
Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones

 , , , , ,
, , , , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
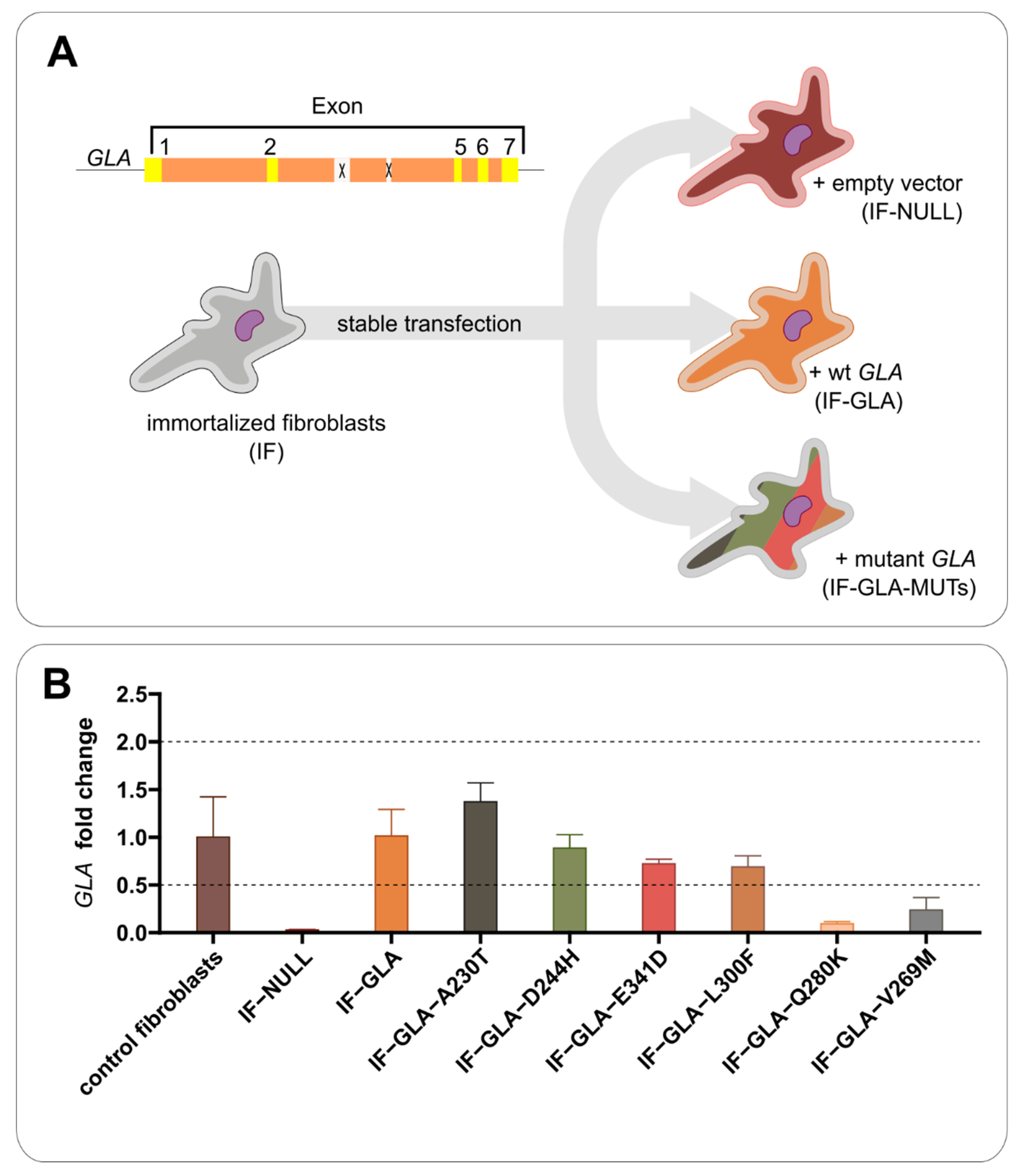
2.1. Establishment of Versatile Cell Models That Do Not Overexpress AGAL
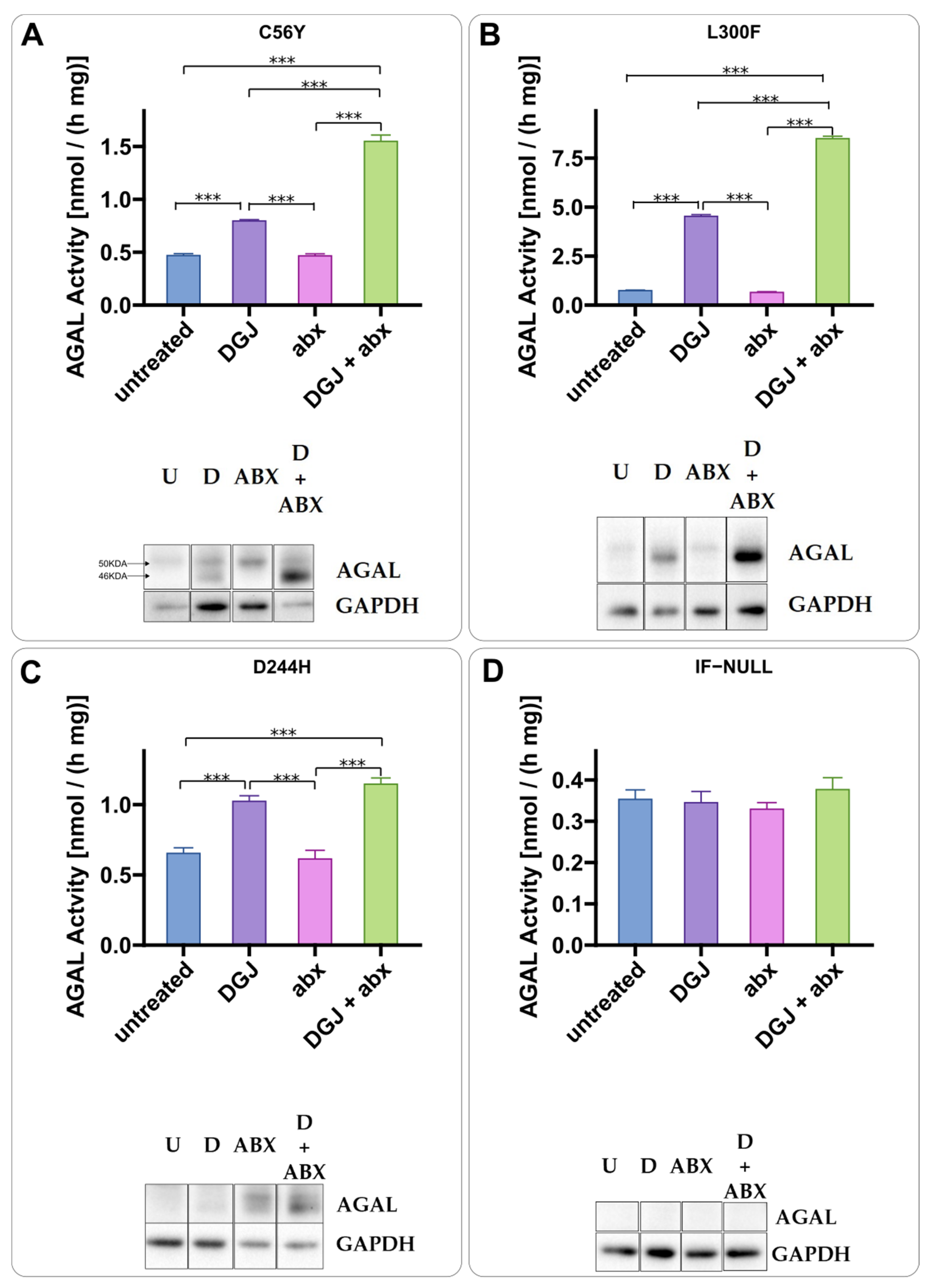
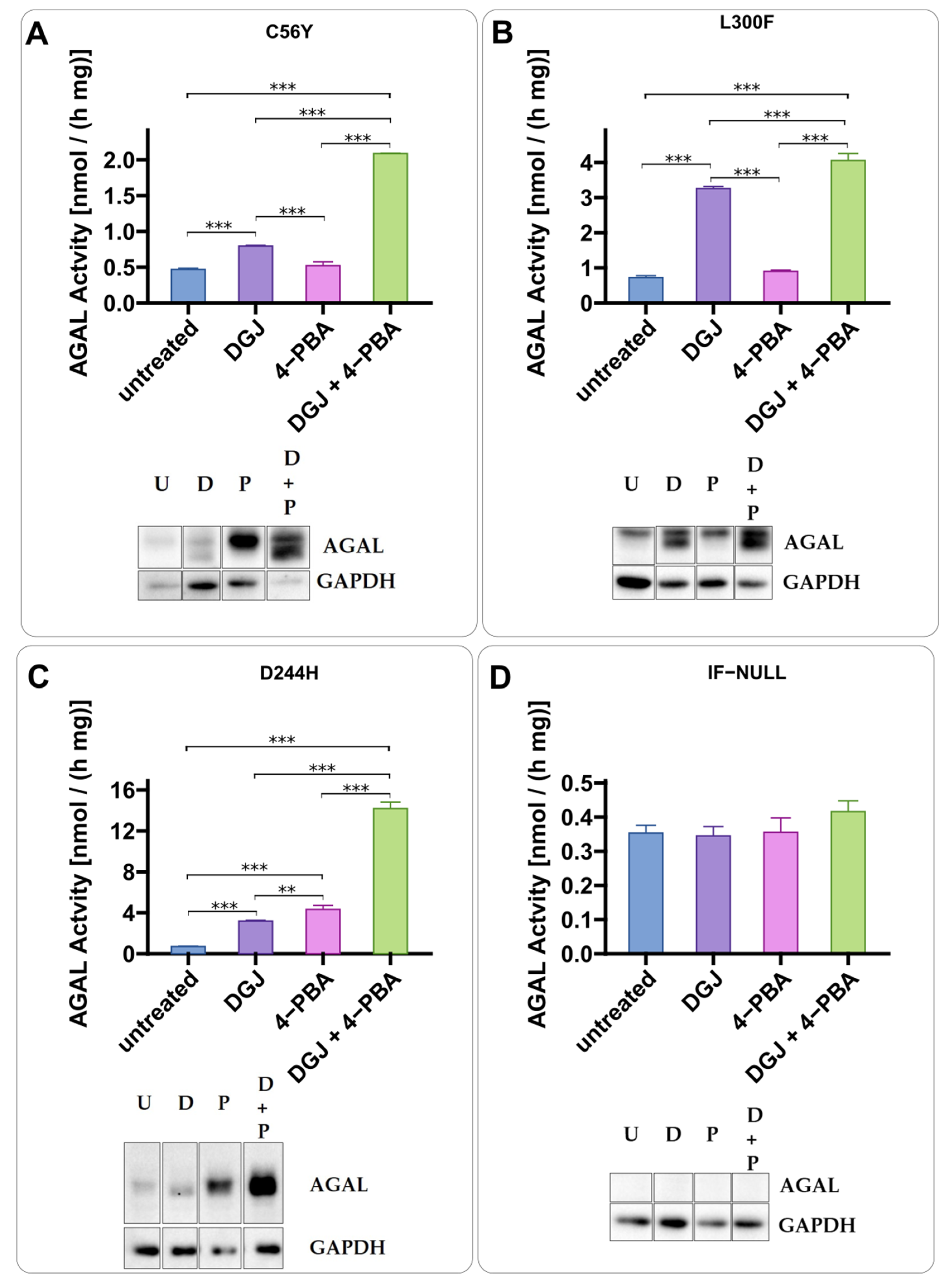
2.2. Intracellular Stabilization of AGAL by DGJ Is Enhanced by Ambroxol and 4-Phenylbutyrate
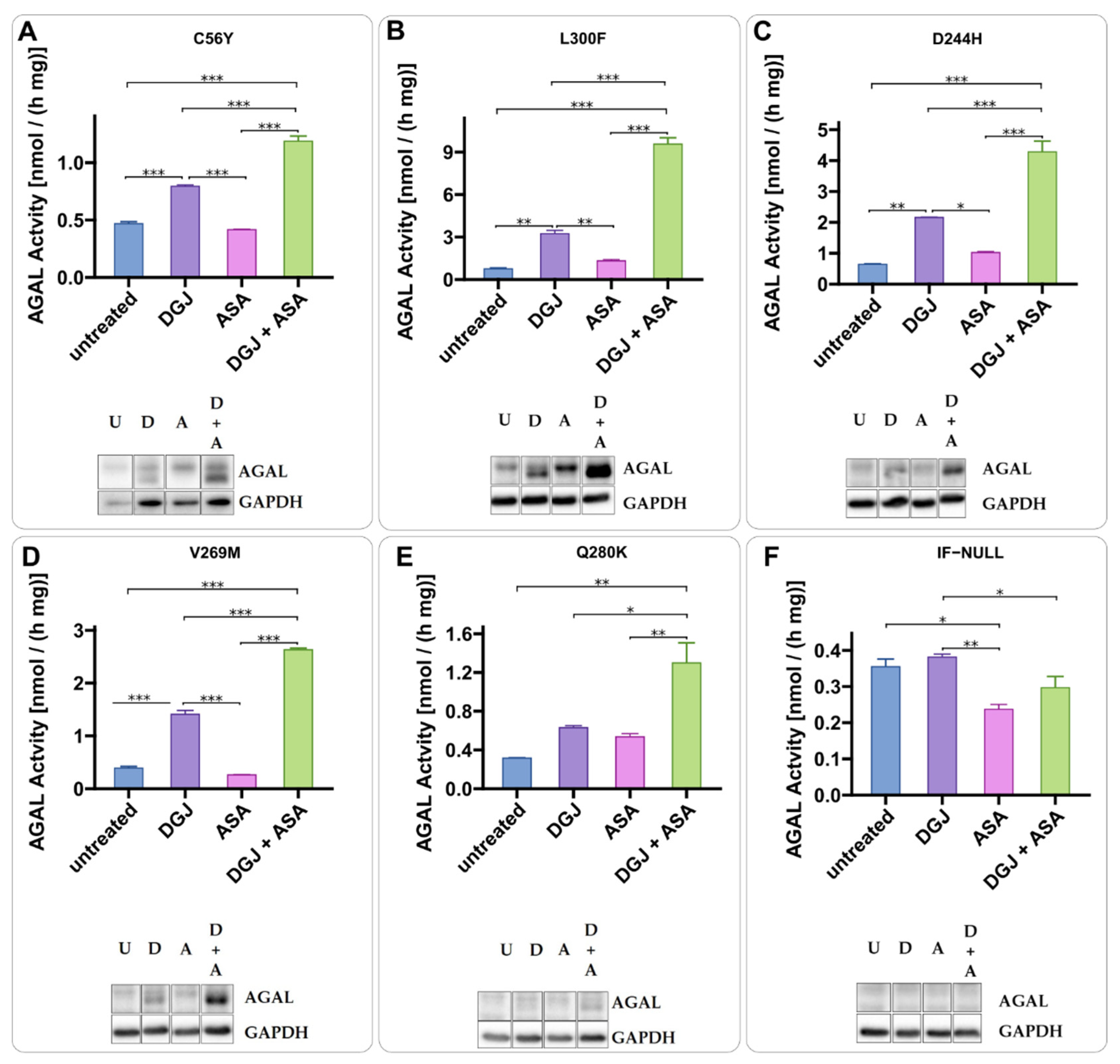
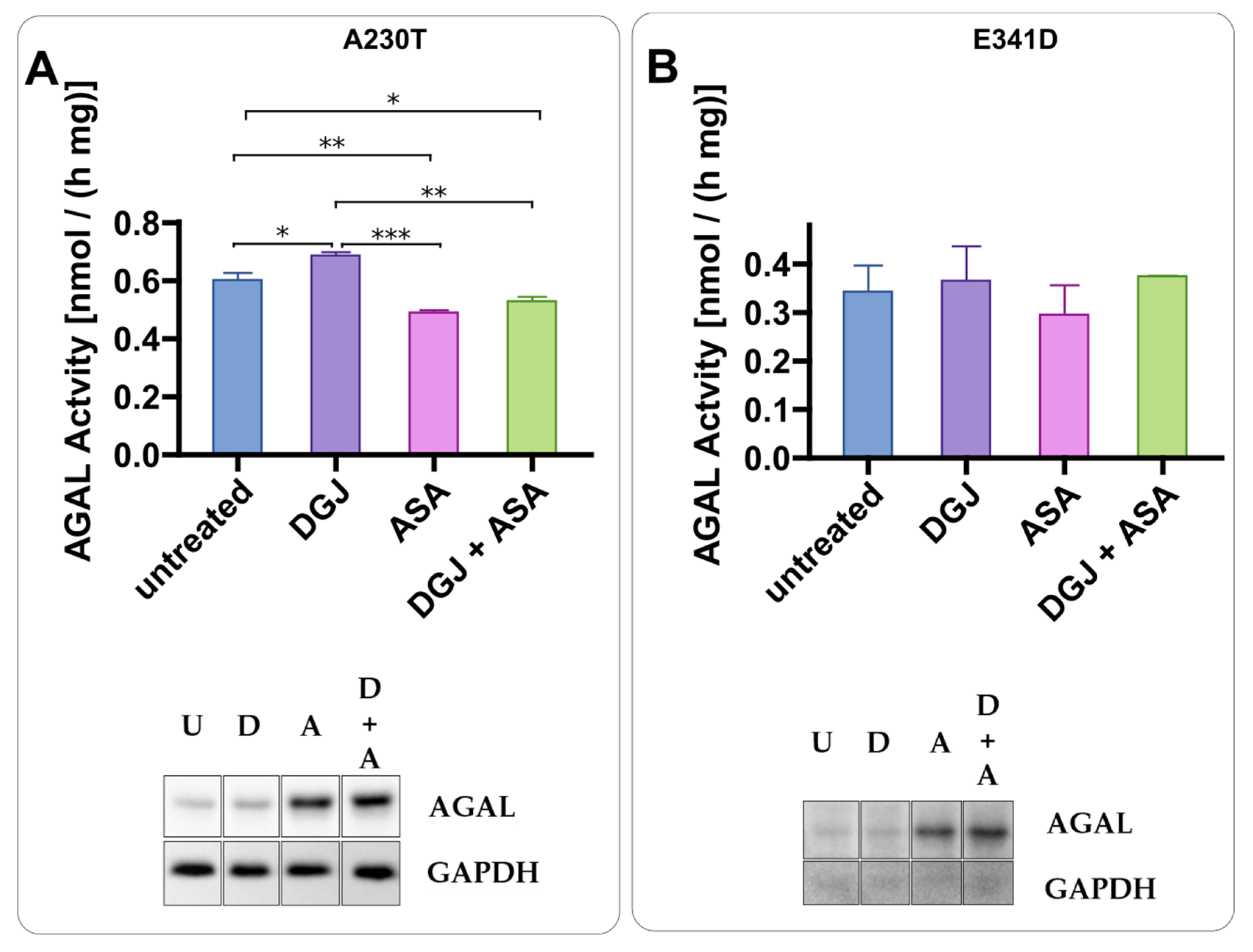
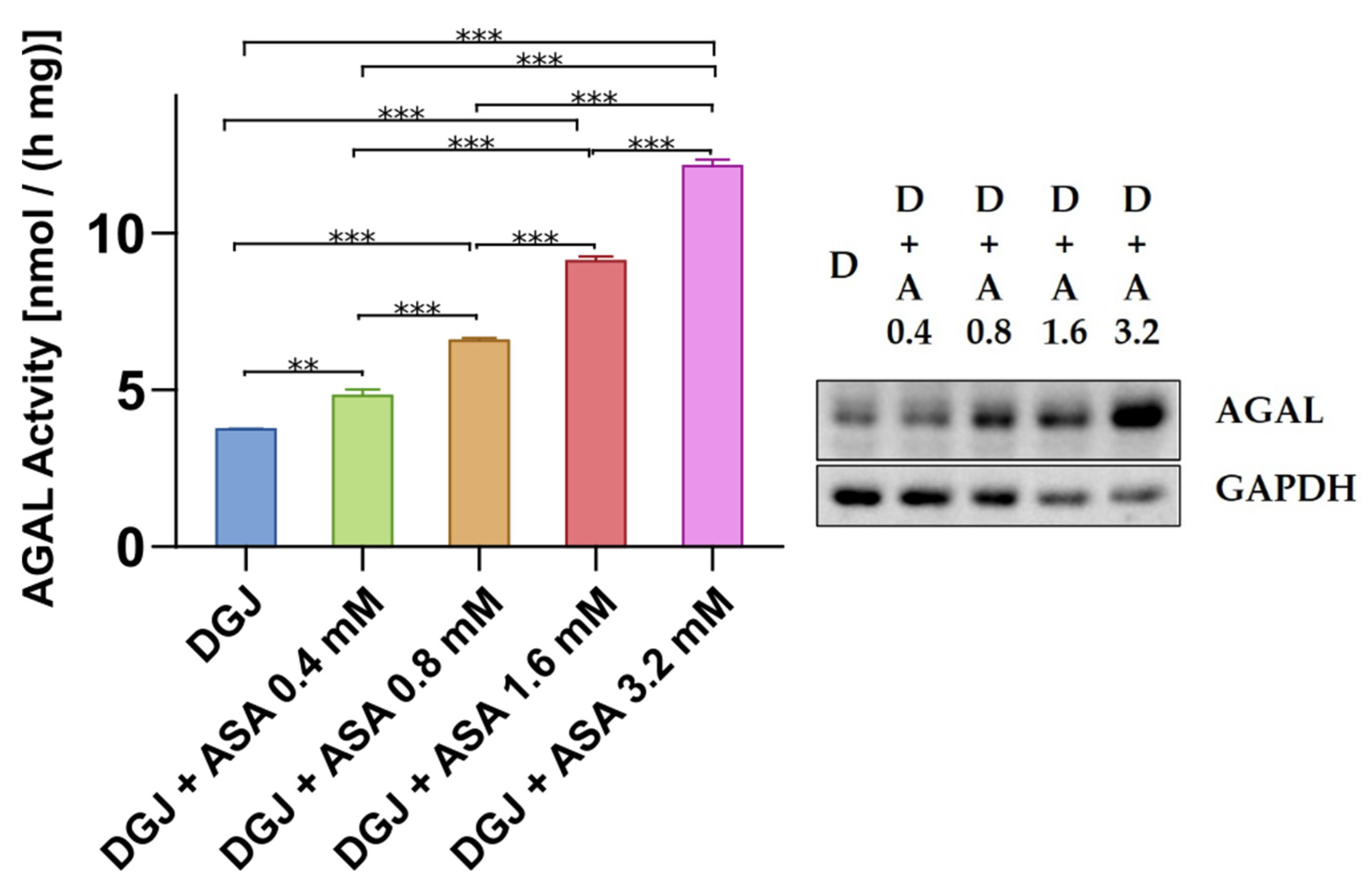
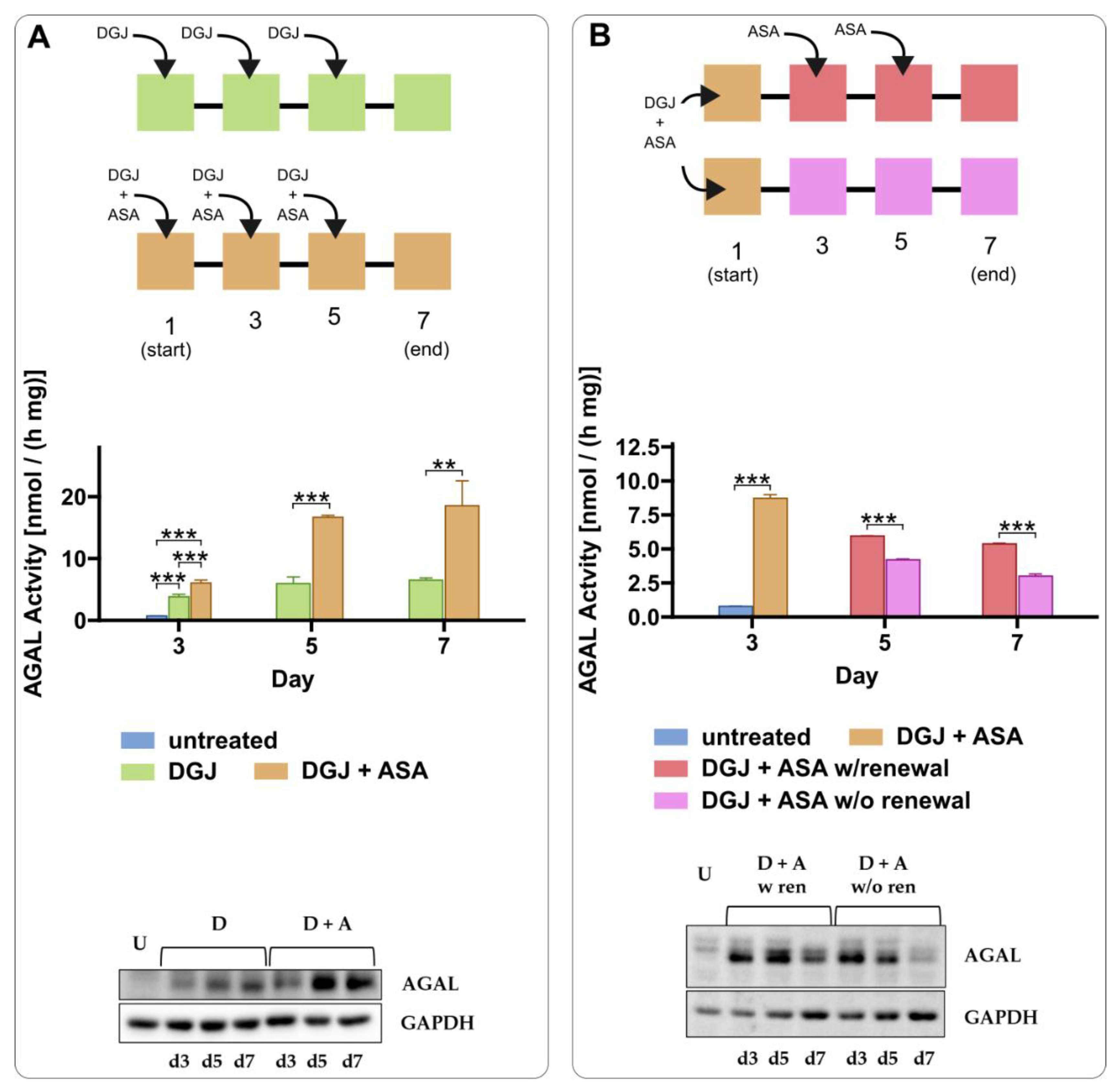
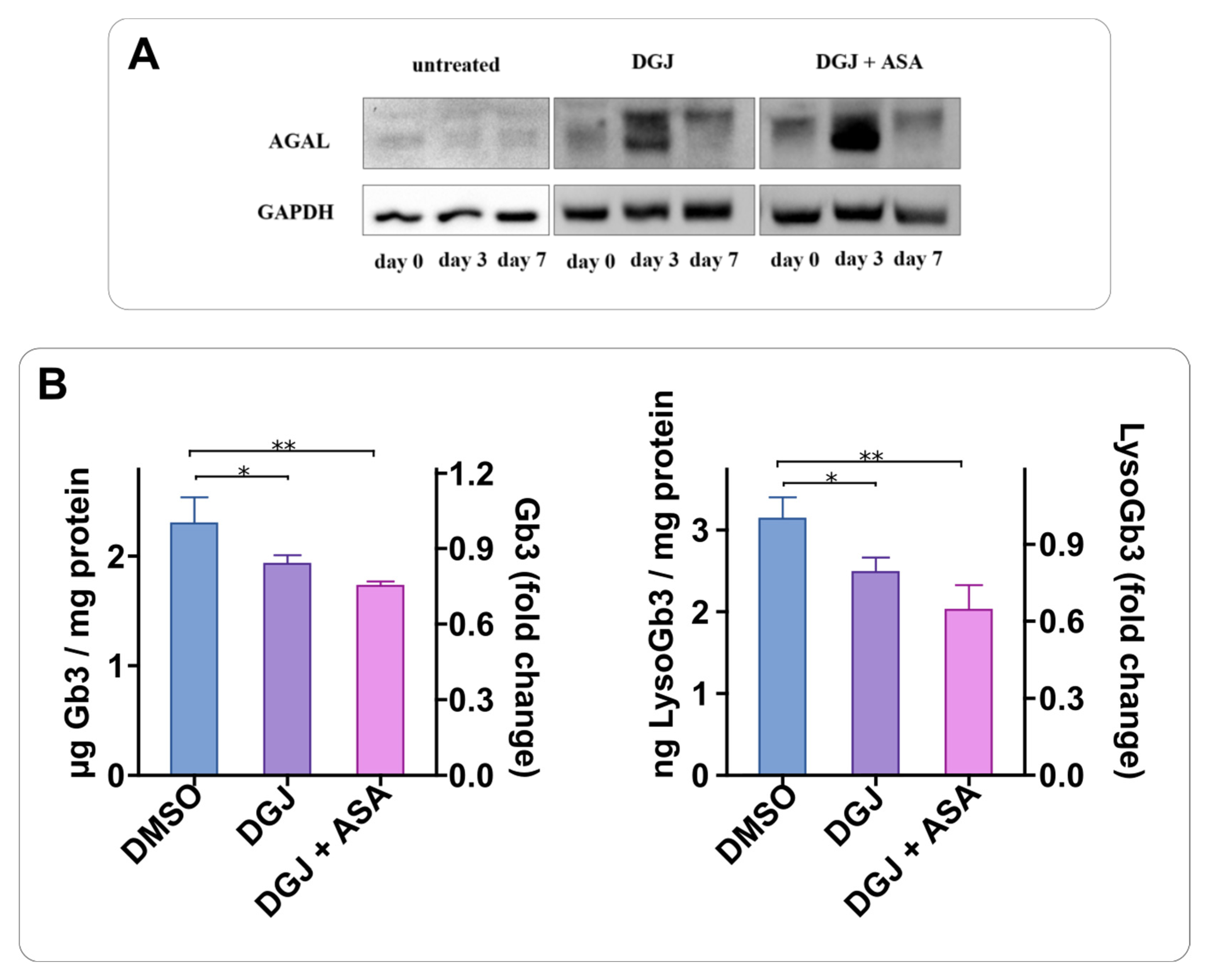
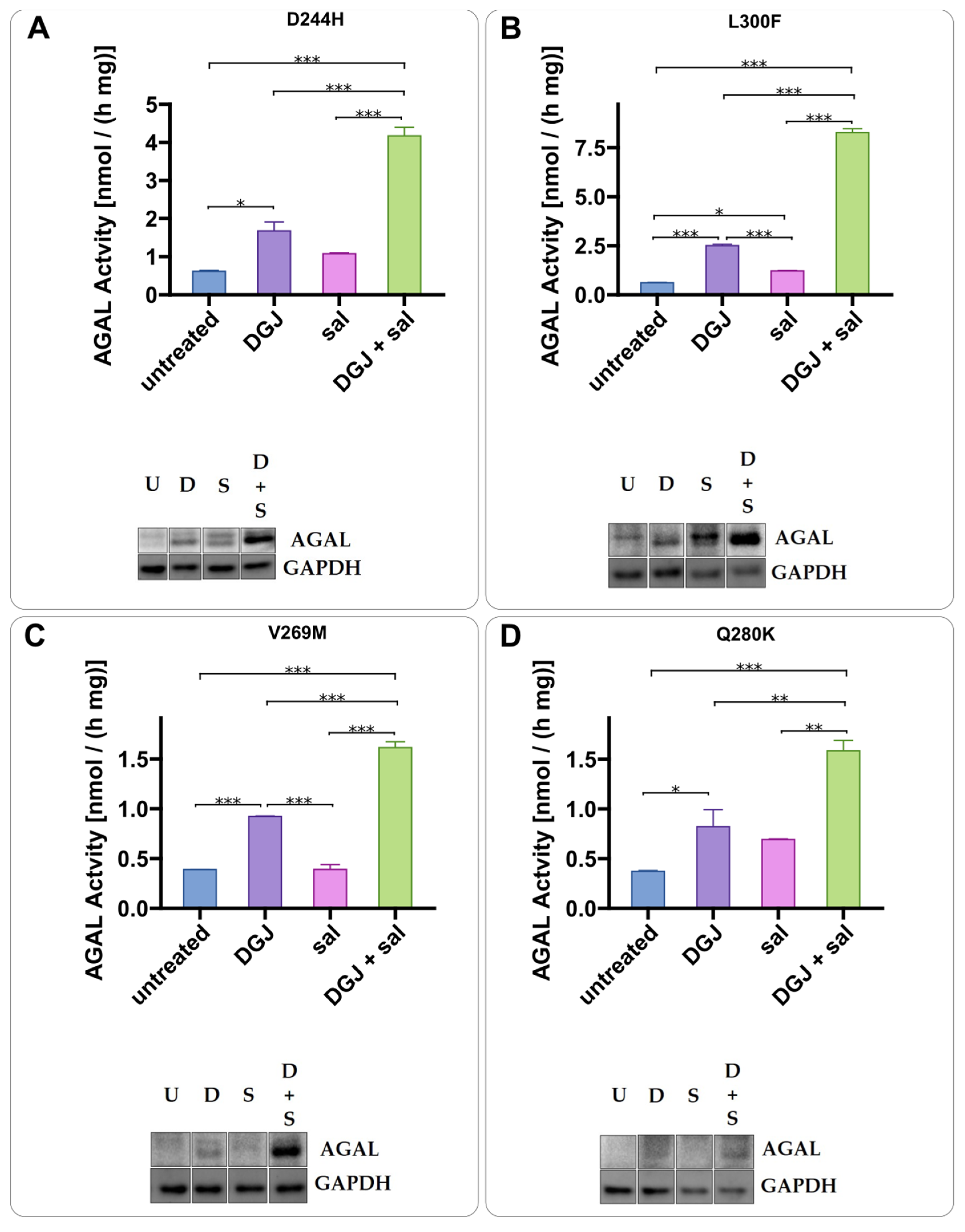
2.3. Intracellular Stabilization of AGAL by DGJ Is Enhanced by Acetylsalicilic Acid
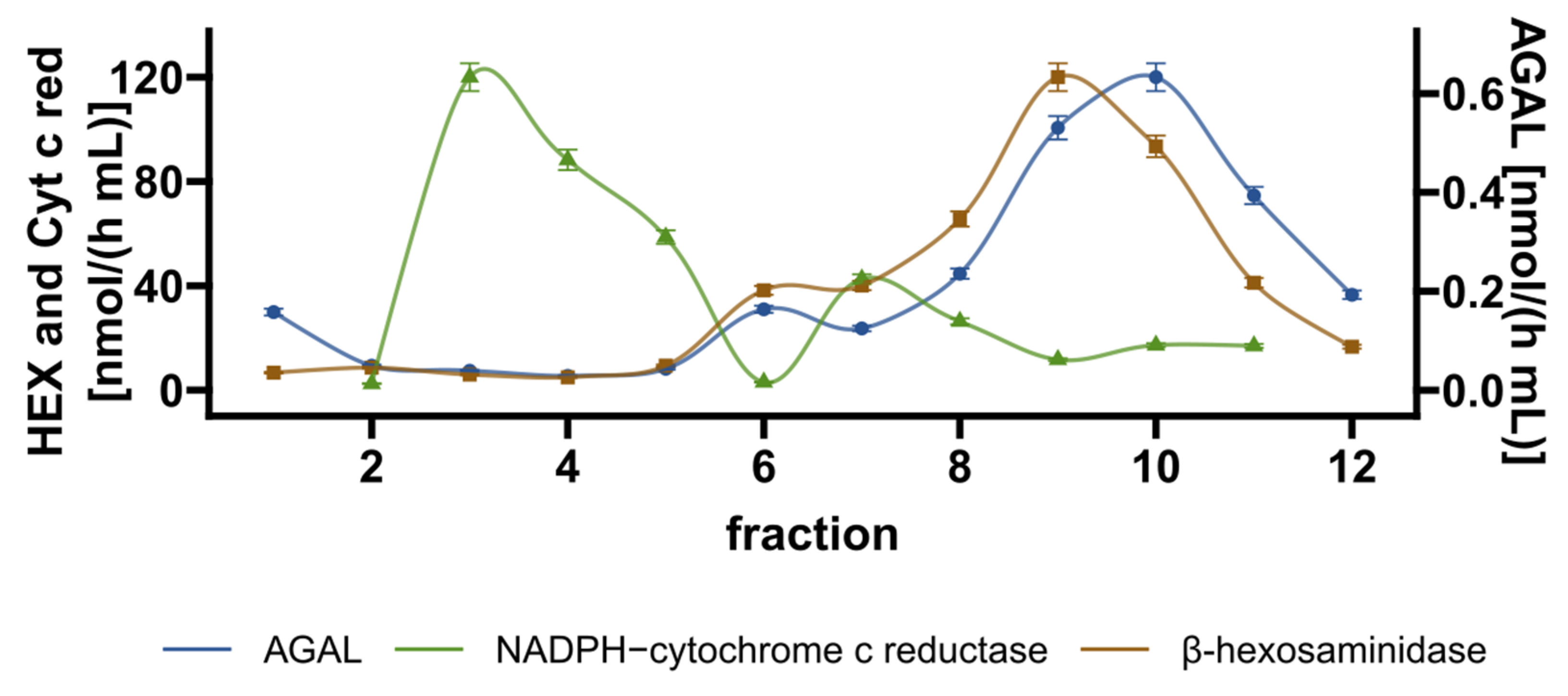
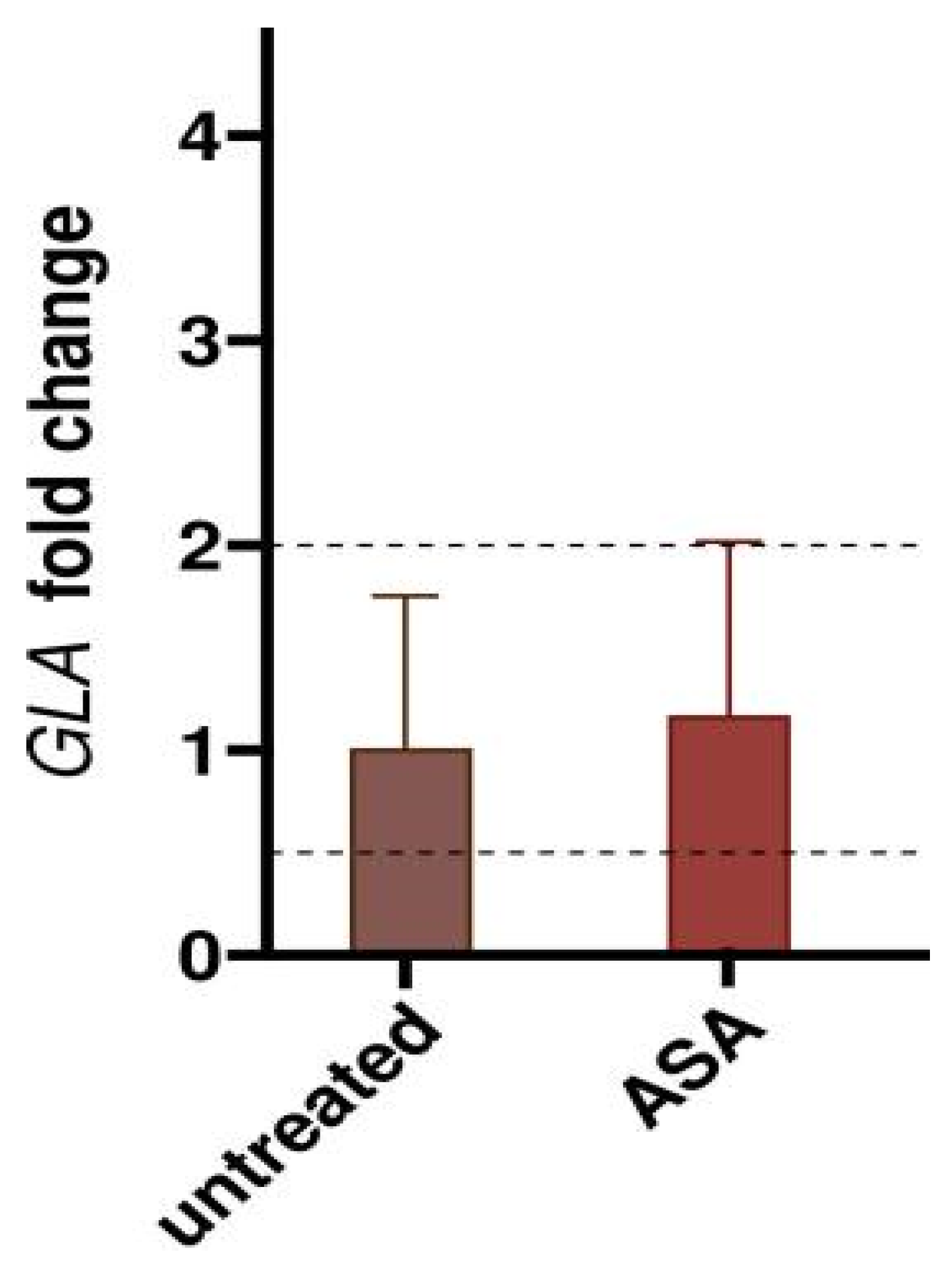
2.4. Mode of Action of Acetylsalicylic Acid
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Cell Cultures and Stable Transfections
5.3. Quantitative Real-Time PCR
5.4. AGAL Enzymatic Activity Assay
5.5. Cell Fractionation
5.6. Gb3 and Lyso-Gb3 Extraction
5.7. Proteomics Analysis
5.8. Bioinformatics Analysis
5.9. Miscellaneous
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABX | ambroxol |
| AGAL | lysosomal alpha-galactosidase |
| ASA | acetylsalicylic acid |
| DGJ | 1-deoxygalactonojirimycin (Migalastat) |
| FD | Fabry Disease |
| IF-GLA-MUTs | immortalized fibroblasts transfected with individual pCMV6-AC plasmids carrying GLA mutants |
| IF-GLA-NULL | immortalized fibroblasts transfected with the empty vector4-PBA: 4-phenylbutyrate |
References
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson–Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 88. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Da Assoc. Médica Bras. 2020, 66, s10–s16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; Adam, D.N. A Review of Fabry Disease. Ski. Ther. Lett. 2018, 23, 4–6. [Google Scholar]
- Mehta, A., Hughes. DA Fabry Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1292/ (accessed on 27 January 2022).
- Andreotti, G.; Monticelli, M.; Cubellis, M.V. Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 2015, 7, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Mauhin, W.; Belmatoug, N.; Garnotel, R.; Bedreddine, N.; Catros, F.; Ancellin, S.; Lidove, O.; Gaches, F. When and How to Diagnose Fabry Disease in Clinical Pratice. Am. J. Med Sci. 2020, 360, 641–649. [Google Scholar] [CrossRef]
- Cairns, T.; Müntze, J.; Gernert, J.; Spingler, L.; Nordbeck, P.; Wanner, C. Hot topics in Fabry disease. Postgrad. Med J. 2018, 94, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Cimmaruta, C.; Liguori, L.; Pantoom, S.; Iwanov, K.; Petters, J.; Hund, C.; Bunschkowski, M.; Hermann, A.; Cubellis, M.V.; et al. Assessment of Gene Variant Amenability for Pharmacological Chaperone Therapy with 1-Deoxygalactonojirimycin in Fabry Disease. Int. J. Mol. Sci. 2020, 21, 956. [Google Scholar] [CrossRef] [Green Version]
- Di Risi, T.; Vinciguerra, R.; Cuomo, M.; Della Monica, R.; Riccio, E.; Cocozza, S.; Imbriaco, M.; Duro, G.; Pisani, A.; Chiariotti, L. DNA methylation impact on Fabry disease. Clin. Epigenetics 2021, 13, 1–9. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Germain, D.P.; Arad, M.; Burlina, A.; Elliott, P.; Falissard, B.; Feldt-Rasmussen, U.; Hilz, M.J.; Hughes, D.A.; Ortiz, A.; Wanner, C.; et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease – A systematic literature review by a European panel of experts. Mol. Genet. Metab. 2019, 126, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Elliott, P.; Falissard, B.; Fomin, V.V.; Hilz, M.J.; Jovanovic, A.; Kantola, I.; Linhart, A.; Mignani, R.; Namdar, M.; et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol. Genet. Metab. Rep. 2019, 19, 100454. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.; Huynh-Do, U.; Krayenbuehl, P.; Beuschlein, F.; Schiffmann, R.; Barbey, F. Fabry disease genotype, phenotype, and migalastat amenability: Insights from a national cohort. J. Inherit. Metab. Dis. 2019, 43, 326–333. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Cammisa, M.; Correra, A.; Andreotti, G.; Cubellis, M.V. Fabry_CEP: A tool to identify Fabry mutations responsive to pharmacological chaperones. Orphanet J. Rare Dis. 2013, 8, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases α-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Kluepfel-Stahl, S.; Cooney, A.M.; Kaneski, C.R.; Quirk, J.M.; Schiffmann, R.; Brady, R.O.; Murray, G.J. Prediction of response of mutated alpha-galactosidase A to a pharmacological chaperone. Pharmacogenetics Genom. 2008, 18, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-H.; Murray, G.J.; Kluepfel-Stahl, S.; Cooney, A.M.; Quirk, J.M.; Schiffmann, R.; Brady, R.O.; Kaneski, C.R. Screening for pharmacological chaperones in Fabry disease. Biochem. Biophys. Res. Commun. 2007, 359, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman, S.C.; Fan, J.-Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening*. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.-M.; Dehn, N.; Cozma, C.; Giese, A.; Rolfs, A. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum. Mutat. 2016, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Suzuki, Y.; Fan, J.-Q. Role of Ser-65 in the Activity of α-Galactosidase A: Characterization of a Point Mutation (S65T) Detected in a Patient with Fabry Disease. Arch. Biochem. Biophys. 2000, 377, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase a that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Citro, V.; De Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry disease with pharmacological chaperones: From in silico predictions to in vitro tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-Y.; Kim, G.-H.; Kim, S.-S.; Ko, J.M.; Lee, J.-J.; Yoo, H.-W. Effects of a chemical chaperone on genetic mutations in α-galactosidase A in Korean patients with Fabry disease. Exp. Mol. Med. 2009, 41, 1–7. [Google Scholar] [CrossRef]
- Giugliani, R.; Waldek, S.; Germain, D.; Nicholls, K.; Bichet, D.; Simosky, J.; Bragat, A.; Castelli, J.; Benjamin, E.; Boudes, P. A Phase 2 study of migalastat hydrochloride in females with Fabry disease: Selection of population, safety and pharmacodynamic effects. Mol. Genet. Metab. 2013, 109, 86–92. [Google Scholar] [CrossRef]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase A Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [Green Version]
- Cubellis, M.V.; Baaden, M.; Andreotti, G. Taming molecular flexibility to tackle rare diseases. Biochimie 2015, 113, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the responsiveness to pharmacological chaperones: Lysosomal human alpha-galactosidase, a case of study. Orphanet J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Tsukimura, T.; Chiba, Y.; Ohno, K.; Saito, S.; Tajima, Y.; Sakuraba, H. Molecular mechanism for stabilization of a mutant α-galactosidase A involving M51I amino acid substitution by imino sugars. Mol. Genet. Metab. 2011, 103, 26–32. [Google Scholar] [CrossRef]
- Citro, V.; Peña-García, J.; Den-Haan, H.; Pérez-Sánchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urquiza, P.; Laín, A.; Sanz-Parra, A.; Moreno, J.; Bernardo-Seisdedos, G.; Dubus, P.; González, E.; Gutiérrez-De-Juan, V.; García, S.; Eraña, H.; et al. Repurposing ciclopirox as a pharmacological chaperone in a model of congenital erythropoietic porphyria. Sci. Transl. Med. 2018, 10, eaat7467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Migalastat: First Global Approval. Drugs 2016, 76, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Pharmacoeconomic Review Report: Migalastat (Galafold): (Amicus Therapeutics): Indication: Fabry Disease [Internet]; Canadian Agency for Drugs and Technologies in Healt: Ottawa, ON, Canada, February 2018.
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; A Sitaraman, S.; Desnick, R.J.; et al. The Pharmacological Chaperone 1-Deoxygalactonojirimycin Reduces Tissue Globotriaosylceramide Levels in a Mouse Model of Fabry Disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Mele, B.H.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [Green Version]
- Lemansky, P.; Bishop, D.; Desnick, R.; Hasilik, A.; von Figura, K. Synthesis and processing of alpha-galactosidase A in human fibroblasts. Evidence for different mutations in Fabry disease. J. Biol. Chem. 1987, 262, 2062–2065. [Google Scholar] [CrossRef]
- Matsuura, F.; Ohta, M.; Ioannou, Y.A.; Desnick, R.J. Human -galactosidase A: Characterization of the N-linked oligosaccharides on the intracellular and secreted glycoforms overexpressed by Chinese hamster ovary cells. Glycobiology 1998, 8, 329–339. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The Molecular Defect Leading to Fabry Disease: Structure of Human α-Galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Segatori, L. Remodeling the Proteostasis Network to Rescue Glucocerebrosidase Variants by Inhibiting ER-Associated Degradation and Enhancing ER Folding. PLoS ONE 2013, 8, e61418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.-M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in Fabry disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.-M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Gläser, A.; Beller, M.; Rolfs, A.; et al. Enzyme Enhancers for the Treatment of Fabry and Pompe Disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, G.H.-F.; Roth, J.; Zuber, C. 4-Phenylbutyrate rescues trafficking incompetent mutant α-galactosidase A without restoring its functionality. Biochem. Biophys. Res. Commun. 2007, 360, 375–380. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Moreau, R. Lipid-regulating properties of butyric acid and 4-phenylbutyric acid: Molecular mechanisms and therapeutic applications. Pharmacol. Res. 2019, 144, 116–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimori, S.; Ohtaka, H.; Koshikawa, Y.; Kawada, K.; Kaneko, M.; Okuma, Y.; Nomura, Y.; Murakami, Y.; Hamana, H. 4-Phenylbutyric acid protects against neuronal cell death by primarily acting as a chemical chaperone rather than histone deacetylase inhibitor. Bioorganic Med. Chem. Lett. 2013, 23, 6015–6018. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Stappers, F.; Brand, E. In Vitro and In Vivo Amenability to Migalastat in Fabry Disease. Mol. Ther. - Methods Clin. Dev. 2020, 19, 24–34. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; Üçeyler, N.; et al. Treatment of Fabry’s Disease With Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar] [CrossRef]
- Alfonso, L.F.; Srivenugopal, K.S.; Bhat, G.J. Does Aspirin Acetylate Multiple Cellular Proteins? Mol. Med. Rep. 2009, 2, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Ayyadevara, S.; Balasubramaniam, M.; Kakraba, S.; Alla, R.; Mehta, J.L.; Reis, R.J.S. Aspirin-Mediated Acetylation Protects Against Multiple Neurodegenerative Pathologies by Impeding Protein Aggregation. Antioxidants Redox Signal. 2017, 27, 1383–1396. [Google Scholar] [CrossRef]
- Morretta, E.; D’Agostino, A.; Cassese, E.; Maglione, B.; Petrella, A.; Schiraldi, C.; Monti, M.C. Label-Free Quantitative Proteomics to Explore the Action Mechanism of the Pharmaceutical-Grade Triticum vulgare Extract in Speeding Up Keratinocyte Healing. Molecules 2022, 27, 1108. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Huang, C.-C.; Taylor, J.M.G.; Misek, D.E.; Kardia, S.L.R.; Giordano, T.; Iannettoni, M.D.; Orringer, M.B.; Hanash, S.M.; et al. Discordant Protein and mRNA Expression in Lung Adenocarcinomas. Mol. Cell. Proteom. 2002, 1, 304–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Pascal, L.; True, L.D.; Campbell, D.S.; Deutsch, E.W.; Risk, M.; Coleman, I.M.; Eichner, L.J.; Nelson, P.S.; Liu, A.Y. Correlation of mRNA and protein levels: Cell type-specific gene expression of cluster designation antigens in the prostate. BMC Genom. 2008, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Gygi, S.P.; Rochon, Y.; Franza, B.R.; Aebersold, R. Correlation between Protein and mRNA Abundance in Yeast. Mol. Cell. Biol. 1999, 19, 1720–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, E.S. Genome-wide Correlation between mRNA and Protein in a Single Cell. Angew. Chem. Int. Ed. 2011, 50, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative Analysis of Proteome and Transcriptome Variation in Mouse. PLoS Genet. 2011, 7, e1001393. [Google Scholar] [CrossRef] [Green Version]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. TheBioGRIDdatabase: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2020, 30, 187–200. [Google Scholar] [CrossRef]
- Deng, W.-G.; Ruan, K.-H.; Du, M.; Saunders, M.A.; Wu, K.K. Aspirin and salicylate bind to immunoglobulin heavy chain binding protein (BiP) and inhibit its ATPase activity in human fibroblasts. FASEB J. 2001, 15, 2463–2470. [Google Scholar] [CrossRef]
- Mele, B.H.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Zheng, W.; Simeonov, A. Drug discovery and development for rare genetic disorders. Am. J. Med Genet. Part A 2017, 173, 2307–2322. [Google Scholar] [CrossRef]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A. Drug repositioning for orphan diseases. Briefings Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, M.; Pottage, J.; Kessler, H.; Fischl, M.; Richman, D.; Merigan, T.; Powderly, W.; Smith, S.; Karim, A.; Sherman, J. The Tolerability and Pharmacokinetics of N-Butyl-deoxynojirimycin in Patients with Advanced HIV Disease (ACTG 100). J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 10, 549–553. [Google Scholar] [CrossRef]
- Hughes, D.A.; Mehta, A.B. Vascular Complications of Fabry Disease: Enzyme Replacement and Other Therapies. Acta Paediatrics 2005, 94, 28–33. [Google Scholar] [CrossRef]
- Sirrs, S.; Bichet, D.; Casey, R.; Clarke, J.; Lemoine, K.; Doucette, S.; West, M. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol. Genet. Metab. 2014, 111, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Arif, H., Aggarwal. Salicylic Acid (Aspirin). In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Aspirin Dosage. Available online: https://www.drugs.com/dosage/aspirin.html (accessed on 28 February 2022).
- Granata, A.; Watson, R.; Collinson, L.M.; Schiavo, G.; Warner, T.T. The Dystonia-associated Protein TorsinA Modulates Synaptic Vesicle Recycling. J. Biol. Chem. 2008, 283, 7568–7579. [Google Scholar] [CrossRef] [Green Version]
- Pu, J.; Schindler, C.; Jia, R.; Jarnik, M.; Backlund, P.; Bonifacino, J.S. BORC, a Multisubunit Complex that Regulates Lysosome Positioning. Dev. Cell 2015, 33, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Vasile, E.; Penman, M.; Novina, C.D.; Dykxhoorn, D.M.; Ungar, D.; Hughson, F.M.; Krieger, M. Genetic Analysis of the Subunit Organization and Function of the Conserved Oligomeric Golgi (COG) Complex. J. Biol. Chem. 2005, 280, 32736–32745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, L.; Park, J.L.; Byun, J.; Pennathur, S.; Kollmeyer, J.; Shayman, J.A. Decreased Nitric Oxide Bioavailability in a Mouse Model of Fabry Disease. J. Am. Soc. Nephrol. 2009, 20, 1975–1985. [Google Scholar] [CrossRef]
- Dhagat, U.; Carbone, V.; Chung, R.; Matsunaga, T.; Endo, S.; Hara, A.; El-Kabbani, O. A Salicylic Acid-Based Analogue Discovered from Virtual Screening as a Potent Inhibitor of Human 20α-Hydroxysteroid Dehydrogenase. Med. Chem. 2007, 3, 546–550. [Google Scholar] [CrossRef]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Gestwicki, J.E. Binding of Human Nucleotide Exchange Factors to Heat Shock Protein 70 (Hsp70) Generates Functionally Distinct Complexes in Vitro. J. Biol. Chem. 2014, 289, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.T.; Plate, L. Revealing functional insights into ER proteostasis through proteomics and interactomics. Exp. Cell Res. 2020, 399, 112417. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Gray, J.; Priestman, D.A.; Wallom, K.-L.; Atkins, J.; Olsen, O.D.; Klein, A.; Drndarski, S.; Petersen, N.H.T.; Ingemann, L.; et al. Heat shock protein–based therapy as a potential candidate for treating the sphingolipidoses. Sci. Transl. Med. 2016, 8, 355ra118. [Google Scholar] [CrossRef] [PubMed]
- Fog, C.K.; Zago, P.; Malini, E.; Solanko, L.M.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.H.; Bembi, B.; et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, N. FDA approves Galafold, a triumph for Amicus. Nat. Biotechnol. 2018, 36, 913. [Google Scholar] [CrossRef]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Müntze, J.; Gensler, D.; Maniuc, O.; Liu, D.; Cairns, T.; Oder, D.; Hu, K.; Lorenz, K.; Frantz, S.; Wanner, C.; et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin. Pharmacol. Ther. 2018, 105, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Mauer, M.; Sokolovskiy, A.; A Barth, J.; Castelli, J.P.; Williams, H.N.; Benjamin, E.R.; Najafian, B. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenableGLAmutations following 6 months of migalastat treatment. J. Med Genet. 2017, 54, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Körver, S.; Feldt-Rasmussen, U.; Svarstad, E.; Kantola, I.; Langeveld, M. Oral Chaperone Therapy Migalastat for the Treatment of Fabry Disease: Potentials and Pitfalls of Real-World Data. Clin. Pharmacol. Ther. 2019, 106, 925–926. [Google Scholar] [CrossRef]
- Miceli, M.; Franci, G.; Dell’Aversana, C.; Ricciardiello, F.; Petraglia, F.; Carissimo, A.; Perone, L.; Maruotti, G.; Savarese, M.; Martinelli, P.; et al. MePR: A Novel Human Mesenchymal Progenitor Model with Characteristics of Pluripotency. Stem Cells Dev. 2013, 22, 2368–2383. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kominami, E.; Ezaki, J.; Muno, D.; Ishido, K.; Ueno, T.; Wolfe, L.S. Specific Storage of Subunit c of Mitochondrial ATP Synthase in Lysosomes of Neuronal Ceroid Lipofuscinosis (Batten’s Disease). J. Biochem. 1992, 111, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Martin, M.V.; Sohl, C.D.; Cheng, Q. Measurement of cytochrome P450 and NADPH–cytochrome P450 reductase. Nat. Protoc. 2009, 4, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, N.; Jubran, J.; Dror, S.; Simonovsky, E.; Basha, O.; Argov, C.; Hekselman, I.; Abu-Qarn, M.; Vinogradov, E.; Mauer, O.; et al. The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nat. Commun. 2021, 12, 2180. [Google Scholar] [CrossRef]
- Gomes, A.V. Genetics of Proteasome Diseases. Scientifica 2013, 2013, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monticelli, M.; Liguori, L.; Allocca, M.; Bosso, A.; Andreotti, G.; Lukas, J.; Monti, M.C.; Morretta, E.; Cubellis, M.V.; Hay Mele, B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones. Int. J. Mol. Sci. 2022, 23, 5105. https://doi.org/10.3390/ijms23095105
Monticelli M, Liguori L, Allocca M, Bosso A, Andreotti G, Lukas J, Monti MC, Morretta E, Cubellis MV, Hay Mele B. Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones. International Journal of Molecular Sciences. 2022; 23(9):5105. https://doi.org/10.3390/ijms23095105
Chicago/Turabian StyleMonticelli, Maria, Ludovica Liguori, Mariateresa Allocca, Andrea Bosso, Giuseppina Andreotti, Jan Lukas, Maria Chiara Monti, Elva Morretta, Maria Vittoria Cubellis, and Bruno Hay Mele. 2022. "Drug Repositioning for Fabry Disease: Acetylsalicylic Acid Potentiates the Stabilization of Lysosomal Alpha-Galactosidase by Pharmacological Chaperones" International Journal of Molecular Sciences 23, no. 9: 5105. https://doi.org/10.3390/ijms23095105