
Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy
 ,
,  , ,
, ,  , and
, and 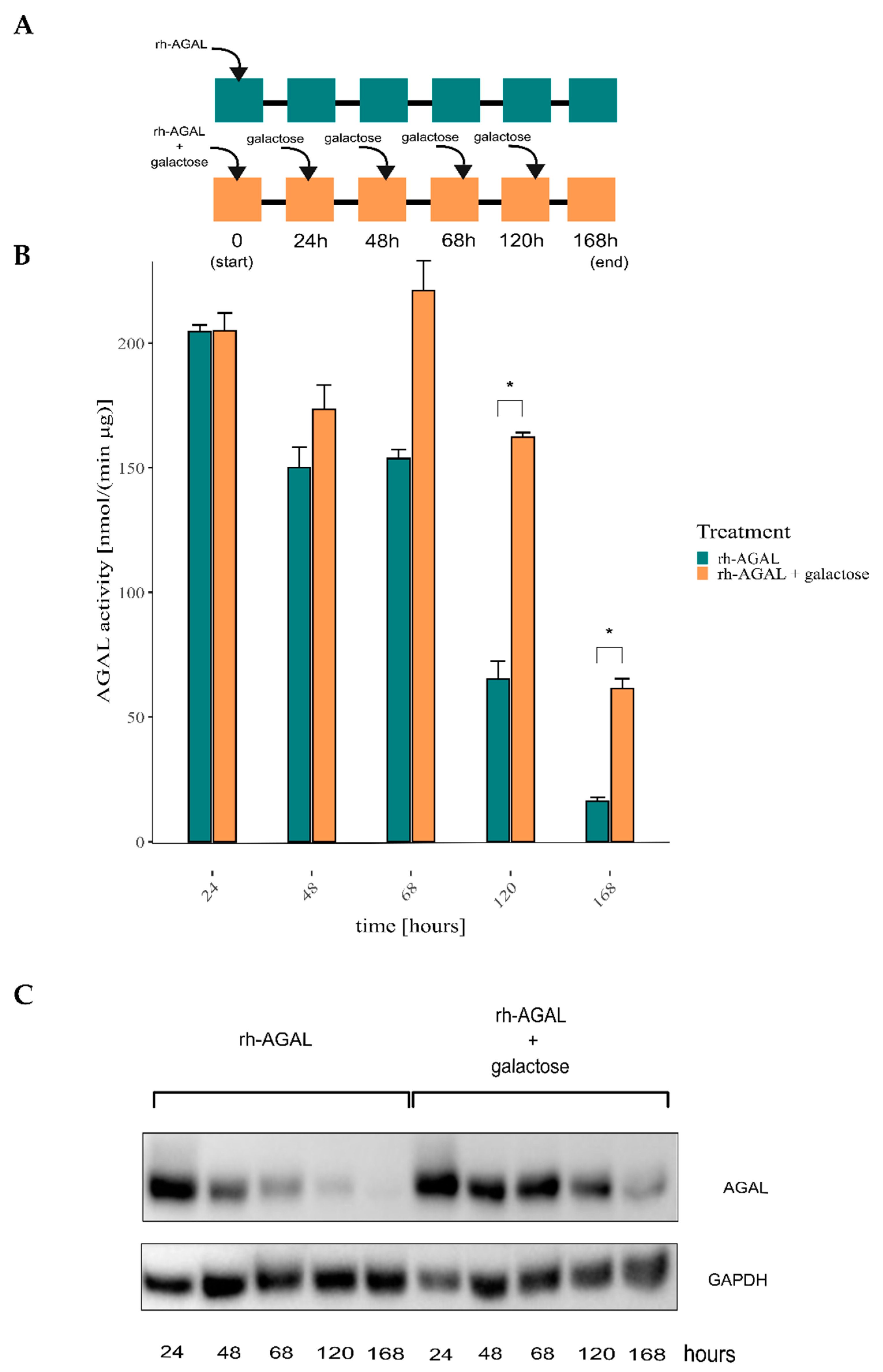{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. ERT May Benefit from a Combination with Galactose Treatment
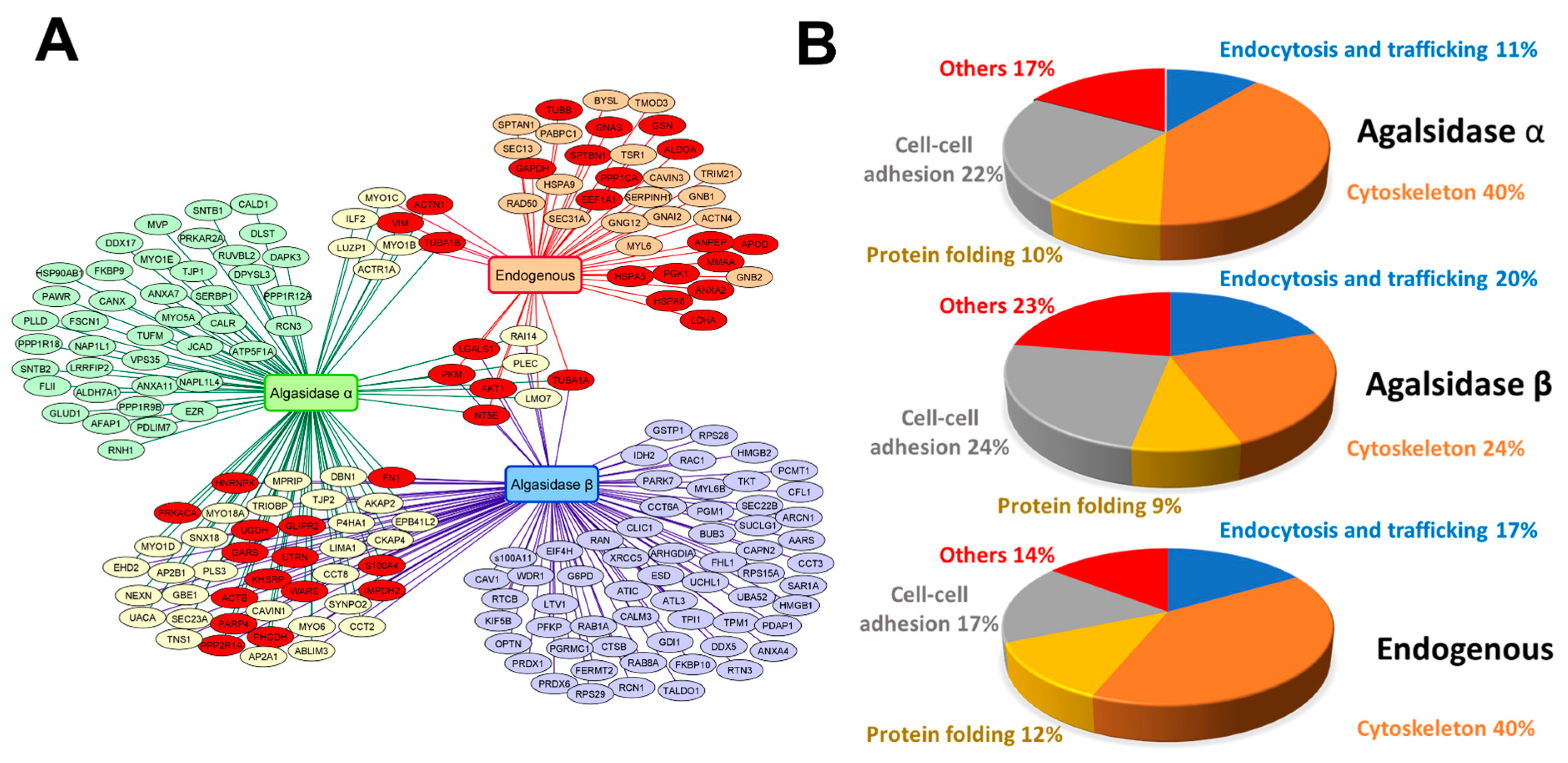
2.2. Identification of AGAL Interactors
3. Materials and Methods
3.1. Cell Cultures
3.2. Enzymatic Activity Assays
3.3. Purification, Identification, and Functional Analysis of AGAL Interactomes
3.4. Miscellaneous
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGAL | α-galactosidase |
| CHO | Chinese hamster ovary cells |
| DGJ | 1-deoxygalactonojirimycin |
| ERT | Enzyme Replacement Therapy |
| FBS | Fetal Bovine Serum |
| FD | Fabry Disease |
| FDA | Food and Drug Administration |
| Gb3 | Globotriaosylceramide |
| IF | FD patient-derived immortalized fibroblasts |
| IF-NULL | FD patient-derived immortalized fibroblasts stably transfected with the empty vector |
| IF-GLA | FD patient-derived immortalized fibroblasts stably transfected with a plasmid encoding wt-GLA |
| PC | Pharmacological Chaperone |
| PCT | Pharmacological Chaperone Therapy |
| rh-AGAL | Recombinant human—α-galactosidase |
References
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson–Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Hughes, D.A. Fabry Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Michaud, M.; Mauhin, W.; Belmatoug, N.; Garnotel, R.; Bedreddine, N.; Catros, F.; Ancellin, S.; Lidove, O.; Gaches, F. When and How to Diagnose Fabry Disease in Clinical Pratice. Am. J. Med. Sci. 2020, 360, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Vittoria, M.; Andreotti, G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinVar—Fabry. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?gr=1&term=GLA%5Bgene%5D+AND+Fabry (accessed on 31 October 2022).
- Galafold Amenability Table. Available online: https://galafoldamenabilitytable.com/reference (accessed on 15 November 2022).
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Mellett, N.; Hein, L.K.; Brooks, D.A.; Meikle, P.J. Absence of α-Galactosidase Cross-Correction in Fabry Heterozygote Cultured Skin Fibroblasts. Mol. Genet. Metab. 2015, 114, 268–273. [Google Scholar] [CrossRef]
- Matalonga, L.; Arias, Á.; Tort, F.; Ferrer-Cortés, X.; Garcia-Villoria, J.; Coll, M.J.; Gort, L.; Ribes, A. Effect of Readthrough Treatment in Fibroblasts of Patients Affected by Lysosomal Diseases Caused by Premature Termination Codons. Neurotherapeutics 2015, 12, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the Treatment of Fabry Disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Felis, A.; Whitlow, M.; Kraus, A.; Warnock, D.G.; Wallace, E. Current and Investigational Therapeutics for Fabry Disease. Kidney Int. Rep. 2019, 5, 407–413. [Google Scholar] [CrossRef]
- Lombardi, S.; Ferrarese, M.; Marchi, S.; Pinton, P.; Pinotti, M.; Bernardi, F.; Branchini, A. Translational Readthrough of GLA Nonsense Mutations Suggests Dominant-Negative Effects Exerted by the Interaction of Wild-Type and Missense Variants. RNA Biol. 2019, 17, 254–263. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Mele, B.H.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a Mutant α-Galactosidase Gene Product for the Late-Onset Cardiac Form of Fabry Disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef]
- Okumiya, T.; Ishii, S.; Takenaka, T.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. Galactose Stabilizes Various Missense Mutants of α-Galactosidase in Fabry Disease. Biochem. Biophys. Res. Commun. 1995, 214, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Chimenti, C.; Ricci, R.; Natale, L.; Russo, M.A.; Pieroni, M.; Eng, C.M.; Desnick, R.J. Improvement in Cardiac Function in the Cardiac Variant of Fabry’s Disease with Galactose-Infusion Therapy. N. Engl. J. Med. 2001, 345, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated Transport and Maturation of Lysosomal α-Galactosidase A in Fabry Lymphoblasts by an Enzyme Inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Jovanovic, A.; Herrmann, K.; Vardarli, I. Chaperone Therapy in Fabry Disease. Int. J. Mol. Sci. 2022, 23, 1887. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef]
- Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; et al. A Biochemical and Pharmacological Comparison of Enzyme Replacement Therapies for the Glycolipid Storage Disorder Fabry Disease. Glycobiology 2003, 13, 305–313. [Google Scholar] [CrossRef]
- Kizhner, T.; Azulay, Y.; Hainrichson, M.; Tekoah, Y.; Arvatz, G.; Shulman, A.; Ruderfer, I.; Aviezer, D.; Shaaltiel, Y. Characterization of a Chemically Modified Plant Cell Culture Expressed Human α-Galactosidase-A Enzyme for Treatment of Fabry Disease. Mol. Genet. Metab. 2015, 114, 259–267. [Google Scholar] [CrossRef]
- Schiffmann, R.; Goker-Alpan, O.; Holida, M.; Giraldo, P.; Barisoni, L.; Colvin, R.B.; Jennette, C.J.; Maegawa, G.; Boyadjiev, S.A.; Gonzalez, D.; et al. Pegunigalsidase Alfa, a Novel PEGylated Enzyme Replacement Therapy for Fabry Disease, Provides Sustained Plasma Concentrations and Favorable Pharmacodynamics: A 1-Year Phase 1/2 Clinical Trial. J. Inherit. Metab. Dis. 2019, 42, 534–544. [Google Scholar] [CrossRef]
- Holida, M.D.; Bernat, J.; Longo, N.; Goker-Alpan, O.; Wallace, E.; Schiffmann, R.; Deegan, P.; Khan, N.; Tøndel, C.; Eyskens, F.; et al. Once Every 4 Weeks—2 Mg/Kg of Pegunigalsidase Alfa for Treating Fabry Disease Preliminary Results of a Phase 3 Study. Mol. Genet. Metab. 2019, 126, S73. [Google Scholar] [CrossRef]
- Xu, S.; Lun, Y.; Brignol, N.; Hamler, R.; Schilling, A.; Frascella, M.; Sullivan, S.; Boyd, R.E.; Chang, K.; Soska, R.; et al. Coformulation of a Novel Human α-Galactosidase a with the Pharmacological Chaperone At1001 Leads to Improved Substrate Reduction in Fabry Mice. Mol. Ther. 2015, 23, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Suzuki, Y.; Fan, J.Q. Role of Ser-65 in the Activity of α-Galactosidase A: Characterization of a Point Mutation (S65T) Detected in a Patient with Fabry Disease. Arch. Biochem. Biophys. 2000, 377, 228–233. [Google Scholar] [CrossRef]
- Guce, A.I.; Clark, N.E.; Rogich, J.J.; Garman, S.C. The Molecular Basis of Pharmacological Chaperoning in Human α-Galactosidase. Chem. Biol. 2011, 18, 1521–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietra, P.J.G.M.; Tager, J.M.; Borst, P. Detection and Properties of an Acid α-Galactosidase (Ceramidetrihexosidase) in Normal Human Urine. BBA Gen. Subj. 1972, 279, 436–445. [Google Scholar] [CrossRef]
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for Protein Stabilizing Drugs with Thermal Shift Assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Lemansky, P.; Bishop, D.F.; Desnick, R.J.; Hasilik, A.; von Figura, K. Synthesis and Processing of α-Galactosidase A in Human Fibroblasts. Evidence for Different Mutations in Fabry Disease. J. Biol. Chem. 1987, 262, 2062–2065. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Liguori, L.; Allocca, M.; Bosso, A.; Andreotti, G.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Hay Mele, B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 5105. [Google Scholar] [CrossRef]
- EMA. Fabrazyme: EPAR—Product Information; EMA (European Medicines Agency): London, UK, 2022. [Google Scholar]
- DrugBank—Galactose. Available online: https://go.drugbank.com/drugs/DB11735 (accessed on 2 November 2022).
- Brasil, S.; Allocca, M.; Magrinho, S.C.M.; Santos, I.; Raposo, M.; Francisco, R.; Pascoal, C.; Martins, T.; Videira, P.A.; Pereira, F.; et al. Systematic Review: Drug Repositioning for Congenital Disorders of Glycosylation (CDG). Int. J. Mol. Sci. 2022, 23, 8725. [Google Scholar] [CrossRef]
- Clinical Trials GOV. Available online: https://clinicaltrials.gov/ (accessed on 16 November 2022).
- Perales-Clemente, E.; Liedtke, K.; Studinski, A.; Radenkovic, S.; Gavrilov, D.; Oglesbee, D.; Matern, D.; Rinaldo, P.; Tortorelli, S.; Morava, E.; et al. A New D-Galactose Treatment Monitoring Index for PGM1-CDG. J. Inherit. Metab. Dis. 2021, 44, 1263–1271. [Google Scholar] [CrossRef]
- Coss-Bu, J.A.; Sunehag, A.L.; Haymond, M.W. Contribution of Galactose and Fructose to Glucose Homeostasis. Metabolism. 2009, 58, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Warnock, D.G.; Bichet, D.G.; Holida, M.; Goker-Alpan, O.; Nicholls, K.; Thomas, M.; Eyskens, F.; Shankar, S.; Adera, M.; Sitaraman, S.; et al. Oral Migalastat HCl Leads to Greater Systemic Exposure and Tissue Levels of Active α-Galactosidase A in Fabry Patients When Co-Administered with Infused Agalsidase. PLoS ONE 2015, 10, e0134341. [Google Scholar] [CrossRef]
- Park, S.Y.; Guo, X. Adaptor Protein Complexes and Intracellular Transport. Biosci. Rep. 2014, 34, 381–390. [Google Scholar] [CrossRef]
- Park, J.; Kim, Y.; Lee, S.; Park, J.J.; Park, Z.Y.; Sun, W.; Kim, H.; Chang, S. SNX18 Shares a Redundant Role with SNX9 and Modulates Endocytic Trafficking at the Plasma Membrane. J. Cell Sci. 2010, 123, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, O.; Tillu, V.A.; Ariotti, N.; Parton, R.G.; Collins, B.M. Cavin Family Proteins and the Assembly of Caveolae. J. Cell Sci. 2015, 128, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Morén, B.; Shah, C.; Howes, M.T.; Schieber, N.L.; McMahon, H.T.; Parton, R.G.; Daumke, O.; Lundmark, R. EHD2 Regulates Caveolar Dynamics via ATP-Driven Targeting and Oligomerization. Mol. Biol. Cell 2012, 23, 1316–1329. [Google Scholar] [CrossRef]
- Lajoie, P.; Nabi, I.R. Lipid Rafts, Caveolae, and Their Endocytosis. Int. Rev. Cell Mol. Biol. 2010, 282, 135–163. [Google Scholar] [CrossRef]
- Prieto-Sánchez, R.M.; Berenjeno, I.M.; Bustelo, X.R. Involvement of the Rho/Rac Family Member RhoG in Caveolar Endocytosis. Oncogene 2006, 25, 2961–2973. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Lu, W.; Zou, S.; Wang, H.; Jiang, Y.; Zhang, X.; Li, P.; Songyang, Z.; Wang, L.; Wang, J.; et al. Rho GDP-Dissociation Inhibitor α Is a Potential Prognostic Biomarker and Controls Telomere Regulation in Colorectal Cancer. Cancer Sci. 2017, 108, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- Heo, S.H.; Kang, E.; Kim, Y.M.; Go, H.; Kim, K.Y.; Jung, J.Y.; Kang, M.; Kim, G.H.; Kim, J.M.; Choi, I.H.; et al. Fabry Disease: Characterisation of the Plasma Proteome Pre- and Post-Enzyme Replacement Therapy. J. Med. Genet. 2017, 54, 771–780. [Google Scholar] [CrossRef] [Green Version]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients with Fabry Disease: A Critical Link to Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [Green Version]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of Proteomic Profiles in PBMC Isolated from Patients with Fabry Disease: Preliminary Findings. Mol. Biosyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A Small Protein with Major Functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef]
- Li, J.; Lu, Q.; Zhang, M. Structural Basis of Cargo Recognition by Unconventional Myosins in Cellular Trafficking. Traffic 2016, 17, 822–838. [Google Scholar] [CrossRef] [Green Version]
- Brandstaetter, H.; Kishi-Itakura, C.; Tumbarello, D.A.; Manstein, D.J.; Buss, F. Loss of Functional MYO1C/Myosin 1c, a Motor Protein Involved in Lipid Raft Trafficking, Disrupts Autophagosome-Lysosome Fusion. Autophagy 2014, 10, 2310–2323. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Huang, J.; He, X.; Hu, M.; Su, S.; Liu, P. Myosin 1b Participated in the Modulation of Hypoxia/Reoxygenation-Caused H9c2 Cell Apoptosis and Autophagy. Anal. Cell. Pathol. 2022, 2022, 5187304. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in Lysosomal Storage Disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jiang, X.; Deng, Q.; Gao, Z.; Tang, X.; Fu, R.; Hu, J.; Li, Y.; Li, L.; Gao, N. Downregulation of MYO1C Mediated by Cepharanthine Inhibits Autophagosome-Lysosome Fusion through Blockade of the F-Actin Network. J. Exp. Clin. Cancer Res. 2019, 38, 457. [Google Scholar] [CrossRef] [Green Version]
- Chibalina, M.V.; Roberts, R.C.; Arden, S.D.; Kendrick-Jones, J.; Buss, F. Rab8-Optineurin-Myosin VI: Analysis of Interactions and Functions in the Secretory Pathway. Methods Enzymol. 2008, 438, 11–24. [Google Scholar]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis Regulators Modulate Proteasomal Activity and Gene Expression to Attenuate Multiple Phenotypes in Fabry Disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, M.; Hay Mele, B.; Allocca, M.; Liguori, L.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Andreotti, G. Curcumin Has Beneficial Effects on Lysosomal Alpha-Galactosidase: Potential Implications for the Cure of Fabry Disease. Int. J. Mol. Sci. 2023, 24, 1095. [Google Scholar] [CrossRef]
- Chen, S.; Yang, S.; Wang, M.; Chen, J.; Huang, S.; Wei, Z.; Cheng, Z.; Wang, H.; Long, M.; Li, P. Curcumin Inhibits Zearalenone-Induced Apoptosis and Oxidative Stress in Leydig Cells via Modulation of the PTEN/Nrf2/Bip Signaling Pathway. Food Chem. Toxicol. 2020, 141, 111385. [Google Scholar] [CrossRef]
- Lee, Y.Q.; Rajadurai, P.; Abas, F.; Othman, I.; Naidu, R. Proteomic Analysis on Anti-Proliferative and Apoptosis Effects of Curcumin Analog, 1,5-Bis(4-Hydroxy-3-Methyoxyphenyl)-1,4-Pentadiene-3-One-Treated Human Glioblastoma and Neuroblastoma Cells. Front. Mol. Biosci. 2021, 8, 645856. [Google Scholar] [CrossRef]
- Hu, C.; Li, M.; Guo, T.; Wang, S.; Huang, W.; Yang, K.; Liao, Z.; Wang, J.; Zhang, F.; Wang, H. Anti-Metastasis Activity of Curcumin against Breast Cancer via the Inhibition of Stem Cell-like Properties and EMT. Phytomedicine 2018, 58, 152740. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.; Zhao, Y.; Li, Y.; Yin, L. Investigating the Therapeutic Potential and Mechanism of Curcumin in Breast Cancer Based on RNA Sequencing and Bioinformatics Analysis. Breast Cancer 2017, 25, 206–212. [Google Scholar] [CrossRef]
- Lakshmanan, A.; Akasov, R.A.; Sholina, N.V.; Demina, P.A.; Generalova, A.N.; Gangadharan, A.; Sardar, D.K.; Lankamsetty, K.B.; Khochenkov, D.A.; Khaydukov, E.V.; et al. Nanocurcumin-Loaded UCNPs for Cancer Theranostics: Physicochemical Properties, in Vitro Toxicity, and in Vivo Imaging Studies. Nanomaterials 2021, 11, 2234. [Google Scholar] [CrossRef]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early Markers of Fabry Disease Revealed by Proteomics. Mol. Biosyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef]
- Wu, W.; Panté, N. Vimentin Plays a Role in the Release of the Influenza A Viral Genome from Endosomes. Virology 2016, 497, 41–52. [Google Scholar] [CrossRef]
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Rab7a Regulates Cell Migration through Rac1 and Vimentin. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 367–381. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of Autophagy as a Common Mechanism in Lysosomal Storage Diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug Repurposing from the Perspective of Pharmaceutical Companies. Br. J. Pharmacol. 2017, 175, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Nosengo, N. Can You Teach Old Drugs New Tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Wouters, O.J.; McKee, M.; Luyten, J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. JAMA J. Am. Med. Assoc. 2020, 323, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhang, J.; Lin, C.; Zhang, L.; Liu, B.; Ouyang, L. Targeting Autophagy-Related Protein Kinases for Potential Therapeutic Purpose. Acta Pharm. Sin. B 2019, 10, 569–581. [Google Scholar] [CrossRef]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective Autophagy of Intracellular Organelles: Recent Research Advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Li, J.; Yang, K.; Cao, D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull. Cancer 2021, 108, 304–322. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Toyota, A.; Goto, M.; Miyamoto, M.; Nagashima, Y.; Iwasaki, S.; Komatsu, T.; Momose, T.; Yoshida, K.; Tsukada, T.; Matsufuji, T.; et al. Novel Protein Kinase CAMP-Activated Catalytic Subunit Alpha (PRKACA) Inhibitor Shows Anti-Tumor Activity in a Fibrolamellar Hepatocellular Carcinoma Model. Biochem. Biophys. Res. Commun. 2022, 621, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Liu, S.; Qin, Z. Extracellular S100A4 as a Key Player in Fibrotic Diseases. J. Cell. Mol. Med. 2020, 24, 5973–5983. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, G.; Citro, V.; De Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry Disease with Pharmacological Chaperones: From in Silico Predictions to in Vitro Tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Scieuzo, C.; Salvia, R.; Franco, A.; Pezzi, M.; Cozzolino, F.; Chicca, M.; Scapoli, C.; Vogel, H.; Monti, M.; Ferracini, C.; et al. An Integrated Transcriptomic and Proteomic Approach to Identify the Main Torymus Sinensis Venom Components. Sci. Rep. 2021, 11, 5032. [Google Scholar] [CrossRef]
- Cozzolino, F.; Vezzoli, E.; Cheroni, C.; Besusso, D.; Conforti, P.; Valenza, M.; Iacobucci, I.; Monaco, V.; Birolini, G.; Bombaci, M.; et al. ADAM10 Hyperactivation Acts on Piccolo to Deplete Synaptic Vesicle Stores in Huntington’s Disease. Hum. Mol. Genet. 2021, 30, 1175–1187. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef]
- Iacobucci, I.; Monaco, V.; Canè, L.; Bibbò, F.; Cioffi, V.; Cozzolino, F.; Guarino, A.; Zollo, M.; Monti, M. Spike S1 Domain Interactome in Non-Pulmonary Systems: A Role beyond the Receptor Recognition. Front. Mol. Biosci. 2022, 9, 975570. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Peña-García, J.; Den-Haan, H.; Pérez-Sánchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS One 2016, 11, e0165463. [Google Scholar] [CrossRef] [Green Version]
- R Core Team R Core Team. R A Lang. Environ. Stat. Comput; R Found. Stat. Comput.: Vienna, Austria, 2021; Available online: http//www.r-project.org (accessed on 2 May 2022).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef] [Green Version]
- Pantoom, S.; Hules, L.; Schöll, C.; Petrosyan, A.; Monticelli, M.; Pospech, J.; Cubellis, M.V.; Hermann, A.; Lukas, J. Mechanistic Insight into the Mode of Action of Acid β-Glucosidase Enhancer Ambroxol. Int. J. Mol. Sci. 2022, 23, 3536. [Google Scholar] [CrossRef] [PubMed]
- Jamalpoor, A.; Gelder, C.A.; Yousef Yengej, F.A.; Zaal, E.A.; Berlingerio, S.P.; Veys, K.R.; Pou Casellas, C.; Voskuil, K.; Essa, K.; Ammerlaan, C.M.; et al. Cysteamine–Bicalutamide Combination Therapy Corrects Proximal Tubule Phenotype in Cystinosis. EMBO Mol. Med. 2021, 13, e13067. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, C.; Witt, M.; Rolfs, A.; Antipova, V.; Wree, A. Gender-specific Effects of Two Treatment Strategies in a Mouse Model of Niemann-pick Disease Type C1. Int. J. Mol. Sci. 2021, 22, 2539. [Google Scholar] [CrossRef]
- Guiraud, S.; Davies, K.E. Pharmacological Advances for Treatment in Duchenne Muscular Dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-Tezacaftor-Ivacaftor: The First Triple-Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulating Therapy. J. Pediatr. Pharmacol. Ther. 2020, 25, 192–197. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Khanna, R.; Schilling, A.; Flanagan, J.J.; Pellegrino, L.J.; Brignol, N.; Lun, Y.; Guillen, D.; Ranes, B.E.; Frascella, M.; et al. Co-Administration with the Pharmacological Chaperone AT1001 Increases Recombinant Human α-Galactosidase a Tissue Uptake and Improves Substrate Reduction in Fabry Mice. Mol. Ther. 2012, 20, 717–726. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobucci, I.; Hay Mele, B.; Cozzolino, F.; Monaco, V.; Cimmaruta, C.; Monti, M.; Andreotti, G.; Monticelli, M. Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy. Int. J. Mol. Sci. 2023, 24, 4548. https://doi.org/10.3390/ijms24054548
Iacobucci I, Hay Mele B, Cozzolino F, Monaco V, Cimmaruta C, Monti M, Andreotti G, Monticelli M. Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy. International Journal of Molecular Sciences. 2023; 24(5):4548. https://doi.org/10.3390/ijms24054548
Chicago/Turabian StyleIacobucci, Ilaria, Bruno Hay Mele, Flora Cozzolino, Vittoria Monaco, Chiara Cimmaruta, Maria Monti, Giuseppina Andreotti, and Maria Monticelli. 2023. "Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy" International Journal of Molecular Sciences 24, no. 5: 4548. https://doi.org/10.3390/ijms24054548