PANC-1 pancreatic cancer. Pharmaceutics 2020, 12, 576. [Google Scholar] [CrossRef] [PubMed]
- Feni, L.; Parente, S.; Robert, C.; Gazzola, S.; Arosio, D.; Piarulli, U.; Neundorf, I. Kiss and run: Promoting effective and targeted cellular uptake of a drug delivery vehicle composed of an integrin-targeting diketopiperazine peptidomimetic and a cell-penetrating peptide. Bioconjugate Chem. 2019, 30, 2011–2022. [Google Scholar] [CrossRef] [PubMed]
- Marelli, M.M.; Manea, M.; Moretti, R.M.; Marzagalli, M.; Limonta, P. Oxime bond-linked daunorubicin-GnRH-III bioconjugates exert antitumor activity in castration-resistant prostate cancer cells via the type I GnRH receptor. Int. J. Oncol. 2015, 46, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ding, J.; Xiao, C.; Li, L.; Zhuang, X.; Chen, X. Versatile preparation of intracellular-acidity-sensitive oxime-linked polysaccharide-doxorubicin conjugate for malignancy therapeutic. Biomaterials 2015, 54, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Fotie, J. The antiprotozoan potential of flavonoids. Pharmacogn. Rev. 2008, 2, 6–19. [Google Scholar]
- Chen, S.; Wang, X.; Cheng, Y.; Gao, H.; Chen, X. A Review of classification, biosynthesis, biological activities and potential applications of flavonoids. Molecules 2023, 28, 4982. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Li, J.; Zhao, X.; Liu, Q.; Song, S.-J. A comprehensive review: Biological activity, modification and synthetic methodologies of prenylated flavonoids. Phytochemistry 2021, 191, 112895. [Google Scholar] [CrossRef]
- Jucá, M.M.; Filho, F.M.S.C.; De Almeida, J.C.; Mesquita, D.D.S.; Barriga, J.R.D.M.; Dias, K.C.F.; Barbosa, T.M.; Vasconcelos, L.C.; Leal, L.K.A.M.; Ribeiro, J.E.; et al. Flavonoids: Biological activities and therapeutic potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef] [PubMed]
- González-Paramás, A.M.; Ayuda-Durán, B.; Martinez, S.; González-Manzano, S.; Santos-Buelga, C. The mechanisms behind the biological activity of flavonoids. Curr. Med. Chem. 2019, 26, 6976–6990. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Alfaro-Aco, R.; Zhang, C.; Russell, R.W.; Petry, S.; Polenova, T. Structural basis of protein condensation on microtubules underlying branching microtubule nucleation. Nat. Commun. 2023, 14, 3682. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.L.; Kavallaris, M.; McCarroll, J.A. Microtubules and their role in cellular stress in cancer. Front. Oncol. 2014, 4, 153. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-Y.; Wang, T.-C.; Lee, C.-H.; Chen, C.-S.; Wang, S.-H.; Lin, Y.-C.; Juang, S.-H. WTC-01, a novel synthetic oxime-flavone compound, destabilizes microtubules in human nasopharyngeal carcinoma cells in vitro and in vivo. Br. J. Pharmacol. 2015, 172, 4671–4683. [Google Scholar] [CrossRef]
- Liu, Z.; Wei, W.; Gan, C.; Huang, Y.; Liu, S.; Cui, J. Synthesis and cytotoxicity of E-hesperetin oximes against SGC-7901. Youji Huaxue 2013, 33, 1970–1974. [Google Scholar] [CrossRef]
- Kocyigit, A.; Koyuncu, I.; Dikilitas, M.; Bahadori, F.; Turkkan, B. Cytotoxic, genotoxic and apoptotic effects of naringenin-oxime relative to naringenin on normal and cancer cell lines. Asian Pac. J. Trop. Biomed. 2016, 6, 872–880. [Google Scholar] [CrossRef]
- Kozłowska, J.; Grela, E.; Baczyńska, D.; Grabowiecka, A.; Anioł, M. Novel O-alkyl derivatives of naringenin and their oximes with antimicrobial and anticancer activity. Molecules 2019, 24, 679. [Google Scholar] [CrossRef]
- Latif, A.D.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, Z.P.; Zupkó, I.; Hunyadi, A. Synthesis and in vitro antitumor activity of naringenin oxime and oxime ether derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef]
- Metodiewa, D.; Kochman, A.; Koceva-Chyła, A. Anticancer potential of N,N-diethylaminoethyl ethers of flavanone oximes: A comparison with mitoxantrone action on rat Yoshida sarcoma cells in vivo. Anticancer Res. 1999, 19, 1249–1254. [Google Scholar]
- Gutam, M.; Mokenapelli, S.; Yerrabelli, J.R.; Banerjee, S.; Roy, P.; Chitneni, P.R. Synthesis and cytotoxicity of novel (E)-2-phenylchroman-4-one-O-((1-substituted-1H-1,2,3-triazol-4-yl)methyl)oxime derivatives. Synth. Commun. 2020, 50, 1883–1891. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Qin, Y.-J.; Zhang, Y.-L.; Li, Y.-J.; Rao, B.; Zhang, Y.-Q.; Yang, M.-R.; Jiang, A.-Q.; Qi, J.-L.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of novel chalcone oxime derivatives as potential tubulin polymerization inhibitors. RSC Adv. 2014, 4, 32263–32275. [Google Scholar] [CrossRef]
- Bukhari, S.N.A. Synthesis and evaluation of new chalcones and oximes as anticancer agents. RSC Adv. 2022, 12, 10307–10320. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-W.; Chen, Y.-L.; Tseng, C.-H.; Liang, C.-C.; Yang, C.-N.; Yao, Y.-C.; Lu, P.-J.; Tzeng, C.-C. Discovery of 4-anilinofuro[2,3-b]quinoline derivatives as selective and orally active compounds against non-small-cell lung cancers. J. Med. Chem. 2011, 54, 4446–4461. [Google Scholar] [CrossRef]
- Chen, I.-L.; Chang, K.-M.; Miaw, C.-L.; Liao, C.-H.; Chen, J.-J.; Wang, T.-C. Synthesis, antiproliferative, and antiplatelet activities of oxime- and amide-containing quinolin-2(1H)-one derivatives. Bioorg. Med. Chem. 2007, 15, 6527–6534. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Leng, J.; Youssif, B.G.M.; Amjad, M.W.; Raja, M.A.G.; Hussain, M.A.; Hussain, Z.; Kazmi, S.N.; Bukhari, S.N.A. Synthesis and mechanistic studies of curcumin analog-based oximes as potential anticancer agents. Chem. Biol. Drug Des. 2017, 90, 443–449. [Google Scholar] [CrossRef]
- Qin, H.-L.; Leng, J.; Zhang, C.-P.; Jantan, I.; Amjad, M.W.; Sher, M.; Naeem-Ul-Hassan, M.; Hussain, M.A.; Bukhari, S.N.A. Synthesis of α,β-unsaturated carbonyl-based compounds, oxime and oxime ether analogs as potential anticancer agents for overcoming cancer multidrug resistance by modulation of efflux pumps in tumor cells. J. Med. Chem. 2016, 59, 3549–3561. [Google Scholar] [CrossRef]
- Zha, G.-F.; Qin, H.-L.; Youssif, B.G.; Amjad, M.W.; Raja, M.A.G.; Abdelazeem, A.H.; Bukhari, S.N.A. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur. J. Med. Chem. 2017, 135, 34–48. [Google Scholar] [CrossRef]
- Roayapalley, P.K.; Dimmock, J.R.; Contreras, L.; Balderrama, K.S.; Aguilera, R.J.; Sakagami, H.; Amano, S.; Sharma, R.K.; Das, U. Design, synthesis and tumour-selective toxicity of novel 1-[3-{3,5-Bis(benzylidene)-4-oxo-1-piperidino}-3-oxopropyl]-4-piperidone oximes and related quaternary ammonium salts. Molecules 2021, 26, 7132. [Google Scholar] [CrossRef]
- Petersen, A.; Konotop, G.; Hanafiah, N.; Hammershøj, P.; Raab, M.; Krämer, A.; Clausen, M. Strategies for improving the solubility and metabolic stability of griseofulvin analogues. Eur. J. Med. Chem. 2016, 116, 210–215. [Google Scholar] [CrossRef]
- Kumar, G.D.; Siva, B.; Bharathi, K.; Devi, A.; Kumar, P.P.; Anusha, K.; Lambhate, S.; Karunakar, T.; Tiwari, A.K.; Babu, K.S. Synthesis and biological evaluation of Schizandrin derivatives as tubulin polymerization inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127354. [Google Scholar] [CrossRef]
- Stancato, L.F.; Silverstein, A.M.; Owens-Grillo, J.K.; Chow, Y.-H.; Jove, R.; Pratt, W.B. The hsp90-binding antibiotic geldanamycin decreases Raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J. Biol. Chem. 1997, 272, 4013–4020. [Google Scholar] [CrossRef]
- Sharma, S.V.; Agatsuma, T.; Nakano, H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998, 16, 2639–2645. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Akinaga, S.; Soga, S.; Sullivan, W.; Stensgard, B.; Toft, D.; Neckers, L.M. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998, 3, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Soga, S.; Neckers, L.M.; Schulte, T.W.; Shiotsu, Y.; Akasaka, K.; Narumi, H.; Agatsuma, T.; Ikuina, Y.; Murakata, C.; Tamaoki, T.; et al. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 1999, 59, 2931–2938. [Google Scholar] [PubMed]
- Soga, S.; Sharma, S.V.; Shiotsu, Y.; Shimizu, M.; Tahara, H.; Yamaguchi, K.; Ikuina, Y.; Murakata, C.; Tamaoki, T.; Kurebayashi, J.; et al. Stereospecific antitumor activity of radicicol oxime derivatives. Cancer Chemother. Pharmacol. 2001, 48, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Agatsuma, T.; Ogawa, H.; Akasaka, K.; Asai, A.; Yamashita, Y.; Mizukami, T.; Akinaga, S.; Saitoh, Y. Halohydrin and oxime derivatives of radicicol: Synthesis and antitumor activities. Bioorg. Med. Chem. 2002, 10, 3445–3454. [Google Scholar] [CrossRef]
- Ikuina, Y.; Amishiro, N.; Miyata, M.; Narumi, H.; Ogawa, H.; Akiyama, T.; Shiotsu, Y.; Akinaga, S.; Murakata, C. Synthesis and antitumor activity of novel O-carbamoylmethyloxime derivatives of radicicol. J. Med. Chem. 2003, 46, 2534–2541. [Google Scholar] [CrossRef]
- Chakravarti, B.; Akhtar, T.; Rai, B.; Yadav, M.; Siddiqui, J.A.; Dwivedi, S.K.D.; Thakur, R.; Singh, A.K.; Singh, A.K.; Kumar, H.; et al. Thioaryl naphthylmethanone oxime ether analogs as novel anticancer agents. J. Med. Chem. 2014, 57, 8010–8025. [Google Scholar] [CrossRef]
- Arnott, J.; Martinkovich, S.; Planey, S.L.; Shah, D. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef]
- Sfogliarini, C.; Pepe, G.; Cesta, C.M.; Allegretti, M.; Locati, M.; Vegeto, E. The immune activity of selective estrogen receptor modulators is gene and macrophage subtype-specific yet converges on Il1b downregulation. Biomed. Pharmacother. 2023, 165, 115008. [Google Scholar] [CrossRef]
- Tienforti, D.; Castellini, C.; Di Giulio, F.; Totaro, M.; Dalmazio, G.; Spagnolo, L.; Muselli, M.; Corona, G.; Baroni, M.G.; Barbonetti, A. Selective modulation of estrogen receptor in obese men with androgen deficiency: A systematic review and meta-analysis. Andrology 2023, 11, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O. Estrogens and estrogen receptor modulators in cancer research and therapy. Cancers 2023, 15, 4318. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kim, H.-I.; An, J.-Y.; Lee, J.; Lee, N.-R.; Heo, J.; Kim, J.-E.; Yu, J.; Lee, Y.S.; Inn, K.-S.; et al. Identification of novel estrogen receptor (ER) agonists that have additional and complementary anti-cancer activities via ER-independent mechanism. Bioorg. Med. Chem. Lett. 2016, 26, 1844–1848. [Google Scholar] [CrossRef]
- Kim, H.-I.; Kim, T.; Kim, J.-E.; Lee, J.; Heo, J.; Lee, N.-R.; Kim, N.-J.; Inn, K.-S. NJK14013, a novel synthetic estrogen receptor-? agonist, exhibits estrogen receptor-independent, tumor cell-specific cytotoxicity. Int. J. Oncol. 2015, 47, 280–286. [Google Scholar] [CrossRef]
- Zuo, D.; Pang, L.; Shen, J.; Guan, Q.; Bai, Z.; Zhang, H.; Li, Y.; Lu, G.; Zhang, W.; Wu, Y. 5-(Furan-2-yl)-4-(3,4,5-trimethoxyphenyl)-3H-1,2-dithiol-3- one oxime (6f), a new synthetic compound, causes human fibrosarcoma HT-1080 cell apoptosis by disrupting tubulin polymerisation and inducing G2/M arrest. Int. J. Oncol. 2017, 50, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent progress on vascular endothelial growth factor receptor inhibitors with dual targeting capabilities for tumor therapy. J. Hematol. Oncol. 2022, 15, 89. [Google Scholar] [CrossRef]
- Gao, H.; Su, P.; Shi, Y.; Shen, X.; Zhang, Y.; Dong, J.; Zhang, J. Discovery of novel VEGFR-2 inhibitors. Part II: Biphenyl urea incorporated with salicylaldoxime. Eur. J. Med. Chem. 2015, 90, 232–240. [Google Scholar] [CrossRef]
- Jo, A.; Kwak, J.-H.; Woo, S.-Y.; Kim, B.-Y.; Son, Y.; Choi, H.-S.; Kim, J.; Kwon, M.; Cho, H.-R.; Eo, S.-K.; et al. Oxime derivative TFOBO promotes cell death by modulating reactive oxygen species and regulating NADPH oxidase activity in myeloid leukemia. Sci. Rep. 2022, 12, 7519. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Wei, W.; Wei, J. Synthesis and biological evaluation of phenylpropanoid derivatives. Med. Chem. Res. 2016, 25, 1074–1086. [Google Scholar] [CrossRef]
- Wipf, P.; Reeves, J.T.; Balachandran, R.; Day, B.W. Synthesis and biological evaluation of structurally highly modified analogues of the antimitotic natural product curacin a. J. Med. Chem. 2002, 45, 1901–1917. [Google Scholar] [CrossRef] [PubMed]
- Nikitjuka, A.; Shestakova, I.; Romanchikova, N.; Jirgensons, A. Synthesis and biological evaluation of aziridin-1-yl oxime-based vorinostat analogs as anticancer agents. Chem. Heterocycl. Compd. 2015, 51, 647–657. [Google Scholar] [CrossRef]
- Baud, M.G.J.; Leiser, T.; Haus, P.; Samlal, S.; Wong, A.C.; Wood, R.J.; Petrucci, V.; Gunaratnam, M.; Hughes, S.M.; Buluwela, L.; et al. Defining the mechanism of action and enzymatic selectivity of Psammaplin a against its epigenetic targets. J. Med. Chem. 2012, 55, 1731–1750. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, H.N.; Khera, R.A. Biological transformations of steroidal compounds: A review. Steroids 2012, 77, 1267–1290. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Qasim, M. A review of natural steroids and their applications. Int. J. Pharm. Sci. Res. 2013, 4, 520–531. [Google Scholar]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef]
- Bansal, R.; Suryan, A. A comprehensive review on steroidal bioconjugates as promising leads in drug discovery. ACS Bio Med. Chem. Au 2022, 2, 340–369. [Google Scholar] [CrossRef]
- Canário, C.; Matias, M.; Brito, V.; Santos, A.O.; Falcão, A.; Silvestre, S.; Alves, G. New estrone oxime derivatives: Synthesis, cytotoxic evaluation and docking studies. Molecules 2021, 26, 2687. [Google Scholar] [CrossRef]
- Jindal, D.P.; Chattopadhaya, R.; Guleria, S.; Gupta, R. Synthesis and antineoplastic activity of 2-alkylaminoethyl derivatives of various steroidal oximes. Eur. J. Med. Chem. 2003, 38, 1025–1034. [Google Scholar] [CrossRef]
- Rodríguez, J.; Nuñez, L.; Peixinho, S.; Jiménez, C. Isolation and synthesis of the first natural 6-hydroximino 4-en-3-one- steroids from the sponges Cinachyrella spp. Tetrahedron Lett. 1997, 38, 1833–1836. [Google Scholar] [CrossRef]
- Deive, N.; Rodríguez, J.; Jiménez, C. Synthesis of cytotoxic 6E-hydroximino-4-ene steroids: Structure/activity studies. J. Med. Chem. 2001, 44, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Huang, L.; Fan, L.; Zhou, A. A facile and efficient synthesis of some (6E)-hydroximino-4-en-3-one steroids, steroidal oximes from Cinachyrella spp. sponges. Steroids 2008, 73, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Berényi, Á.; Minorics, R.; Iványi, Z.; Ocsovszki, I.; Ducza, E.; Thole, H.; Messinger, J.; Wölfling, J.; Mótyán, G.; Mernyák, E.; et al. Synthesis and investigation of the anticancer effects of estrone-16-oxime ethers in vitro. Steroids 2013, 78, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.C.; Bansal, R. Synthesis and antiproliferative activity of some androstene oximes and their O-alkylated derivatives. Arch. Pharm. 2014, 347, 193–199. [Google Scholar] [CrossRef]
- Mernyák, E.; Fiser, G.; Szabó, J.; Bodnár, B.; Schneider, G.; Kovács, I.; Ocsovszki, I.; Zupkó, I.; Wölfling, J. Synthesis and in vitro antiproliferative evaluation of d-secooxime derivatives of 13β- and 13α-estrone. Steroids 2014, 89, 47–55. [Google Scholar] [CrossRef]
- Chen, S.-R.; Shen, F.-J.; Feng, G.-L.; Yuan, R.-X. Synthesis and anticancer activity of 4-azasteroidal-20-oxime derivatives. J. Chem. Res. 2015, 39, 527–530. [Google Scholar] [CrossRef]
- Ajduković, J.J.; Jakimov, D.S.; Rárová, L.; Strnad, M.; Dzichenka, Y.U.; Usanov, S.; Škorić, D.; Jovanović-Šanta, S.S.; Sakač, M.N. Novel alkylaminoethyl derivatives of androstane 3-oximes as anticancer candidates: Synthesis and evaluation of cytotoxic effects. RSC Adv. 2021, 11, 37449–37461. [Google Scholar] [CrossRef] [PubMed]
- Kolsi, L.E.; Leal, A.S.; Yli-Kauhaluoma, J.; Liby, K.T.; Moreira, V.M. Dehydroabietic oximes halt pancreatic cancer cell growth in the G1 phase through induction of p27 and downregulation of cyclin D1. Sci. Rep. 2018, 8, 15923. [Google Scholar] [CrossRef]
- Hou, Q.; He, C.; Lao, K.; Luo, G.; You, Q.; Xiang, H. Design and synthesis of novel steroidal imidazoles as dual inhibitors of AR/CYP17 for the treatment of prostate cancer. Steroids 2019, 150, 108384. [Google Scholar] [CrossRef]
- Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Paluszczak, J.; Szaefer, H.; Narożna, M.; Zaprutko, L.; Baer-Dubowska, W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HepG2 hepatoma cells. Bioorg. Chem. 2019, 93, 103326. [Google Scholar] [CrossRef]
- D’Yakonov, V.A.; Tuktarova, R.A.; Dzhemileva, L.U.; Ishmukhametova, S.R.; Yunusbaeva, M.M.; Dzhemilev, U.M. Catalytic cyclometallation in steroid chemistry V: Synthesis of hybrid molecules based on steroid oximes and (5Z,9Z)-tetradeca-5,9-dienedioic acid as potential anticancer agents. Steroids 2018, 138, 14–20. [Google Scholar] [CrossRef]
- Acharya, P.C.; Bansal, R. Synthesis of androstene oxime-nitrogen mustard bioconjugates as potent antineoplastic agents. Steroids 2017, 123, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Bu, M.; Cao, T.; Li, H.; Guo, M.; Yang, B.B.; Zeng, C.; Zhou, Y.; Zhang, N.; Hu, L. Synthesis and biological evaluation of novel steroidal 5α,8α-epidioxyandrost-6-ene-3β-ol-17-(O-phenylacetamide)oxime derivatives as potential anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Pavlíčková, V.; Jurášek, M.; Rimpelová, S.; Záruba, K.; Sedlák, D.; Šimková, M.; Kodr, D.; Staňková, E.; Fähnrich, J.; Rottnerová, Z.; et al. Oxime-based 19-nortestosterone–pheophorbide a conjugate: Bimodal controlled release concept for PDT. J. Mater. Chem. B 2019, 7, 5465–5477. [Google Scholar] [CrossRef] [PubMed]
- Alderden, R.A.; Hall, M.D.; Hambley, T.W.; Hou, G.-L.; Govind, N.; Xantheas, S.S.; Wang, X.-B.; Wu, Y.; Lai, R.Y.; Xiao, H.; et al. The discovery and development of cisplatin. J. Chem. Educ. 2006, 83, 728. [Google Scholar] [CrossRef]
- Kelland, L.R.; Sharp, S.Y.; O’neill, C.F.; Raynaud, F.I.; Beale, P.J.; Judson, I.R. Mini-review: Discovery and development of platinum complexes designed to circumvent cisplatin resistance. J. Inorg. Biochem. 1999, 77, 111–115. [Google Scholar] [CrossRef]
- Correa-Morales, J.E.; Giraldo-Moreno, S.; Mantilla-Manosalva, N.; Cuellar-Valencia, L.; Borja-Montes, O.F.; Bedoya-Muñoz, L.J.; Iriarte-Aristizábal, M.F.; Quintero-Muñoz, E.; Zuluaga-Liberato, A.M. Prevention and treatment of cisplatin-induced ototoxicity in adults: A systematic review. Clin. Otolaryngol. 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, A.; Syed, M.P.; Low, C.A.; Owonikoko, T.K. Cisplatin-Induced Ototoxicity: A concise review of the burden, prevention, and interception strategies. JCO Oncol. Pract. 2023, 19, 278–283. [Google Scholar] [CrossRef]
- Sharon, Y.; Motiei, M.; Tzror-Azankot, C.; Sadan, T.; Popovtzer, R.; Rosenbaum, E. A ‘golden’ alternative for prevention of cisplatin nephrotoxicity in bladder cancer. Cancer Nanotechnol. 2023, 14, 72. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Belsky, V.K.; Tudela, D. Unusual reactivity mode of coordinated oximes: Platinum(iv)-assisted ring closure by reaction with acetone. Inorg. Chem. 1996, 35, 510–513. [Google Scholar] [CrossRef]
- Kukushkin, V.Y.; Izotova, Y.A.; Tudela, D.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Organometallic and coordination complexes. Platinum(II) complexes of propanone oxime. Inorg. Synth. 2004, 34, 81–85. [Google Scholar]
- Kukushkin, V.Y.; Nishioka, T.; Tudela, D.; Isobe, K.; Kinoshita, I. Hydrogen-bonding patterns in oxime/oximato platinum(II) species providing the formation of one-dimensional chains, two-dimensional networks, and cages. Inorg. Chem. 1997, 36, 6157–6165. [Google Scholar] [CrossRef]
- Scaffidi-Domianello, Y.Y.; Legin, A.A.; Jakupec, M.A.; Arion, V.B.; Kukushkin, V.Y.; Galanski, M.S.; Keppler, B.K. Synthesis, characterization, and cytotoxic activity of novel potentially pH-Sensitive nonclassical platinum(II) complexes featuring 1,3-dihydroxyacetone oxime ligands. Inorg. Chem. 2011, 50, 10673–10681. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi-Domianello, Y.Y.; Legin, A.A.; Jakupec, M.A.; Roller, A.; Kukushkin, V.Y.; Galanski, M.S.; Keppler, B.K. Novel oximato-bridged platinum(II) di- and trimer(s): Synthetic, structural, and in vitro anticancer activity studies. Inorg. Chem. 2012, 51, 7153–7163. [Google Scholar] [CrossRef]
- Bartel, C.; Bytzek, A.K.; Scaffidi-Domianello, Y.Y.; Grabmann, G.; Jakupec, M.A.; Hartinger, C.G.; Galanski, M.S.; Keppler, B.K. Cellular accumulation and DNA interaction studies of cytotoxic trans-platinum anticancer compounds. JBIC J. Biol. Inorg. Chem. 2012, 17, 465–474. [Google Scholar] [CrossRef]
- Hyeraci, M.; Colalillo, M.; Labella, L.; Marchetti, F.; Samaritani, S.; Scalcon, V.; Rigobello, M.P.; Via, L.D. Platinum(II) complexes bearing triphenylphosphine and chelating oximes: Antiproliferative effect and biological profile in resistant cells. ChemMedChem 2020, 15, 1464–1472. [Google Scholar] [CrossRef]
- Eddings, D.; Barnes, C.; Gerasimchuk, N.; Durham, P.; Domasevich, K. First bivalent palladium and platinum cyanoximates: Synthesis, characterization, and biological activity. Inorg. Chem. 2004, 43, 3894–3909. [Google Scholar] [CrossRef]
- Sharma, S.; Chamola, G. Systematic review of organometallic half-sandwich complexes: Medicinal application and biological evaluation of anticancer activity. AIP Conf Proc. 2023, 2735, 030014. [Google Scholar] [CrossRef]
- Zhang, J.-J.; Xu, Q.-J.; Schmidt, C.; Abu el Maaty, M.A.; Song, J.; Yu, C.; Zhou, J.; Han, K.; Sun, H.; Casini, A.; et al. Elucidating the multimodal anticancer mechanism of an organometallic terpyridine platinum(II) N-heterocyclic carbene complex against triple-negative breast cancer in vitro and in vivo. J. Med. Chem. 2023, 66, 3995–4008. [Google Scholar] [CrossRef]
- Samiee, S.; Shiralinia, A.; Hoveizi, E.; Gable, R.W. A new family of oxime palladacycles mixed with unsymmetrical phosphorus ylides; synthesis, structural, cytotoxicity and catalytic activity studies. J. Organomet. Chem. 2019, 900, 120927. [Google Scholar] [CrossRef]
- Samiee, S.; Shiralinia, A.; Hoveizi, E.; Gable, R.W. Mono- and dinuclear oxime palladacycles bearing diphosphine ligands: An unusual coordination mode for dppe. Appl. Organomet. Chem. 2019, 33, e5098. [Google Scholar] [CrossRef]
- Palepu, N.R.; Adhikari, S.; Verma, A.K.; Shepherd, S.L.; Phillips, R.M.; Kaminsky, W.; Kollipara, M.R. Half-sandwich ruthenium, rhodium and iridium complexes featuring oxime ligands: Structural studies and preliminary investigation of in vitro and in vivo anti-tumour activities. Appl. Organomet. Chem. 2017, 31, e3640. [Google Scholar] [CrossRef]
- Benabdelouahab, Y.; Muñoz-Moreno, L.; Frik, M.; de la Cueva-Alique, I.; El Amrani, M.A.; Contel, M.; Bajo, A.M.; Cuenca, T.; Royo, E. Hydrogen bonding and anticancer properties of water-soluble chiral p-cymene RuII compounds with amino-oxime ligands. Eur. J. Inorg. Chem. 2015, 2015, 2295–2307. [Google Scholar] [CrossRef]
- El-Tabl, A.S.; El-Waheed, M.M.A.; Shakdofa, M.M.; El-Fadl, N.A.A. Synthesis, spectroscopic characterization and antitumor activity of new metal complexes of isonicotinoylhydrazide oxime. Main Group Chem. 2013, 12, 153–168. [Google Scholar] [CrossRef]
- El-Tabl, A.S.; El-Waheed, M.M.A.; Wahba, M.A.; El-Fadl, N.A.E.-H.A. Synthesis, characterization, and anticancer activity of new metal complexes derived from 2-hydroxy-3-((hydroxyimino)-4-oxopentan-2-ylidene)benzohydrazide. Bioinorg. Chem. Appl. 2015, 126023. [Google Scholar] [CrossRef]
- Samy, F.; Shebl, M. Synthesis, spectroscopic, biological, and theoretical studies of new complexes from (E)-3-(2-(5,6-diphenyl-1,2,4-triazin-3-yl)hydrazono)butan-2-one oxime. Appl. Organomet. Chem. 2020, 34, e5502. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fotie, J.; Matherne, C.M.; Mather, J.B.; Wroblewski, J.E.; Johnson, K.; Boudreaux, L.G.; Perez, A.A. The Fundamental Role of Oxime and Oxime Ether Moieties in Improving the Physicochemical and Anticancer Properties of Structurally Diverse Scaffolds. Int. J. Mol. Sci. 2023, 24, 16854. https://doi.org/10.3390/ijms242316854
Fotie J, Matherne CM, Mather JB, Wroblewski JE, Johnson K, Boudreaux LG, Perez AA. The Fundamental Role of Oxime and Oxime Ether Moieties in Improving the Physicochemical and Anticancer Properties of Structurally Diverse Scaffolds. International Journal of Molecular Sciences. 2023; 24(23):16854. https://doi.org/10.3390/ijms242316854
Chicago/Turabian StyleFotie, Jean, Caitlyn M. Matherne, Jasmine B. Mather, Jordan E. Wroblewski, Khaitlynn Johnson, Lara G. Boudreaux, and Alba A. Perez. 2023. "The Fundamental Role of Oxime and Oxime Ether Moieties in Improving the Physicochemical and Anticancer Properties of Structurally Diverse Scaffolds" International Journal of Molecular Sciences 24, no. 23: 16854. https://doi.org/10.3390/ijms242316854