
Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery

 , ,
, , 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Different Types of Pediatric Brain Tumors and Their Molecular Subtypes
2.1. Medulloblastoma
2.1.1. Molecular Subtypes of Medulloblastoma
SHH Subtype
Wnt Subtype
Group 3 and Group 4
2.2. Gliomas
2.2.1. High-Grade Gliomas
2.2.2. Low-Grade Gliomas
2.3. Ependymoma
3. Conventional Therapeutic Approaches
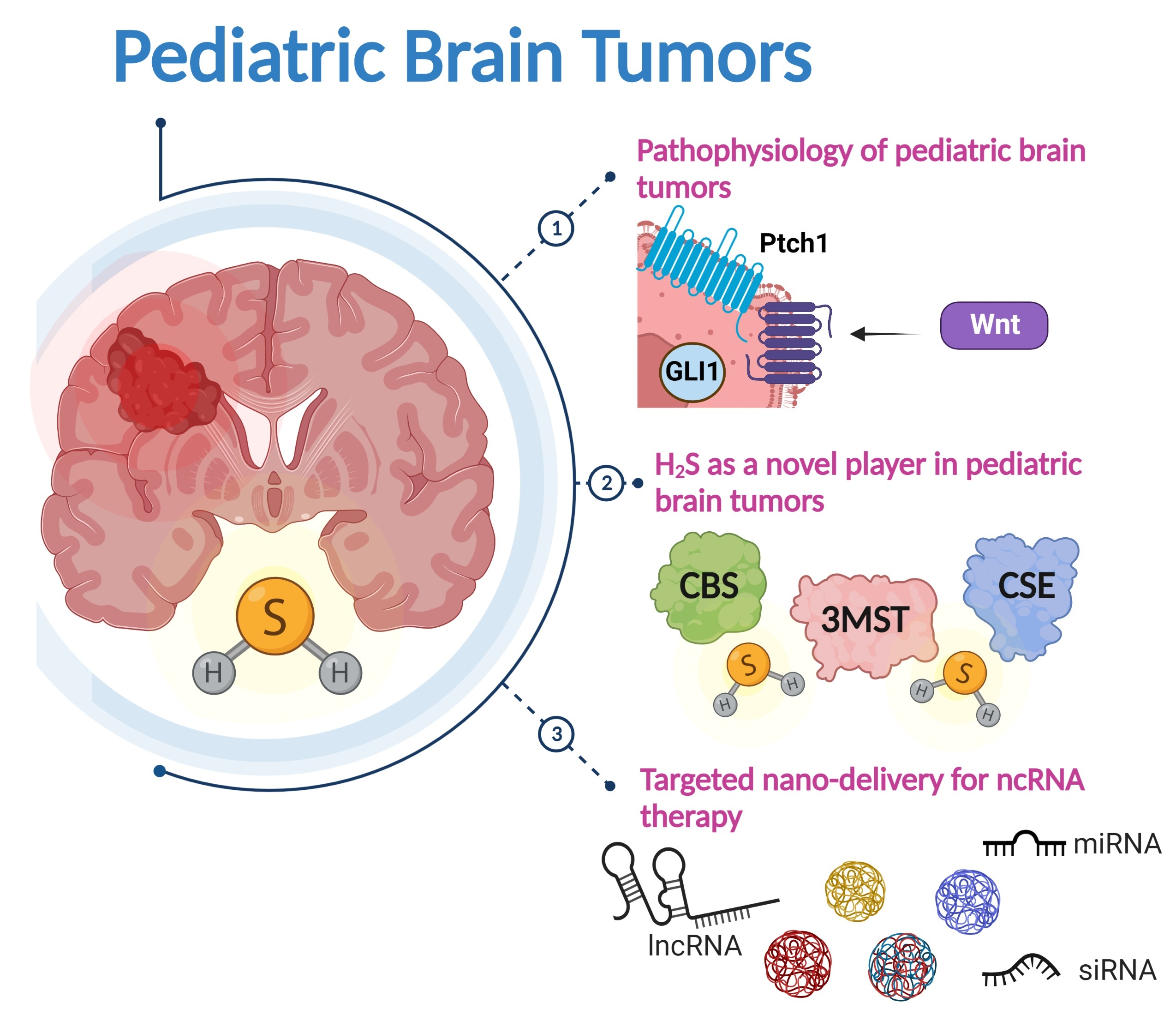
4. The Role of Hydrogen Sulfide in Brain Tumors
4.1. Biosynthesis of H2S
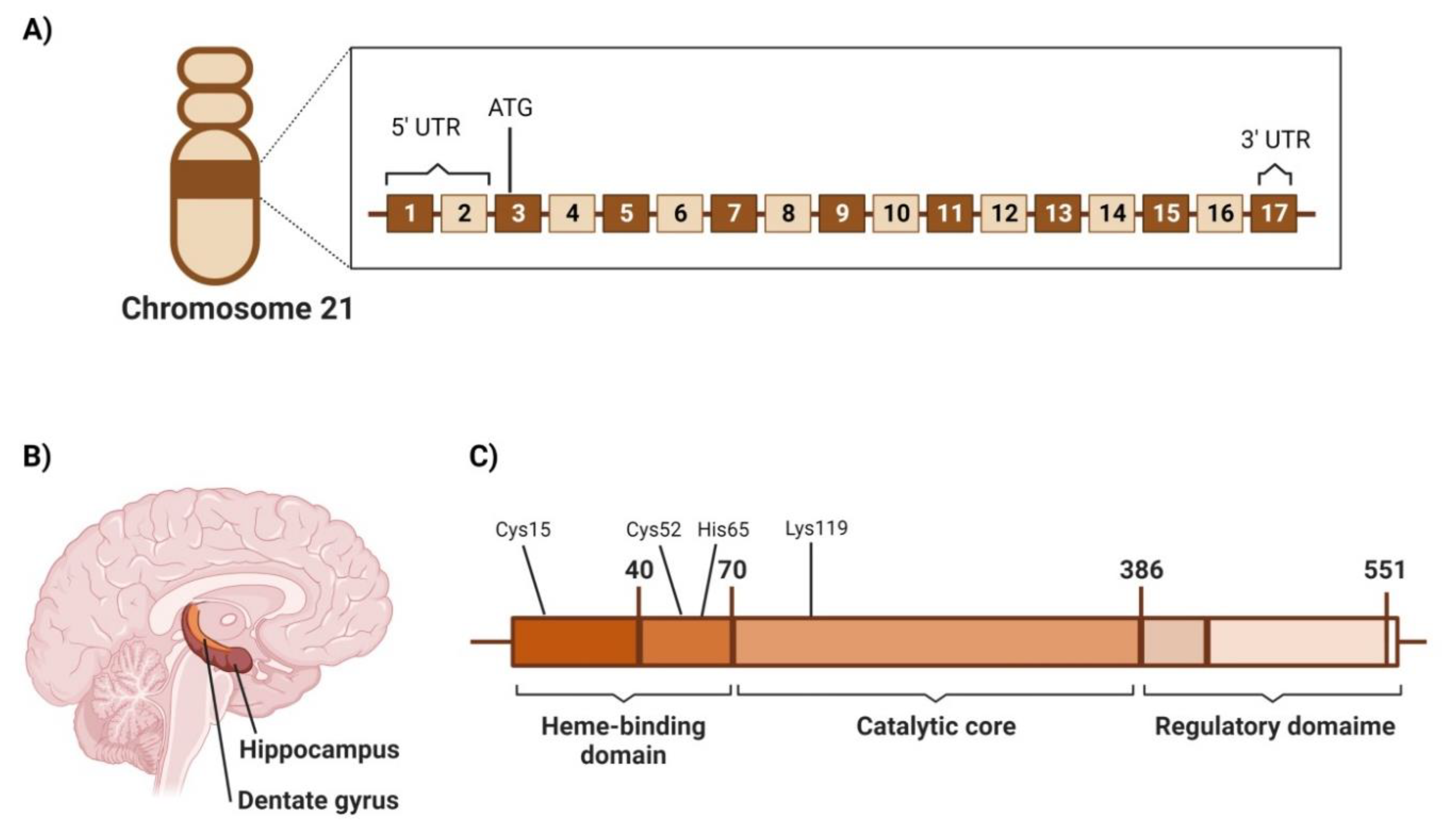
4.2. Cystathionine β-Synthetase (CBS)
4.2.1. Genetic Location
4.2.2. Structure
4.2.3. Area of High Expression
4.2.4. CBS Screening in Brain Tumors and Its Significance in Disease Progression
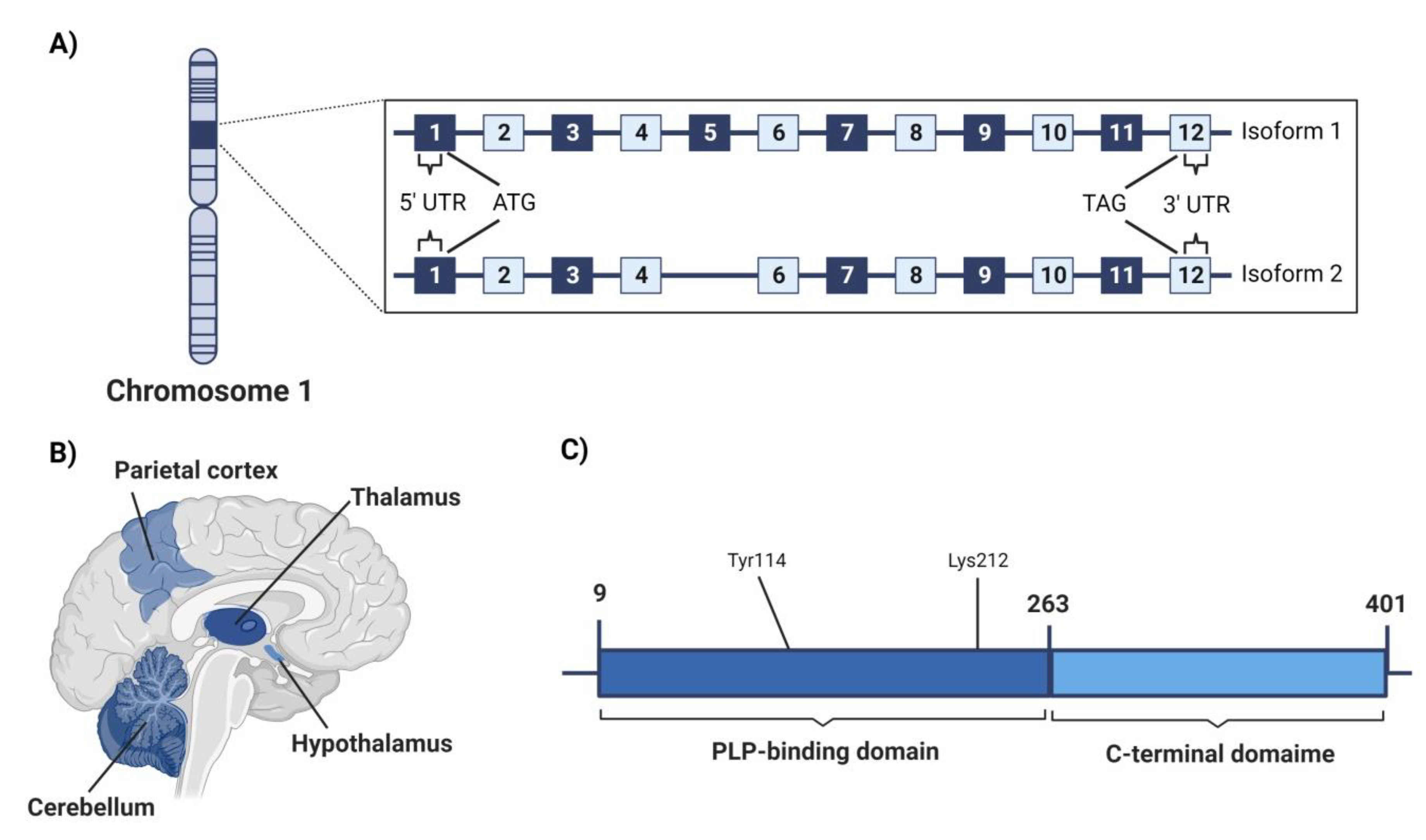
4.3. Cystathionine γ-Lyase (CSE)
4.3.1. Genetic Location
4.3.2. Structure
4.3.3. Area of High Expression
4.3.4. CSE Screening in Brain Tumors and Significance in Disease Progression
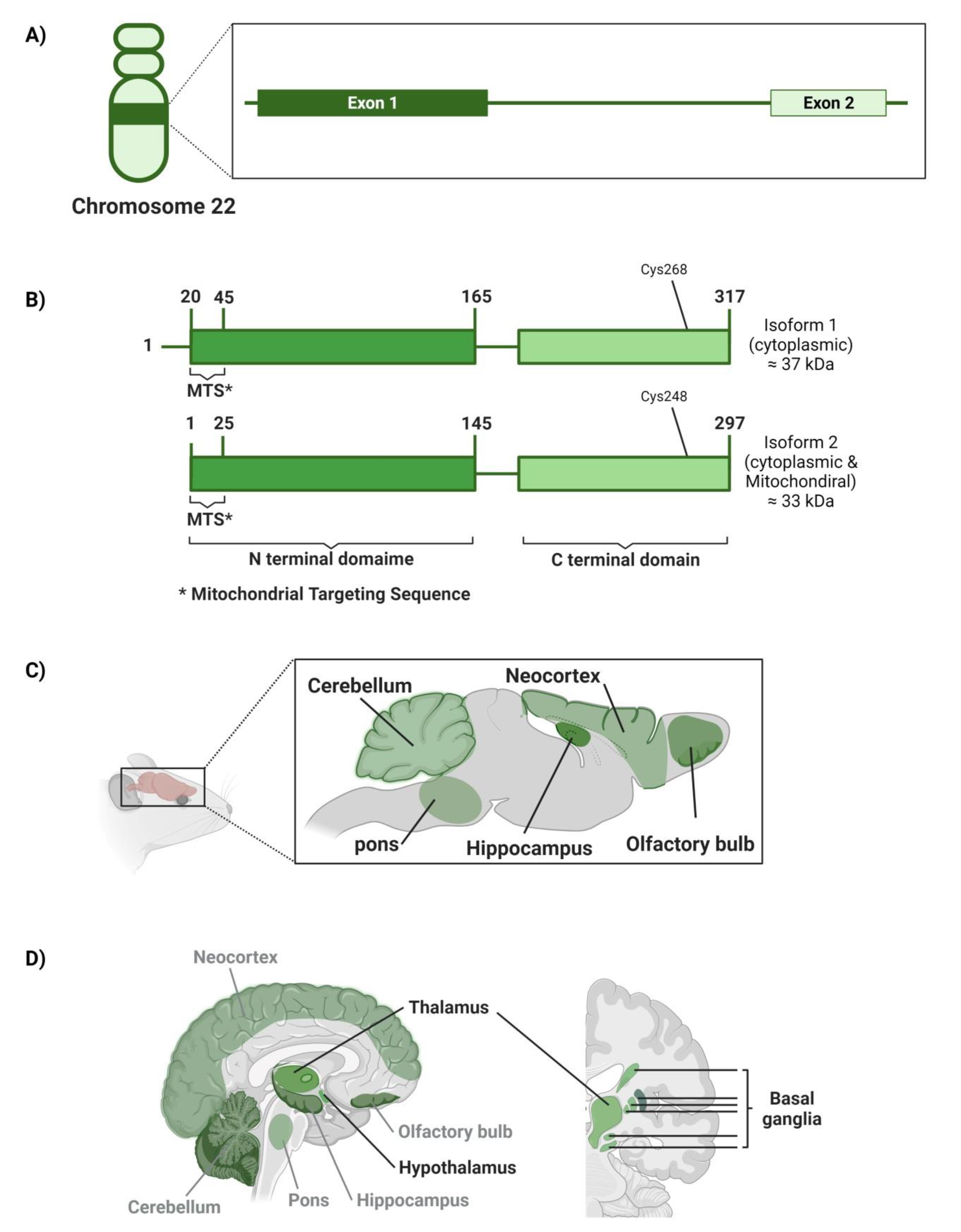
4.4. Mercaptopyruvate Sulfurtransferase (3MST)
4.4.1. Genetic Location
4.4.2. Structure
4.4.3. Area of High Expression
4.4.4. MST Screening in Brain Tumors and Its Significance in Disease Progression
5. Modern RNA-Based Therapeutic Modalities
5.1. Targeted Nano-Delivery of Therapeutic RNAs in Brain Tumor Therapy
5.2. Therapeutic RNAs Loaded Nanocarriers in Brain Tumors
6. Conclusions and Future Recommendations
Author Contributions
Funding
Conflicts of Interest
References
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA A Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Brain and Other Nervous System Statistics|American Cancer Society Cancer Facts & Statistics. Available online: https://cancerstatisticscenter.cancer.org (accessed on 20 September 2022).
- Salander, P.; Bergenheim, T.; Henriksson, R. The creation of protection and hope in patients with malignant brain tu-mours. Soc. Sci. Med. 1996, 42, 985–996. [Google Scholar] [CrossRef]
- Short, P.F.; Vasey, J.J.; Tunceli, K. Employment pathways in a large cohort of adult cancer survivors. Cancer 2005, 103, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T.; Liu, Q.; Yasui, Y.; Huang, S.; Ness, K.K.; Leisenring, W.; Hudson, M.M.; Donaldson, S.S.; King, A.A.; Stovall, M.; et al. Long-Term Outcomes Among Adult Survivors of Childhood Central Nervous System Malignancies in the Childhood Cancer Survivor Study. JNCI J. Natl. Cancer Inst. 2009, 101, 946–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.; Karajannis, M.A. Pediatric Brain Tumors: An Update. Curr. Probl. Pediatr. Adolesc. Health Care 2016, 46, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Pfister, S.M. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibi-tion. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Grammel, D.; Warmuth-Metz, M.; Von Bueren, A.O.; Kool, M.; Pietsch, T.; Kretzschmar, H.A.; Rowitch, D.H.; Rutkowski, S.; Pfister, S.M.; Schüller, U. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012, 123, 601–614. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics Identifies Medulloblastoma Subgroups That Are Enriched for Specific Genetic Alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. 1 Developmental roles and clinical significance of Hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [CrossRef]
- Epstein, D.; Marti, E.; Scott, M.; McMahon, A. Antagonizing cAMP-dependent protein kinase A in the dorsal CNS activates a conserved Sonic hedgehog signaling pathway. Development 1996, 122, 2885–2894. [Google Scholar] [CrossRef]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmycupregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Stephen, D.; Joyner, A.; Curran, T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene 2005, 24, 4026–4036. [Google Scholar] [CrossRef] [Green Version]
- Brugières, L.; Remenieras, A.; Pierron, G.; Varlet, P.; Forget, S.; Byrde, V.; Bombled, J.; Puget, S.; Caron, O.; Dufour, C.; et al. High Frequency of Germline SUFU Mutations in Children with Desmoplastic/Nodular Medulloblastoma Younger Than 3 Years of Age. J. Clin. Oncol. 2012, 30, 2087–2093. [Google Scholar] [CrossRef] [Green Version]
- Svärd, J.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, Å.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic Elimination of Suppressor of Fused Reveals an Essential Repressor Function in the Mammalian Hedgehog Signaling Pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef]
- Pietsch, T.; Waha, A.; Koch, A.; Kraus, J.; Albrecht, S.; Tonn, J.; Sörensen, N.; Berthold, F.; Henk, B.; Schmandt, N.; et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997, 57, 2085–2088. [Google Scholar] [PubMed]
- Ellison, D.W.; Kocak, M.; Dalton, J.; Megahed, H.; Lusher, M.E.; Ryan, S.L.; Zhao, W.; Nicholson, S.L.; Taylor, R.E.; Bailey, S.; et al. Definition of Disease-Risk Stratification Groups in Childhood Medulloblastoma Using Combined Clinical, Pathologic, and Molecular Variables. J. Clin. Oncol. 2011, 29, 1400–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, D.W.; Onilude, O.E.; Lindsey, J.C.; Lusher, M.E.; Weston, C.L.; Taylor, R.E.; Pearson, A.D.; Clifford, S.C. β-Catenin Status Predicts a Favorable Outcome in Childhood Medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J. Clin. Oncol. 2005, 23, 7951–7957. [Google Scholar] [CrossRef] [PubMed]
- Li, K.K.-W.; Lau, K.-M.; Ng, H.-K. Signaling pathway and molecular subgroups of medulloblastoma. Int. J. Clin. Exp. Pathol. 2013, 6, 1211–1222. [Google Scholar] [PubMed]
- Skowron, P.; Ramaswamy, V.; Taylor, M.D. Genetic and molecular alterations across medulloblastoma subgroups. Klin. Wochenschr. 2015, 93, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Eberhart, C.G.; Tihan, T.; Burger, P.C. Nuclear Localization and Mutation of β-Catenin in Medulloblastomas. J. Neuropathol. Exp. Neurol. 2000, 59, 333–337. [Google Scholar] [CrossRef] [Green Version]
- da Silva, R.; Marie, S.K.N.; Uno, M.; Matushita, H.; Wakamatsu, A.; Rosemberg, S.; Oba-Shinjo, S.M. CTNNB1, AXIN1 and APC expression analysis of different medulloblastoma variants. Clinics 2013, 68, 167–172. [Google Scholar] [CrossRef]
- Eberhart, C.G.; Kratz, J.; Wang, Y.; Summers, K.; Stearns, D.; Cohen, K.; Dang, C.V.; Burger, P.C. Histopathological and Molecular Prognostic Markers in Medulloblastoma. J. Neuropathol. Exp. Neurol. 2004, 63, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Mahler-Araujo, B.M.; Sankila, A.; Chimelli, L.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. APC Mutations in Sporadic Medulloblastomas. Am. J. Pathol. 2000, 156, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Geron, L.; Salomão, K.B.; Borges, K.S.; Andrade, A.F.; Corrêa, C.A.P.; Scrideli, C.A.; Tone, L.G. Molecular characterization of Wnt pathway and function of β-catenin overexpression in medulloblastoma cell lines. Cytotechnology 2018, 70, 1713–1722. [Google Scholar] [CrossRef]
- Kratz, J.E.; Stearns, D.; Huso, D.L.; Slunt, H.H.; Price, D.L.; Borchelt, D.R.; Eberhart, C.G. Expression of stabilized β-catenin in differentiated neurons of transgenic mice does not result in tumor formation. BMC Cancer 2002, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guessous, F.; Li, Y.; Abounader, R. Signaling pathways in medulloblastoma. J. Cell Physiol. 2008, 217, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Adamson, D.C.; Shi, Q.; Wortham, M.; Northcott, P.A.; Di, C.; Duncan, C.G.; Li, J.; McLendon, R.E.; Bigner, D.D.; Taylor, M.D.; et al. OTX2 Is Critical for the Maintenance and Progression of Shh-Independent Medulloblastomas. Cancer Res. 2010, 70, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, C.; Liao, S.; Adamson, D.C.; Parrett, T.J.; Broderick, D.K.; Shi, Q.; Lengauer, C.; Cummins, J.M.; Velculescu, V.E.; Fults, D.W.; et al. Identification of OTX2 as a Medulloblastoma Oncogene Whose Product can be Targeted by All-Trans Retinoic Acid. Cancer Res. 2005, 65, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Bunt, J.; Hasselt, N.A.; Zwijnenburg, D.A.; Koster, J.; Versteeg, R.; Kool, M. OTX2 sustains a bivalent-like state of OTX2-bound promoters in medulloblastoma by maintaining their H3K27me3 levels. Acta Neuropathol. 2013, 125, 385–394. [Google Scholar] [CrossRef]
- Forget, A.; Martignetti, L.; Puget, S.; Calzone, L.; Brabetz, S.; Picard, D.; Montagud, A.; Liva, S.; Sta, A.; Dingli, F.; et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell 2018, 34, 379–395.e7. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Jeon, H.-Y.; Joo, K.M.; Kim, J.-K.; Jin, J.; Kim, S.H.; Kang, B.G.; Beck, S.; Lee, S.J.; Kim, J.K.; et al. Frizzled 4 Regulates Stemness and Invasiveness of Migrating Glioma Cells Established by Serial Intracranial Transplantation. Cancer Res. 2011, 71, 3066–3075. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Yang, C.; Iyer, R.R.; Yu, A.C.H.; Yong, R.L.; Park, D.M.; Weil, R.J.; Ikejiri, B.; Brady, R.O.; Lonser, R.R.; Zhuang, Z. β-Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc. Natl. Acad. Sci. USA 2012, 109, 6963–6968. [Google Scholar] [CrossRef] [Green Version]
- Pu, P.; Zhang, Z.; Kang, C.; Jiang, R.; Jia, Z.; Wang, G.; Jiang, H. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009, 16, 351–361. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, H.; Chen, Y.; Cheng, X. Significance of beta-catenin and Cyclin D1 express in glio-ma. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2009, 25, 1010–1012. [Google Scholar] [PubMed]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; et al. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2011, 11, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Visioli, A.; Giani, F.; Trivieri, N.; Palumbo, O.; Restelli, S.; Dezi, F.; Mazza, T.; Fusilli, C.; Legnani, F.; et al. Wnt5a Drives an Invasive Phenotype in Human Glioblastoma Stem-like Cells. Cancer Res. 2017, 77, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic Amplification of Multiple Receptor Tyrosine Kinase Genes in Glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.J.; Acquaviva, J.; Chi, D.; Lessard, J.; Zhu, H.; Woolfenden, S.; Bronson, R.T.; Pfannl, R.; White, F.; Housman, D.E.; et al. Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene 2011, 31, 3039–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Tao, B.; Chen, Y.; Guo, Z.; Yang, X.; Peng, L.; Xia, X.; Chen, L. B7-H3 Regulates Glioma Growth and Cell Invasion Through a JAK2/STAT3/Slug-Dependent Signaling Pathway. OncoTargets Ther. 2020, 13, 2215–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, K.M.; Hoeman, C.M.; Becher, O. Children are not just little adults: Recent advances in understanding of diffuse intrinsic pontine glioma biology. Pediatr. Res. 2014, 75, 205–209. [Google Scholar] [CrossRef]
- Werbrouck, C.; Evangelista, C.C.; Lobón-Iglesias, M.-J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Gamble, L.D.; Upton, D.H.; Ung, C.; Yu, D.M.T.; Ehteda, A.; Pandher, R.; Mayoh, C.; Hébert, S.; Jabado, N.; et al. Dual targeting of polyamine synthesis and uptake in diffuse intrinsic pontine gliomas. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Williams, J.R.; Young, C.C.; Vitanza, N.A.; McGrath, M.; Feroze, A.H.; Browd, S.R.; Hauptman, J.S. Progress in diffuse intrinsic pontine glioma: Advocating for stereotactic biopsy in the standard of care. Neurosurg. Focus 2020, 48, E4. [Google Scholar] [CrossRef]
- El-Hashash, A.H.K. Histone H3K27M Mutation in Brain Tumors. Histone Mutat. Cancer 2021, 1283, 43–52. [Google Scholar] [CrossRef]
- Ryall, S.; Tabori, U.; Hawkins, C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol. Commun. 2020, 8, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Et the St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Pfister, S.M.; Jones, D.T.W. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol. 2015, 16, e293–e302. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Donahoe, J.; Brown, T.; James, C.D.; Perry, A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol. Appl. Neurobiol. 2000, 26, 361–367. [Google Scholar] [CrossRef]
- Lau, N.; Feldkamp, M.M.; Roncari, L.; Loehr, A.H.; Shannon, P.; Gutmann, D.H.; Guha, A. Loss of Neurofibromin Is Associated with Activation of RAS/MAPK and PI3-K/AKT Signaling in a Neurofibromatosis 1 Astrocytoma. J. Neuropathol. Exp. Neurol. 2000, 59, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Sievert, A.J.; Fisher, M.J. Pediatric Low-Grade Gliomas. J. child Neurol. 2009, 24, 1397–1408. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.M.; Bell, W.R.; Yuan, M.; Harris, L.; Poore, B.; Arnold, A.; Engle, E.L.; Asnaghi, L.; Lim, M.; Raabe, E.H.; et al. PD-L1 Expression in Pediatric Low-Grade Gliomas Is Independent of BRAF V600E Mutational Status. J. Neuropathol. Exp. Neurol. 2020, 79, 74–85. [Google Scholar] [CrossRef]
- Jang, B.-S.; Kim, I.A. A radiosensitivity gene signature and PD-L1 predict the clinical outcomes of patients with lower grade glioma in TCGA. Radiother. Oncol. 2018, 128, 245–253. [Google Scholar] [CrossRef]
- Mei, J.; Cai, Y.; Xu, R.; Yang, X.; Zhou, W.; Wang, H.; Liu, C. Characterization of the Clinical Significance of PD-1/PD-Ls Expression and Methylation in Patients with Low-Grade Glioma. Technol. Cancer Res. Treat. 2021, 20. [Google Scholar] [CrossRef]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Roliński, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 Blockade and Stereotactic Radiation Produce Long-Term Survival in Mice with Intracranial Gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Latif, M.; Youness, R.A. Why natural killer cells in triple negative breast cancer? World J. Clin. Oncol. 2020, 11, 464–476. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Modena, P.; Lualdi, E.; Facchinetti, F.; Veltman, J.; Reid, J.F.; Minardi, S.; Janssen, I.; Giangaspero, F.; Forni, M.; Finocchiaro, G.; et al. Identification of Tumor-Specific Molecular Signatures in Intracranial Ependymoma and Association with Clinical Characteristics. J. Clin. Oncol. 2006, 24, 5223–5233. [Google Scholar] [CrossRef]
- Conover, J.C.; Doetsch, F.; García-Verdugo, J.M.; Gale, N.W.; Yancopoulos, G.D.; Alvarez-Buylla, A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat. Neurosci. 2000, 3, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Hitoshi, S.; Alexson, T.; Tropepe, V.; Donoviel, D.; Elia, A.J.; Nye, J.S.; Conlon, R.A.; Mak, T.W.; Bernstein, A.; van der Kooy, D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002, 16, 846–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Merino, B.; Hubank, M.; Revesz, T.; Harkness, W.; Hayward, R.; Thompson, D.; Darling, J.L.; Thomas, D.G.; Warr, T.J. Microarray analysis of pediatric ependymoma identifies a cluster of 112 candidate genes including four transcripts at 22q12.1-q13.3. Neuro-Oncology 2005, 7, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bont, J.M.; Packer, R.; Michiels, E.; Boer, M.D.; Pieters, R. Biological background of pediatric medulloblastoma and ependymoma: A review from a translational research perspective. Neuro-Oncology 2008, 10, 1040–1060. [Google Scholar] [CrossRef] [Green Version]
- de Bont, J.M.; van Doorn, J.; Reddingius, R.E.; Graat, G.H.; Passier, M.M.; Boer, M.L.D.; Pieters, R. Various components of the insulin-like growth factor system in tumor tissue, cerebrospinal fluid and peripheral blood of pediatric medulloblastoma and ependymoma patients. Int. J. Cancer 2008, 123, 594–600. [Google Scholar] [CrossRef]
- Chintagumpala, M.; Gajjar, A. Brain Tumors. Pediatr. Clin. North Am. 2015, 62, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Wells, E.M.; Packer, R.J. Pediatric Brain Tumors. Contin. Lifelong Learn. Neurol. 2015, 21, 373–396. [Google Scholar] [CrossRef] [PubMed]
- Fattet, S.; Haberler, C.; Legoix, P.; Varlet, P.; Lellouch-Tubiana, A.; Lair, S.; Delattre, O. Beta-catenin status in paediatric medulloblastomas: Correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J. Pathol. A J. Pathol. Soc. Gt. Britain Irel. 2009, 218, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neglia, J.P.; Robison, L.L.; Stovall, M.; Liu, Y.; Packer, R.J.; Hammond, S.; Yasui, Y.; Kasper, C.E.; Mertens, A.C.; Donaldson, S.S.; et al. New Primary Neoplasms of the Central Nervous System in Survivors of Childhood Cancer: A Report from the Childhood Cancer Survivor Study. JNCI: J. Natl. Cancer Inst. 2006, 98, 1528–1537. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of Medulloblastoma with Hedgehog Pathway Inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I Study of Vismodegib in Children with Recurrent or Refractory Medulloblastoma: A Pediatric Brain Tumor Consortium Study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Lassaletta, A.; Scheinemann, K.; Zelcer, S.M.; Hukin, J.; Wilson, B.A.; Jabado, N.; Carret, A.S.; Lafay-Cousin, L.; Larouche, V.; Hawkins, C.E.; et al. Phase II Weekly Vinblastine for Chemotherapy-Naïve Children with Progressive Low-Grade Glioma: A Canadian Pediatric Brain Tumor Consortium Study. J. Clin. Oncol. 2016, 34, 3537–3543. [Google Scholar] [CrossRef]
- Bouffet, E.; Jakacki, R.; Goldman, S.; Hargrave, D.; Hawkins, C.; Shroff, M.; Hukin, J.; Bartels, U.; Foreman, N.; Kellie, S.; et al. Phase II Study of Weekly Vinblastine in Recurrent or Refractory Pediatric Low-Grade Glioma. J. Clin. Oncol. 2012, 30, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.C.; Siffert, J.; Hukin, J. Clinical Manifestations of Childhood Ependymoma:A Multitude of Syndromes. Pediatr. Neurosurg. 1998, 28, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Youness, R.A.; Gad, A.Z.; Sanber, K.; Ahn, Y.J.; Lee, G.-J.; Khallaf, E.; Hafez, H.M.; Motaal, A.A.; Ahmed, N.; Gad, M.Z. Targeting hydrogen sulphide signaling in breast cancer. J. Adv. Res. 2021, 27, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2015, 15, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youness, R.A.; Assal, R.A.; Motaal, A.A.; Gad, M.Z. A novel role of sONE/NOS3/NO signaling cascade in mediating hydrogen sulphide bilateral effects on triple negative breast cancer progression. Nitric Oxide 2018, 80, 12–23. [Google Scholar] [CrossRef]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.-Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A Review of Hydrogen Sulfide Synthesis, Metabolism, and Measurement: Is Modulation of Hydrogen Sulfide a Novel Therapeutic for Cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Guidotti, T.L. Hydrogen Sulfide. Int. J. Toxicol. 2010, 29, 569–581. [Google Scholar] [CrossRef]
- Nagpure, B.V.; Bian, J.-S. Brain, Learning, and Memory: Role of H2S in Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2015, 230, 193–215. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Kraus, J.P.; Oliveriusová, J.; Sokolová, J.; Krausa, E.; Vlčekc, Č.; De Franchis, R.; MacLean, K.N.; Baoa, L.; Bukovskáa, G.; Pattersond, D.; et al. The Human Cystathionine β-Synthase (CBS) Gene: Complete Sequence, Alternative Splicing, and Polymorphisms. Genomics 1998, 52, 312–324. [Google Scholar] [CrossRef]
- Chasse, J.; Paly, E.; Paris, D.; Paul, V.; Sinet, P.; Kamoun, P.; London, J. Genomic Organization of the Human Cystathionine β-Synthase Gene: Evidence for Various cDNAs. Biochem. Biophys. Res. Commun. 1995, 211, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Konrad, M.A.; Matherly, L.H.; Taub, J.W. Transcriptional regulation of the human cystathionine β-synthase −1b basal promoter: Synergistic transactivation by transcription factors NF-Y and Sp1/Sp3. Biochem. J. 2001, 357, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Renga, B. Hydrogen Sulfide Generation in Mammals: The Molecular Biology of Cystathionine-β-Synthase (CBS) and Cystathionine-γ-Lyase (CSE). Inflamm. Allergy-Drug Targets 2011, 10, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Skovby, F.; Kraus, J.P.; Rosenberg, L.E. Biosynthesis and proteolytic activation of cystathionine beta-synthase in rat liver. J. Biol. Chem. 1984, 259, 588–593. [Google Scholar] [CrossRef]
- Majtan, T.; Pey, A.L.; Gimenez-Mascarell, P.; Martínez-Cruz, L.A.; Szabo, C.; Kožich, V.; Kraus, J.P. Potential pharmacological chaperones for cystathionine beta-synthase-deficient homocystinuria. Handb. Exp. Pharmacol. 2018, 245, 345–383. [Google Scholar] [CrossRef]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of human cystathionine beta-synthase: A unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [Green Version]
- Robert, K.; Vialard, F.; Thiery, E.; Toyama, K.; Sinet, P.-M.; Janel, N.; London, J. Expression of the Cystathionine β Synthase (CBS) Gene During Mouse Development and Immunolocalization in Adult Brain. J. Histochem. Cytochem. 2003, 51, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Kandil, S.; Brennan, L.; McBean, G.J. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-γ-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem. Int. 2010, 56, 611–619. [Google Scholar] [CrossRef]
- Chertok, V.M.; Kotsyuba, A.E.; Kotsyuba, E.P. Cystathionine β-synthase in structural elements of the human brain and spinal cord. Cell Tissue Biol. 2011, 5, 573–579. [Google Scholar] [CrossRef]
- Quere, I.; Paul, V.; Rouillac, C.; Janbon, C.; London, J.; Demaille, J.; Kamoun, P.; Dufier, J.-L.; Abitbol, M.; Chassé, J.-F. Spatial and Temporal Expression of the Cystathionine β-Synthase Gene During Early Human Development. Biochem. Biophys. Res. Commun. 1999, 254, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The Therapeutic Potential of Cystathionine β-Synthetase/Hydrogen Sulfide Inhibition in Cancer. Antioxidants Redox Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, N.; Sarfraz, Y.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Suematsu, M.; Zagzag, D.; Semenza, G.L. Decreased Expression of Cystathionine β-Synthase Promotes Glioma Tumorigenesis. Mol. Cancer Res. 2014, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuhra, K.; Panagaki, T.; Randi, E.B.; Augsburger, F.; Blondel, M.; Friocourt, G.; Herault, Y.; Szabo, C. Mechanism of cystathionine-β-synthase inhibition by disulfiram: The role of bis(N,N-diethyldithiocarbamate)-copper(II). Biochem. Pharmacol. 2020, 182, 114267. [Google Scholar] [CrossRef]
- Druzhyna, N.; Szczesny, B.; Olah, G.; Módis, K.; Asimakopoulou, A.; Pavlidou, A.; Szoleczky, P.; Gerö, D.; Yanagi, K.; Törö, G.; et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine β-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol. Res. 2016, 113, 18–37. [Google Scholar] [CrossRef]
- Mu, T.; Chu, T.; Li, W.; Dong, Q.; Liu, Y. N1, N12-Diacetylspermine Is Elevated in Colorectal Cancer and Promotes Proliferation through the miR-559/CBS Axis in Cancer Cell Lines. J. Oncol. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Shen, Y.; Shen, Z.; Guo, L.; Zhang, Q.; Wang, Z.; Miao, L.; Wang, M.; Wu, J.; Guo, W.; Zhu, Y. MiR-125b-5p is involved in oxygen and glucose deprivation injury in PC-12 cells via CBS/H 2 S pathway. Nitric Oxide 2018, 78, 11–21. [Google Scholar] [CrossRef]
- Zhou, Y.-F.; Song, S.-S.; Tian, M.-X.; Tang, Z.; Wang, H.; Fang, Y.; Qu, W.-F.; Jiang, X.-F.; Tao, C.-Y.; Huang, R.; et al. Cystathionine β-synthase mediated PRRX2/IL-6/STAT3 inactivation suppresses Tregs infiltration and induces apoptosis to inhibit HCC carcinogenesis. J. Immunother. Cancer 2021, 9, e003031. [Google Scholar] [CrossRef]
- Youness, R.; Assal, R.; Khallaf, E.; Hafez, H.; Abdelmotaal, A.; Gad, M. Dual targeting of cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) by miR-4317 displays a synergistic efficacy in repressing breast cancer progression. Ann. Oncol. 2017, 28, v584. [Google Scholar] [CrossRef]
- Lu, Y.; O’Dowd, B.F.; Orrego, H.; Israel, Y. Cloning and nucleotide sequence of human liver cDNA encoding for cystathionine γ-lyase. Biochem. Biophys. Res. Commun. 1992, 189, 749–758. [Google Scholar] [CrossRef]
- Levonen, A.-L.; Lapatto, R.; Saksela, M.; Raivio, K.O. Human cystathionine γ-lyase: Developmental and in vitro expression of two isoforms. Biochem. J. 2000, 347, 291–295. [Google Scholar] [CrossRef]
- Sun, Q.; Collins, R.; Huang, S.; Holmberg-Schiavone, L.; Anand, G.S.; Tan, C.H.; Sivaraman, J. Structural basis for the inhibition mechanism of human cystathionine γ-lyase, an enzyme responsible for the production of H2S. J. Biol. Chem. 2009, 284, 3076–3085. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.P.; Hašek, J.; Kožich, V.; Collard, R.; Venezia, S.; Janošíková, B.; Wang, J.; Stabler, S.P.; Allen, R.H.; Jakobs, C.; et al. Cystathionine γ-lyase: Clinical, metabolic, genetic, and structural studies. Mol. Genet. Metab. 2009, 97, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Wróbel, M.; Czubak, J.; Bronowicka-Adamska, P.; Jurkowska, H.; Adamek, D.; Papla, B. Is Development of High-Grade Gliomas Sulfur-Dependent? Molecules 2014, 19, 21350–21362. [Google Scholar] [CrossRef] [Green Version]
- Cano-Galiano, A.; Oudin, A.; Fack, F.; Allega, M.-F.; Sumpton, D.; Martinez-Garcia, E.; Dittmar, G.; Hau, A.-C.; De Falco, A.; Herold-Mende, C.; et al. Cystathionine-γ-lyase drives antioxidant defense in cysteine-restricted IDH1-mutant astrocytomas. Neuro-Oncology Adv. 2021, 3, vdab057. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, L.; Han, X.; Zhou, Y.; Zhang, T.; Wang, L.; Hong, T.; Zhang, W.; Guo, X.-X.; Sun, J.; et al. Discovery of a Bioactive Inhibitor with a New Scaffold for Cystathionine γ-Lyase. J. Med. Chem. 2019, 62, 1677–1683. [Google Scholar] [CrossRef]
- Wang, L.; Shi, H.; Zhang, X.; Zhang, X.; Liu, Y.; Kang, W.; Shi, X.; Wang, T. I157172, a novel inhibitor of cystathionine γ-lyase, inhibits growth and migration of breast cancer cells via SIRT1-mediated deacetylation of STAT3. Oncol. Rep. 2019, 41, 427–436. [Google Scholar] [CrossRef]
- Yang, G.; Pei, Y.; Cao, Q.; Wang, R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J. Cell Physiol. 2012, 227, 3192–3200. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, X.; Chen, H.-P.; Li, L.; Xie, W.; Lan, G.; Zhao, Z.-W.; Zheng, X.-L.; Wang, Z.-B.; Tang, C.-K. MicroRNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine γ-lyase in THP-1 macrophages. Atherosclerosis 2016, 250, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, S.; Jia, Q.; Zhang, A.; Li, Y.; Zhu, Y.; Lv, S.; Zhang, J. The microRNA in ventricular remodeling: The miR-30 family. Biosci. Rep. 2019, 39, BSR20190788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forgan, L.G.; Donald, J.A. Hydrogen Sulfide. Handb. Horm. 2016, 609-e103C-2. [Google Scholar] [CrossRef]
- Mercaptopyruvate sulfurtransferase as a defense against cyanide toxication: Molecular properties and mode of detoxification. Histol. Histopathol. 1999, 14, 1277–1286. [CrossRef]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)—Hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2018, 154, 104083. [Google Scholar] [CrossRef]
- Yadav, P.K.; Vitvitsky, V.; Carballal, S.; Seravalli, J.; Banerjee, R. Thioredoxin regulates human mercaptopyruvate sul-furtransferase at physiologically-relevant concentrations. J. Biol. Chem. 2020, 295, 6299–6310. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, N.; Tanaka, M.; Tanaka, Y.; Ito, T. Novel Characterization of Antioxidant Enzyme, 3-Mercaptopyruvate Sulfurtransferase-Knockout Mice: Overexpression of the Evolutionarily-Related Enzyme Rhodanese. Antioxidants 2019, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOxproduction. J. Cereb. Blood Flow Metab. 2018, 175, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate Sulfurtransferase Produces Hydrogen Sulfide and Bound Sulfane Sulfur in the Brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef]
- Pedre, B.; Dick, T.P. 3-Mercaptopyruvate sulfurtransferase: An enzyme at the crossroads of sulfane sulfur trafficking. Biol. Chem. 2021, 402, 223–237. [Google Scholar] [CrossRef]
- Jurkowska, H.; Placha, W.; Nagahara, N.; Wróbel, M. The expression and activity of cystathionine-γ-lyase and 3-mercaptopyruvate sulfurtransferase in human neoplastic cell lines. Amino Acids 2011, 41, 151–158. [Google Scholar] [CrossRef]
- Wróbel, M.; Bronowicka-Adamska, P.; Bentke, A. Hydrogen sulfide generation from L-cysteine in the human glioblastoma-astrocytoma U-87 MG and neuroblastoma SHSY5Y cell lines. Acta Biochim. Pol. 2017, 64, 171–176. [Google Scholar] [CrossRef]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and Mechanistic Characterization of Selective Inhibitors of H2S-producing Enzyme: 3-Mercaptopyruvate Sulfurtransferase (3MST) Targeting Active-site Cysteine Persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef] [Green Version]
- Bantzi, M.; Augsburger, F.; Loup, J.; Berset, Y.; Vasilakaki, S.; Myrianthopoulos, V.; Mikros, E.; Szabo, C.; Bochet, C.G. Novel Aryl-Substituted Pyrimidones as Inhibitors of 3-Mercaptopyruvate Sulfurtransferase with Antiproliferative Efficacy in Colon Cancer. J. Med. Chem. 2021, 64, 6221–6240. [Google Scholar] [CrossRef]
- Selem, N.; Nafae, H.; Youness, R.; Gad, M. 28P Hijacking CCAT1/miR-17-5p axis alleviates immune checkpoint blockers resistance in PDL1+ TNBC patients. Ann. Oncol. 2021, 32, S12. [Google Scholar] [CrossRef]
- Nafea, H.; Youness, R.A.; Abou-Aisha, K.; Gad, M.Z. LncRNA HEIH/miR-939-5p interplay modulates triple-negative breast cancer progression through NOS2-induced nitric oxide production. J. Cell Physiol. 2021, 236, 5362–5372. [Google Scholar] [CrossRef]
- Shaalan, Y.M.; Handoussa, H.; Youness, R.A.; Assal, R.A.; El-Khatib, A.H.; Linscheid, M.W.; El Tayebi, H.M.; Abdelaziz, A.I. Destabilizing the interplay between miR-1275 and IGF2BPs by Tamarix articulata and quercetin in hepatocellular carcinoma. Nat. Prod. Res. 2017, 32, 2217–2220. [Google Scholar] [CrossRef]
- Youness, R.A.; Gad, M.Z. Long non-coding RNAs: Functional regulatory players in breast cancer. Non-coding RNA Res. 2019, 4, 36–44. [Google Scholar] [CrossRef]
- Abdel-Latif, M.; Riad, A.; Soliman, R.A.; Elkhouly, A.M.; Nafae, H.; Gad, M.Z.; Youness, R.A. MALAT-1/p53/miR-155/miR-146a ceRNA circuit tuned by methoxylated quercitin glycoside alters immunogenic and oncogenic profiles of breast cancer. Mol. Cell Biochem. 2022, 477, 1281–1293. [Google Scholar] [CrossRef]
- Youness, R.; Gomaa, A. 128P Ex-vivo co-blockade of CD-155/TIGIT and PD-1/PD-L1 using CCAT-1, H19 and MALAT-1 LncRNAs in hepatocellular carcinoma. Ann. Oncol. 2021, 32, S1433. [Google Scholar] [CrossRef]
- Awad, A.R.; Youness, R.A.; Ibrahim, M.; Motaal, A.A.; El-Askary, H.I.; Assal, R.; Gad, M.Z. An acetylated derivative of vitexin halts MDA-MB-231 cellular progression and improves its immunogenic profile through tuning miR- 20a-MICA/B axis. Nat. Prod. Res. 2021, 35, 3126–3130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-R.; Jiang, Y.-Z.; Xu, X.-E.; Yu, K.-D.; Jin, X.; Hu, X.; Zuo, W.-J.; Hao, S.; Wu, J.; Liu, G.-Y.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Nafea, H.; Youness, R.; Abou-Aisha, K.; Gad, M. 27P MiR-939-5p exhibits tumour suppressor activity and immune surveillance manipulation in triple-negative breast cancer. Ann. Oncol. 2021, 32, S11. [Google Scholar] [CrossRef]
- Khater, N.; Youness, R.; Habashy, D.; Gad, M. 51P A novel crosstalk between pyridoxal 5’-phosphate (PLP)-dependent enzymes, CBS and CSE, modulated by MALAT-1/STAT-3 axis. Ann. Oncol. 2021, 32, S1361. [Google Scholar] [CrossRef]
- Soliman, R.A.; Youness, R.A.; Manie, T.M.; Khallaf, E.; El-Shazly, M.; Abdelmohsen, M.; Gad, M.Z. Uncoupling tumor necrosis factor-α and interleukin-10 at tumor immune microenvironment of breast cancer through miR-17-5p/MALAT-1/H19 circuit. Biocell 2022, 46, 769. Available online: https://www.researchgate.net/profile/Mohamed-Gad-33/publication/356356342_Uncoupling_tumor_necrosis_factor-a_and_interleu-kin-10_at_tumor_immune_microenvironment_of_breast_cancer_through_miR-17-5pMALAT-1H19_circuit/links/6196709261f0987720af9e67/Uncoupling (accessed on 20 September 2022). [CrossRef]
- Ramzy, A.; Youness, R.; Manie, T.; Sebak, A.; Mansour, S. 40P MALAT-1/miR-30a-5p competing endogenous (ceRNA) network releases the brakes of immune surveillance in breast cancer through its quadruple targets: PD-L1, MIF, IL-10 and TNF-α. Ann. Oncol. 2021, 32, S1357–S1358. [Google Scholar] [CrossRef]
- Selem, N.; Nafae, H.; Youness, R.; Gad, M. 32P Immunoregulatory loop between let-7a and CCAT1 lncRNA coordinated by c-Myc underlies the PD-1/PD-L1 immunoresistance in triple negative breast cancer patients. Ann. Oncol. 2021, 32, S1355. [Google Scholar] [CrossRef]
- Youness, R.A.; Gad, M.Z. Short Non-coding RNAs: Promising Biopharmaceutical Weapons in Breast Carcinogenesis. Adv. Cancer Nanotheranostics Exp. Pers. Med. 2020, 116–130. [Google Scholar] [CrossRef]
- Eltahtawy, O.; Gomaa, A.; Youness, R. 30P Differential expression of miR-873, miR-181a and miR-17-5p and their correlation with immune checkpoints PD-L1 and CD155 among hepatocellular carcinoma patients. Ann. Oncol. 2021, 32, S1385–S1386. [Google Scholar] [CrossRef]
- Wang, M.; Chen, Y.; Cai, W.; Feng, H.; Du, T.; Liu, W.; Jiang, H.; Pasquarelli, A.; Weizmann, Y.; Wang, X. In situ self-assembling Au-DNA complexes for targeted cancer bioimaging and inhibition. Proc. Natl. Acad. Sci. USA 2020, 117, 308–316. [Google Scholar] [CrossRef]
- Pantier, R.; Chhatbar, K.; Quante, T.; Skourti-Stathaki, K.; Cholewa-Waclaw, J.; Alston, G.; Alexander-Howden, B.; Lee, H.Y.; Cook, A.G.; Spruijt, C.G.; et al. SALL4 controls cell fate in response to DNA base composition. Mol. Cell 2021, 81, 845–858.e8. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.G.R.; Coutinho, A.J.; Pinheiro, M.; Neves, A.R. Nanoparticles for Targeted Brain Drug Delivery: What Do We Know? Int. J. Mol. Sci. 2021, 22, 11654. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.; Koziara, J.M.; Mumper, R.J.; Allen, D.D. Nanoparticle Surface Charges Alter Blood–Brain Barrier Integrity and Permeability. J. Drug Target. 2008, 12, 635–641. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Hanada, S.; Fujioka, K.; Inoue, Y.; Kanaya, F.; Manome, Y.; Yamamoto, K. Cell-Based in Vitro Blood–Brain Barrier Model Can Rapidly Evaluate Nanoparticles’ Brain Permeability in Association with Particle Size and Surface Modification. Int. J. Mol. Sci. 2014, 15, 1812–1825. [Google Scholar] [CrossRef] [Green Version]
- Jo, D.H.; Kim, J.H.; Lee, T.G.; Kim, J.H. Size, surface charge, and shape determine therapeutic effects of nanoparticles on brain and retinal diseases. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1603–1611. [Google Scholar] [CrossRef]
- El-Shafie, S.; Fahmy, S.A.; Ziko, L.; Elzahed, N.; Shoeib, T.; Kakarougkas, A. Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics 2020, 12, 863. [Google Scholar] [CrossRef]
- Yang, Z.; Gao, W.; Liu, Y.-J.; Pang, N.; Qi, X.-R. Delivering siRNA and Chemotherapeutic Molecules Across BBB and BTB for Intracranial Glioblastoma Therapy. Mol. Pharm. 2017, 14, 1012–1022. [Google Scholar] [CrossRef]
- Ravi, V.; Madhankumar, A.B.; Abraham, T.; Slagle-Webb, B.; Connor, J.R. Liposomal delivery of ferritin heavy chain 1 (FTH1) siRNA in patient xenograft derived glioblastoma initiating cells suggests different sensitivities to radiation and distinct survival mechanisms. PLoS ONE 2019, 14, e0221952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Huang, Q.; Wang, F.; Zhang, X.; Hu, J.; Tan, Y.; Huang, N.; Wang, Z.; Wang, Z.; Cheng, Y. Targeted shRNA-loaded liposome complex combined with focused ultrasound for blood brain barrier disruption and suppressing glioma growth. Cancer Lett. 2018, 418, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Azzazy, H.M.E.-S.; Sawy, A.M.; Abdelnaser, A.; Meselhy, M.R.; Shoeib, T.; Fahmy, S.A. Peganum harmala Alkaloids and Tannic Acid Encapsulated in PAMAM Dendrimers: Improved Anticancer Activities as Compared to Doxorubicin. ACS Appl. Polym. Mater. 2022, 4, 7228–7239. [Google Scholar] [CrossRef]
- Qiu, J.; Kong, L.; Cao, X.; Li, A.; Wei, P.; Wang, L.; Mignani, S.; Caminade, A.-M.; Majoral, J.-P.; Shi, X. Enhanced Delivery of Therapeutic siRNA into Glioblastoma Cells Using Dendrimer-Entrapped Gold Nanoparticles Conjugated with β-Cyclodextrin. Nanomaterials 2018, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Liang, Y.; Sang, C.; Mei, C.; Li, X.; Chen, T. Therapeutic nanosystems co-deliver anticancer drugs and oncogene SiRNA to achieve synergetic precise cancer chemo-gene therapy. J. Mater. Chem. B 2018, 6, 3013–3022. [Google Scholar] [CrossRef]
- Danhier, F.; Messaoudi, K.; Lemaire, L.; Benoit, J.-P.; Lagarce, F. Combined anti-Galectin-1 and anti-EGFR siRNA-loaded chitosan-lipid nanocapsules decrease temozolomide resistance in glioblastoma: In vivo evaluation. Int. J. Pharm. 2015, 481, 154–161. [Google Scholar] [CrossRef]
- Van Woensel, M.; Wauthoz, N.; Rosière, R.; Mathieu, V.; Kiss, R.; Lefranc, F.; Steelant, B.; Dilissen, E.; Van Gool, S.W.; Mathivet, T.; et al. Development of siRNA-loaded chitosan nanoparticles targeting Galectin-1 for the treatment of glioblastoma multiforme via intranasal administration. J. Control. Release 2016, 227, 71–81. [Google Scholar] [CrossRef]
- Boche, M.; Pokharkar, V. Quetiapine Nanoemulsion for Intranasal Drug Delivery: Evaluation of Brain-Targeting Efficiency. AAPS PharmSciTech 2017, 18, 686–696. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Schuh, R.S.; Michels, L.R.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Lenz, G.S.; de Oliveira, F.H.; Venturin, G.; Greggio, S.; et al. Nasal Administration of Cationic Nanoemulsions as CD73-siRNA Delivery System for Glioblastoma Treatment: A New Therapeutical Approach. Mol. Neurobiol. 2020, 57, 635–649. [Google Scholar] [CrossRef]
- Ye, C.; Pan, B.; Xu, H.; Zhao, Z.; Shen, J.; Lu, J.; Yu, R.; Liu, H. Co-delivery of GOLPH3 siRNA and gefitinib by cationic lipid-PLGA nanoparticles improves EGFR-targeted therapy for glioma. Klin. Wochenschr. 2019, 97, 1575–1588. [Google Scholar] [CrossRef]
- Linder, B.; Weirauch, U.; Ewe, A.; Uhmann, A.; Seifert, V.; Mittelbronn, M.; Harter, P.N.; Aigner, A.; Kögel, D. Therapeutic Targeting of Stat3 Using Lipopolyplex Nanoparticle-Formulated siRNA in a Syngeneic Orthotopic Mouse Glioma Model. Cancers 2019, 11, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboeita, N.M.; Fahmy, S.A.; El-Sayed, M.M.H.; Azzazy, H.M.E.-S.; Shoeib, T. Enhanced Anticancer Activity of Nedaplatin Loaded onto Copper Nanoparticles Synthesized Using Red Algae. Pharmaceutics 2022, 14, 418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fu, X.; Jia, J.; Wikerholmen, T.; Xi, K.; Kong, Y.; Wang, J.; Chen, H.; Ma, Y.; Li, Z.; et al. Glioblastoma Therapy Using Codelivery of Cisplatin and Glutathione Peroxidase Targeting siRNA from Iron Oxide Nanoparticles. ACS Appl. Mater. Interfaces 2020, 12, 43408–43421. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhao, Z.; Zhang, J.; Xue, W.; Tong, H.; Liu, H.; Zhang, W. Albumin-stabilized manganese-based nanocomposites with sensitive tumor microenvironment responsivity and their application for efficient SiRNA delivery in brain tumors. J. Mater. Chem. B 2020, 8, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.Y.; Alnakhli, M.; Bhardwaj, R.; Apostolou, S.; Sinha, S.; Fraser, C.; Kuchel, T.; Kuss, B.; Voelcker, N.H. Delivery of siRNA in vitro and in vivo using PEI-capped porous silicon nanoparticles to silence MRP1 and inhibit proliferation in glioblastoma. J. Nanobiotechnology 2018, 16, 1–17. [Google Scholar] [CrossRef]
- Li, D.; Gao, C.; Kuang, M.; Xu, M.; Wang, B.; Luo, Y.; Teng, L.; Xie, J. Nanoparticles as Drug Delivery Systems of RNAi in Cancer Therapy. Molecules 2021, 26, 2380. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor | % Prevalence in Children | Age (Years) | WHO Grade | 5 Years Overall Survival | Prognosis | Predisposed Genetic Mutation | Standard Treatment | Suspected Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| 2% | >3 years old | - | 95% | Very good |
| Surgery, Radiation, chemotherapy |
| [7,9,74,75,76] |
| 6% | All over Age spectrum | - | 50–75% with metastasis or <50% with TP53 mutation | Poor |
|
| Small molecule inhibitors harbor better results for adults harbor than children | [7,9,77,78,79] |
| 5% | Mostly infants and toddlers | - | 50% | Extremely poor |
| Surgery, Radiation, chemotherapy | Carries worst prognosis with current therapies | [7,9] |
| 7% | Mostly children and teenagers | - | 90% or 50–75% according to mutation | Intermediate |
| Surgery, Radiation, chemotherapy | No breakthrough in treatment options | [7,9,77] |
| |||||||||
| |||||||||
| 1.5% | 0–19 | III | 32% | Poor prognosis | Thalamic tumors have FGFR1 mutations + H3.3 point mutations |
|
| [7,9,80] |
| 2.9% | 0–19 | IV | 18% | Poor prognosis | DNA methylations | |||
| |||||||||
| 15.6% | 0–14 | I | 97% | Excellent | BRAF V600E |
|
| [7,9,81,82] |
| |||||||||
| N/A | Infants and young children | II | N/A | Worst prognosis | Balanced Genome |
|
| [7,9,83] |
| N/A | All over age spectrum | III | N/A | Worst prognosis | Chromotripsis, RELA fusion | |||
| Tumor Type | Before 2016 | After 2016 | ||
|---|---|---|---|---|
| Standard Treatment | Outcomes | New Therapeutic Approaches | Outcomes | |
| Medulloblastoma | Surgery, radiation, and chemotherapy |
|
| Better therapeutic effects in adults versus children. |
| Glioma |
|
| Targeted therapies such as erlotinib and ematinib. | Minimal benefits for pediatric AA and GBM. |
| Ependymoma |
|
| Epigenetic demethylation drugs. | Under clinical research |
| Class | Inhibitor | Reference |
|---|---|---|
| Pharmacological inhibitors | Aminooxyacetic acid (AOAA) | [105] |
| Copper diethyldithiocarbamate [Cu(DDC)2]—disulfiram metabolite | [106] | |
| Benserazide | [107] | |
| miRNAs | miR-559 | [108] |
| miR-125b-5p | [109] | |
| miR-24-3p | [110] | |
| miR-4317 | [111] | |
| shRNAs | shCBS#07 | [104] |
| Class | Inhibitor | Reference |
|---|---|---|
| Pharmacological inhibitors | propargylglycine (PAG) | [119] |
| Aminooxyacetic acid (AOAA) | [105] | |
| aurintricarboxylic acid (NSC4056) | [120] | |
| β-cyanoalanine (BCA) | [105] | |
| L-aminoethoxyvinylglycine (AVG) | [105] | |
| Hydroxylamine | [105] | |
| I157172 | [121] | |
| miRNAs | miR-21 | [122] |
| miR-186 | [123] | |
| miR-30 | [124] | |
| miR-4317 | [111] |
| Class | Inhibitor | Reference |
|---|---|---|
| Pharmacological inhibitors | HMPSNE: 2-[(4-hydroxy-6-methylpyrimidin-2-yl) sulfanyl]-1-(naphthalen-1-yl)ethan-1-one | [135] |
| 2-(2-Naphthalen-1-yl-2-oxo-ethylsulfanyl)-3H-pyrimidin-4-one | [136] |
| Nanoplatform | Therapeutic RNA Target | Surface Decoration | Chemotherapeutic Agent | Advantages | Ref |
|---|---|---|---|---|---|
| Cationic liposomes | VEGF | Angiopep-2 and neuro pilin-1 receptor | docetaxel |
| [161] |
| Liposomes | FTH1 | - | - |
| [162] |
| Liposomes | CD13 receptors | Asn-Gly-Arg (NGR) peptide-targeting ligand | - |
| [163] |
| β-cyclodextrin (β-CD)-coated PAMAM | Bcl-2 VEGF | - | - |
| [165] |
| PAMAM dendrimers | c-Myc | arginine-glycine-aspartic acid (RGD) peptide | doxorubicin (DOX) loaded selenium NPs |
| [166] |
| Chitosan-Lipids NPs | Galectin-1 EGFR | - | - |
| [167] |
| Chitosan NPs | Galectin-1 | - | - |
| [168] |
| Lipid-based nanoemulsions | CD37 | - | - |
| [170] |
| Cationic lipid-poly (lactic-co-glycolic acid) (PLGA) NPs | Golgi phosphoprotein 3 (GOLPH3) | Angiopep-2 | Gefitinib |
| [171] |
| Lipopolyplexes | STAT3 | - |
| [172] | |
| Iron oxide NPs (IONPs) | GPX4 | - | Cisplatin |
| [173] |
| Manganese-based NPs | VEGF | Bovine serum albumin | - |
| [175] |
| Porous silica NPs | MRP-1 | PEI |
| [176] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahmy, S.A.; Dawoud, A.; Zeinelabdeen, Y.A.; Kiriacos, C.J.; Daniel, K.A.; Eltahtawy, O.; Abdelhalim, M.M.; Braoudaki, M.; Youness, R.A. Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery. Cancers 2022, 14, 5244. https://doi.org/10.3390/cancers14215244
Fahmy SA, Dawoud A, Zeinelabdeen YA, Kiriacos CJ, Daniel KA, Eltahtawy O, Abdelhalim MM, Braoudaki M, Youness RA. Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery. Cancers. 2022; 14(21):5244. https://doi.org/10.3390/cancers14215244
Chicago/Turabian StyleFahmy, Sherif Ashraf, Alyaa Dawoud, Yousra Ahmed Zeinelabdeen, Caroline Joseph Kiriacos, Kerolos Ashraf Daniel, Omar Eltahtawy, Miriam Mokhtar Abdelhalim, Maria Braoudaki, and Rana A. Youness. 2022. "Molecular Engines, Therapeutic Targets, and Challenges in Pediatric Brain Tumors: A Special Emphasis on Hydrogen Sulfide and RNA-Based Nano-Delivery" Cancers 14, no. 21: 5244. https://doi.org/10.3390/cancers14215244