A Stilbenoid Isorhapontigenin as a Potential Anti-Cancer Agent against Breast Cancer through Inhibiting Sphingosine Kinases/Tubulin Stabilization

 , , and
, , and
Abstract
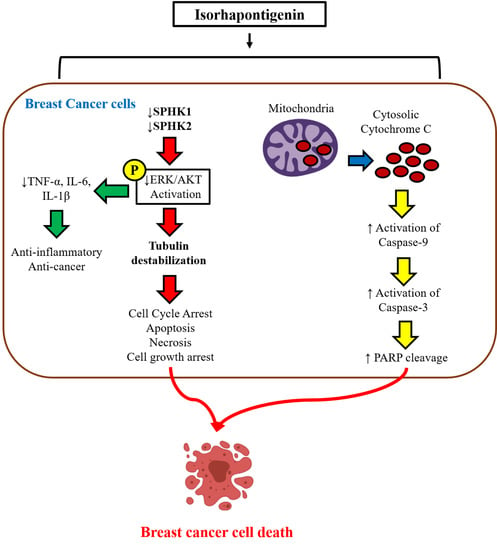
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Western Blot Analysis
2.4. BrdU Proliferation Staining Assay and Immunofluorescence (IF) Labeling
2.5. Receptor X-Ray Structure
2.6. Ligand Preparation
2.7. Structure-Based Virtual Screening
2.8. SPHK Enzyme Activity Assay
2.9. ROS Production Assay
2.10. Measurement of Cell Viability (MTT Assay)
2.11. BrdU and Sulforhodamine B (SRB) Proliferation Assay
2.12. LDH Assay for Cytotoxicity
2.13. Annexin V/PI Staining
2.14. Transfection with SPHK1 & SPHK2 siRNA
2.15. Measurement of TNF-α, IL-6, and IL-1β Production
2.16. Cell Cycle Analysis
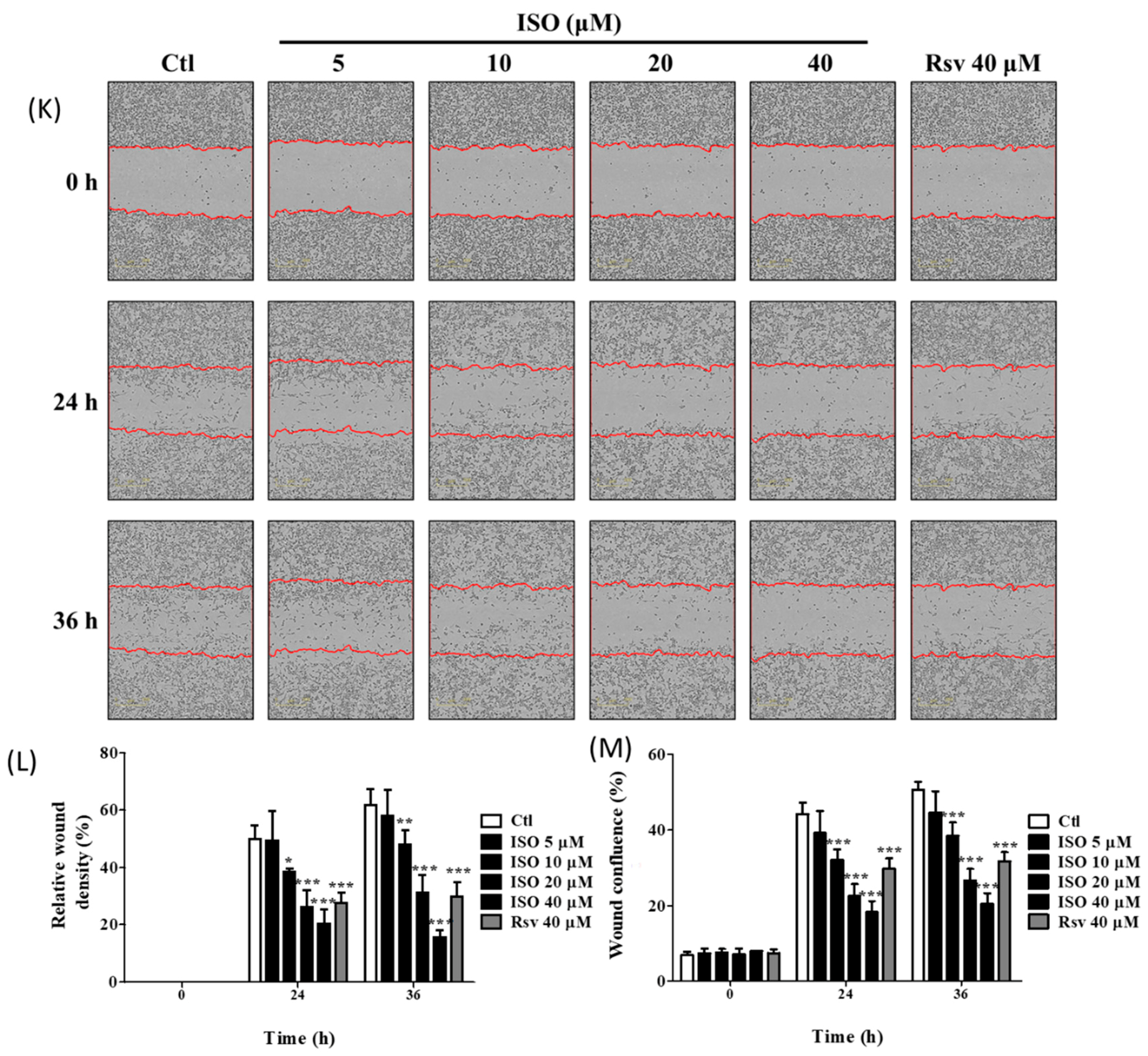
2.17. Cell Migration Assay
2.18. Statistical Analysis
3. Results
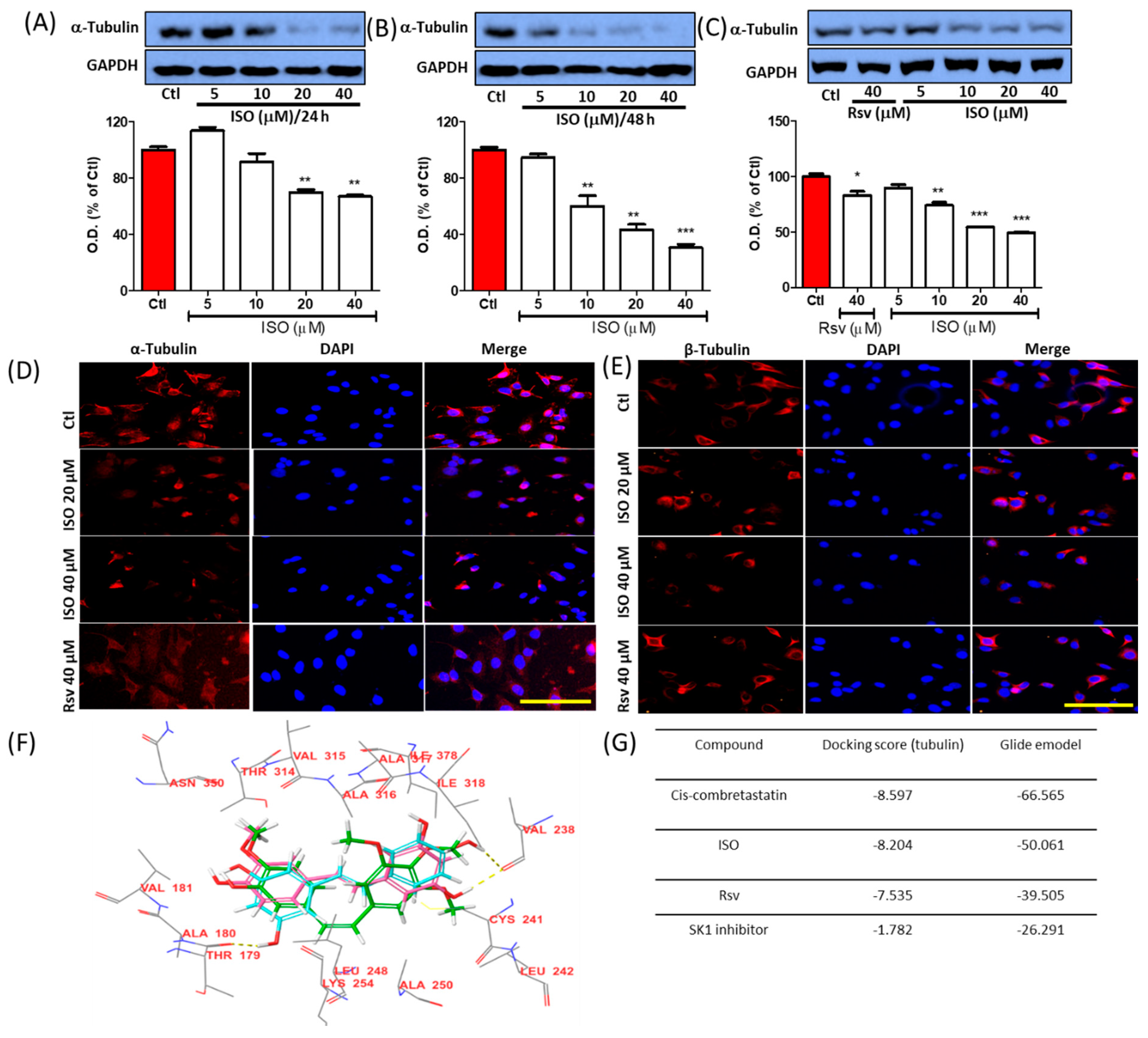
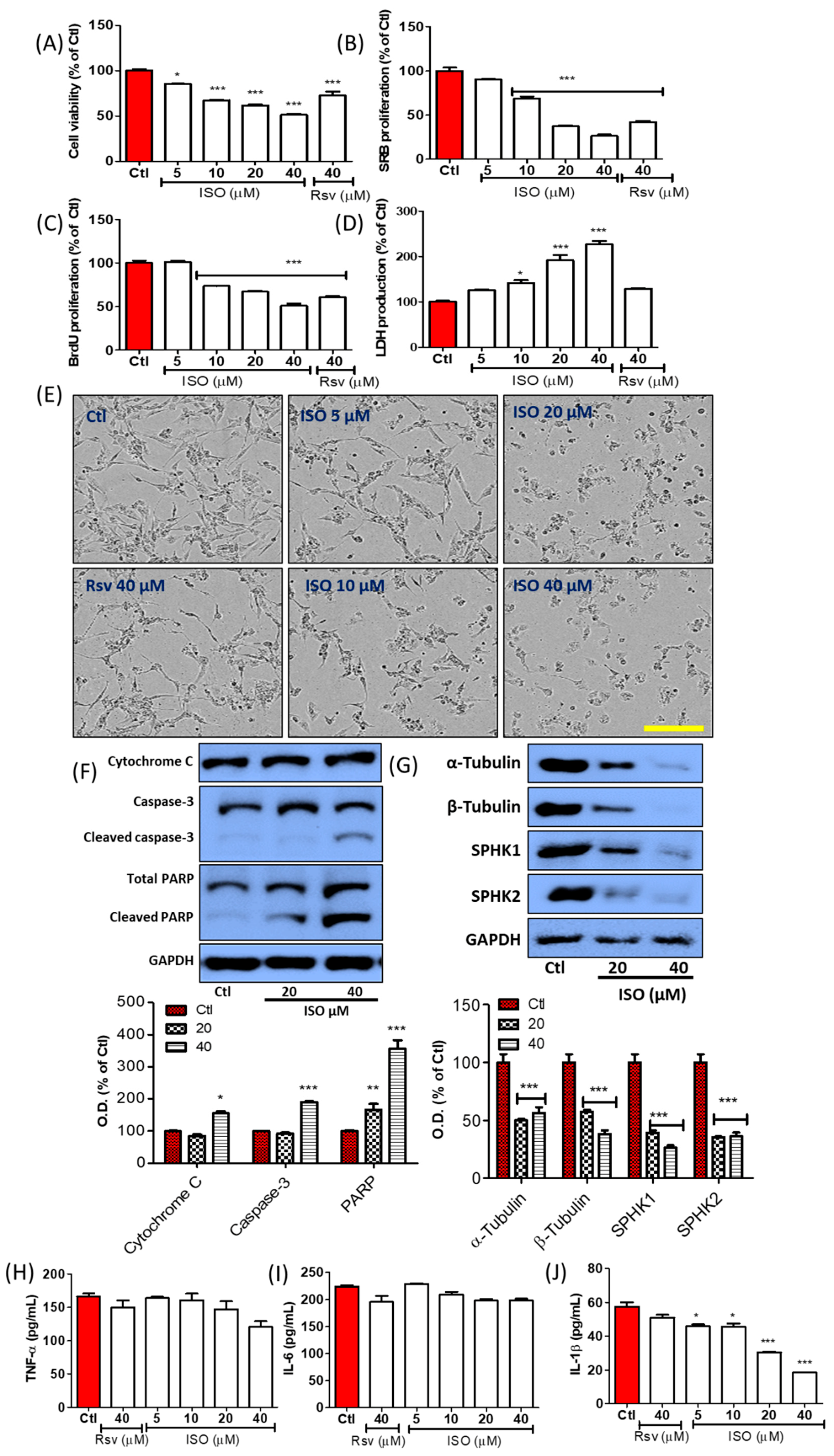
3.1. ISO Induces Tubulin Destabilization
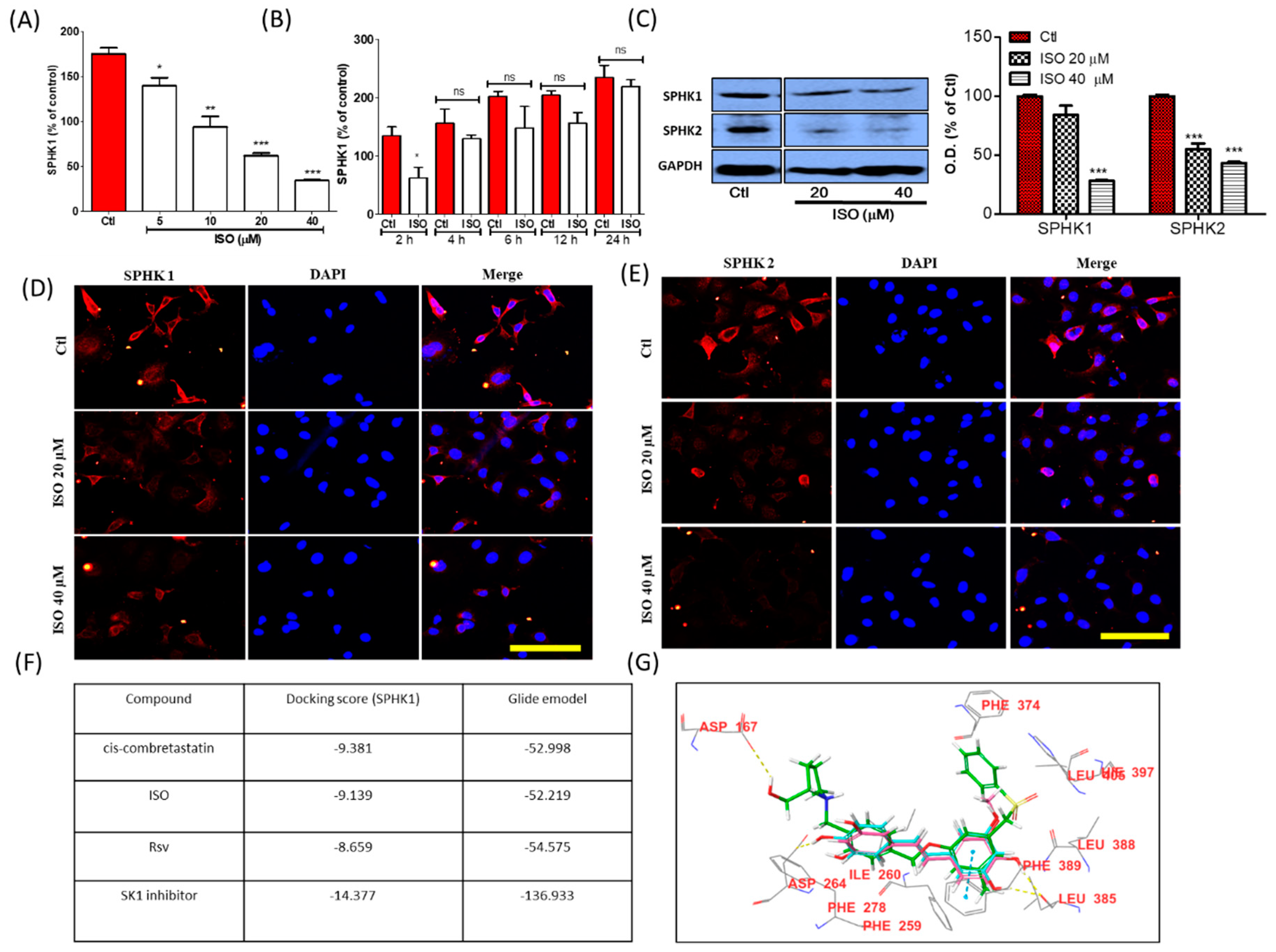
3.2. ISO Alters the Activity and Expression of Sphingosine Kinases (SPHK) in MCF7 Cells
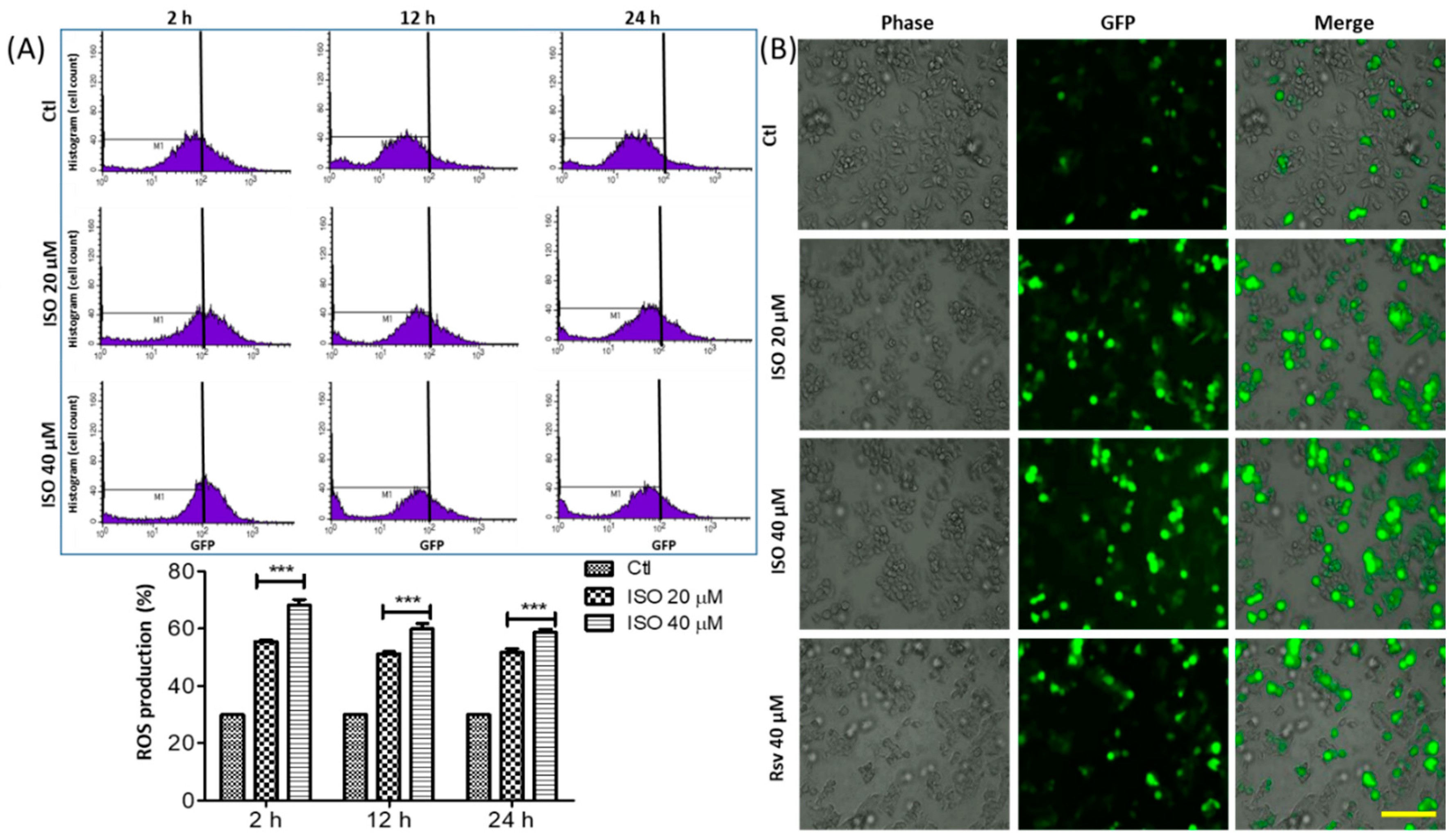
3.3. ISO Treatment Induced ROS Production in MCF7 Cells
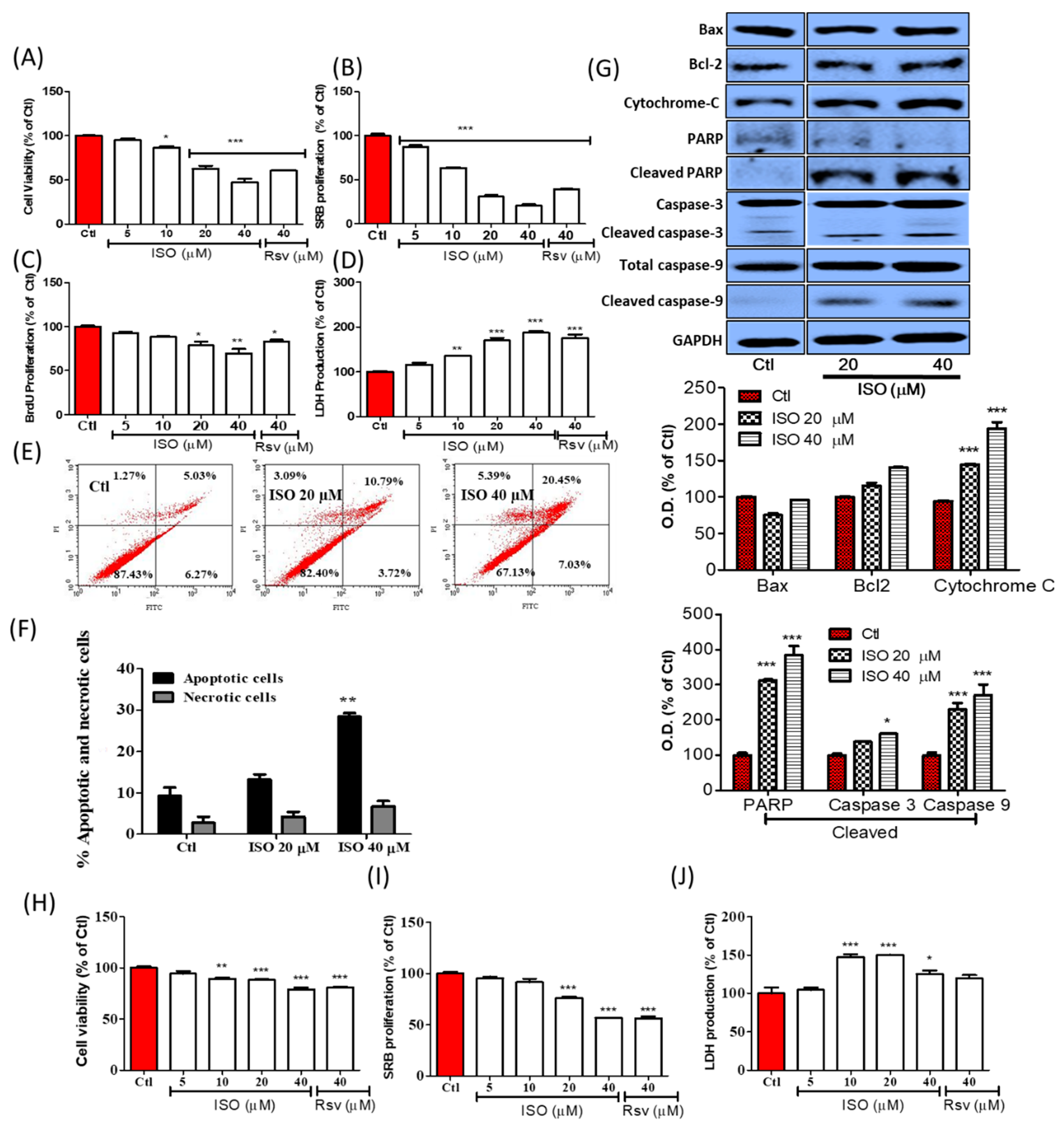
3.4. ISO Induced Breast Cancer Cell Death through Caspase-Dependent Pathways
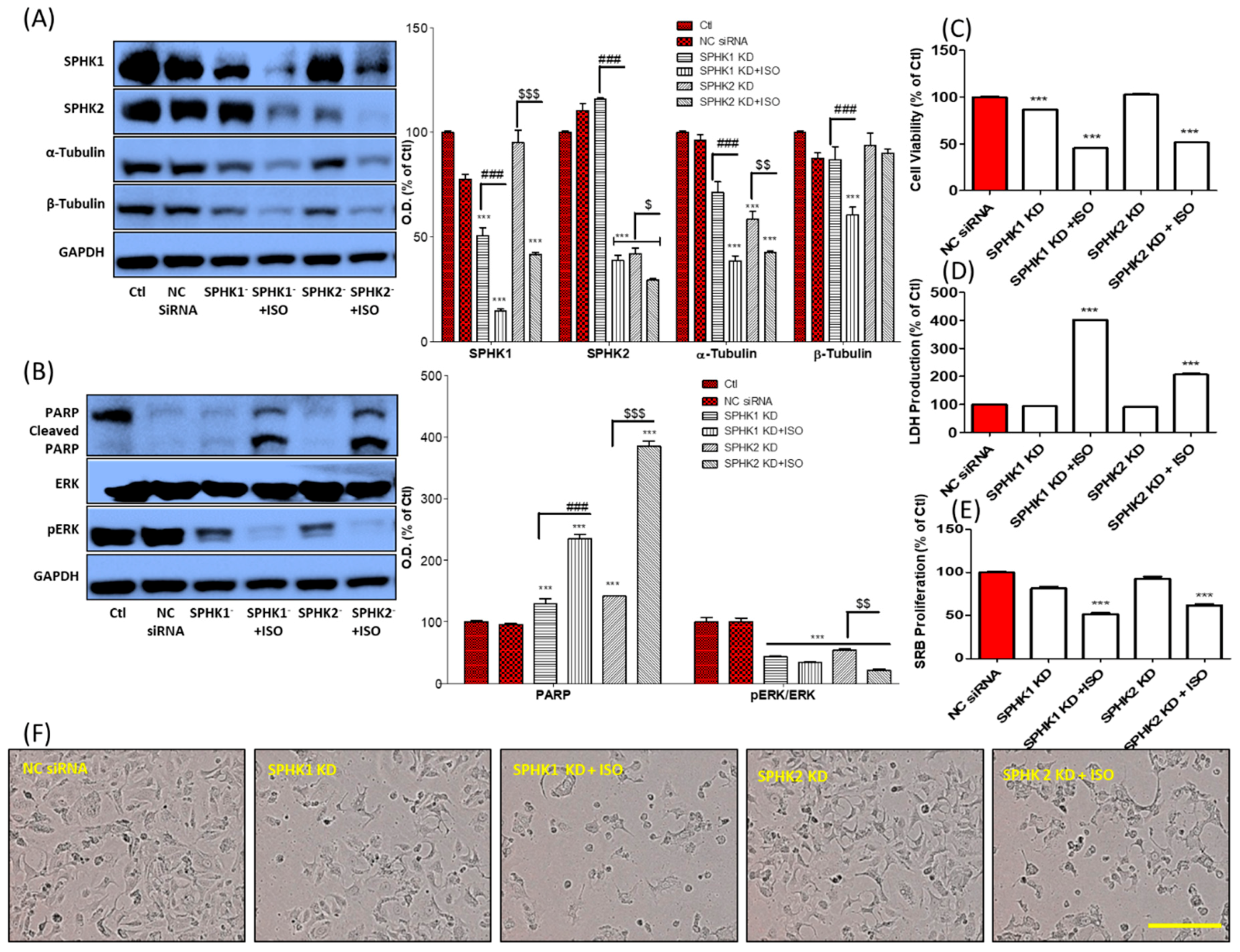
3.5. ISO-Mediated SPHK1/2 Inhibition Is Responsible for Tubulin Destabilization, Cell Death, and Growth Arrest in MCF7 Cells
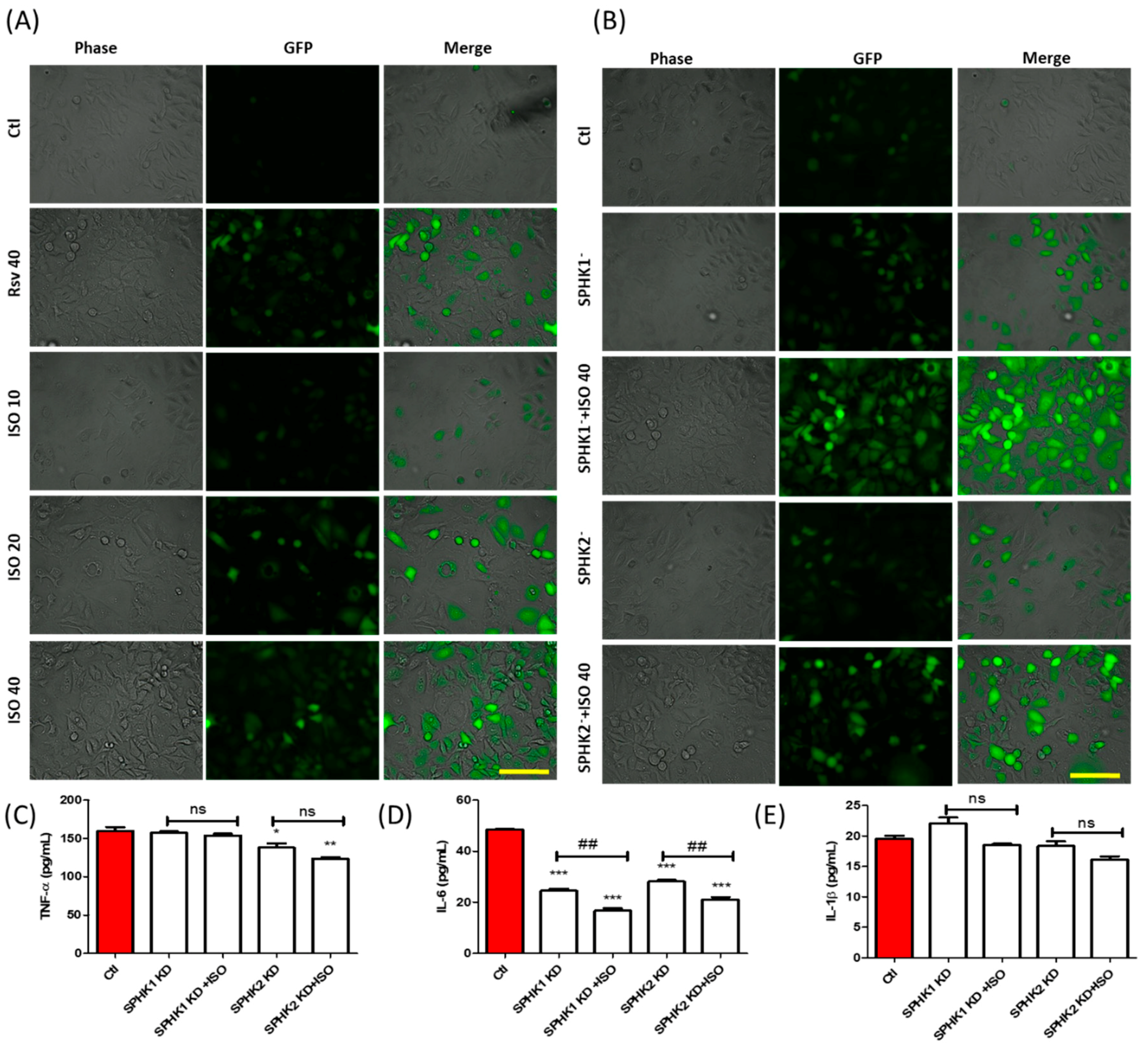
3.6. ISO-Mediated ROS Production and Anti-Inflammation in MCF7 Cells Could Be through SPHK1/SPHK2 Inhibition
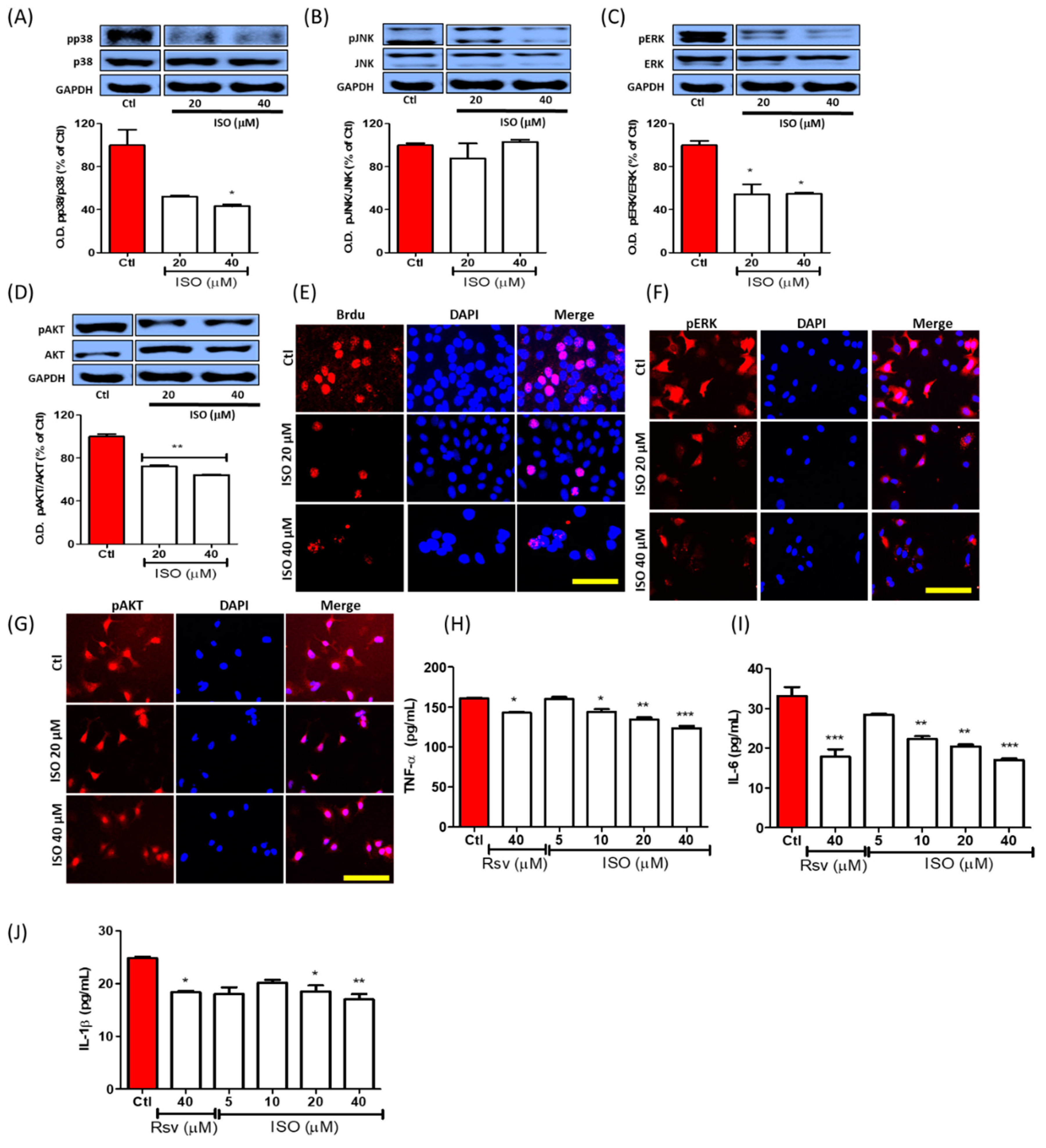
3.7. ISO Inhibited Phosphorylation of the MAPK/Pi3K Signaling Pathway and Pro-Inflammatory Cytokine Production in MCF7 Cells
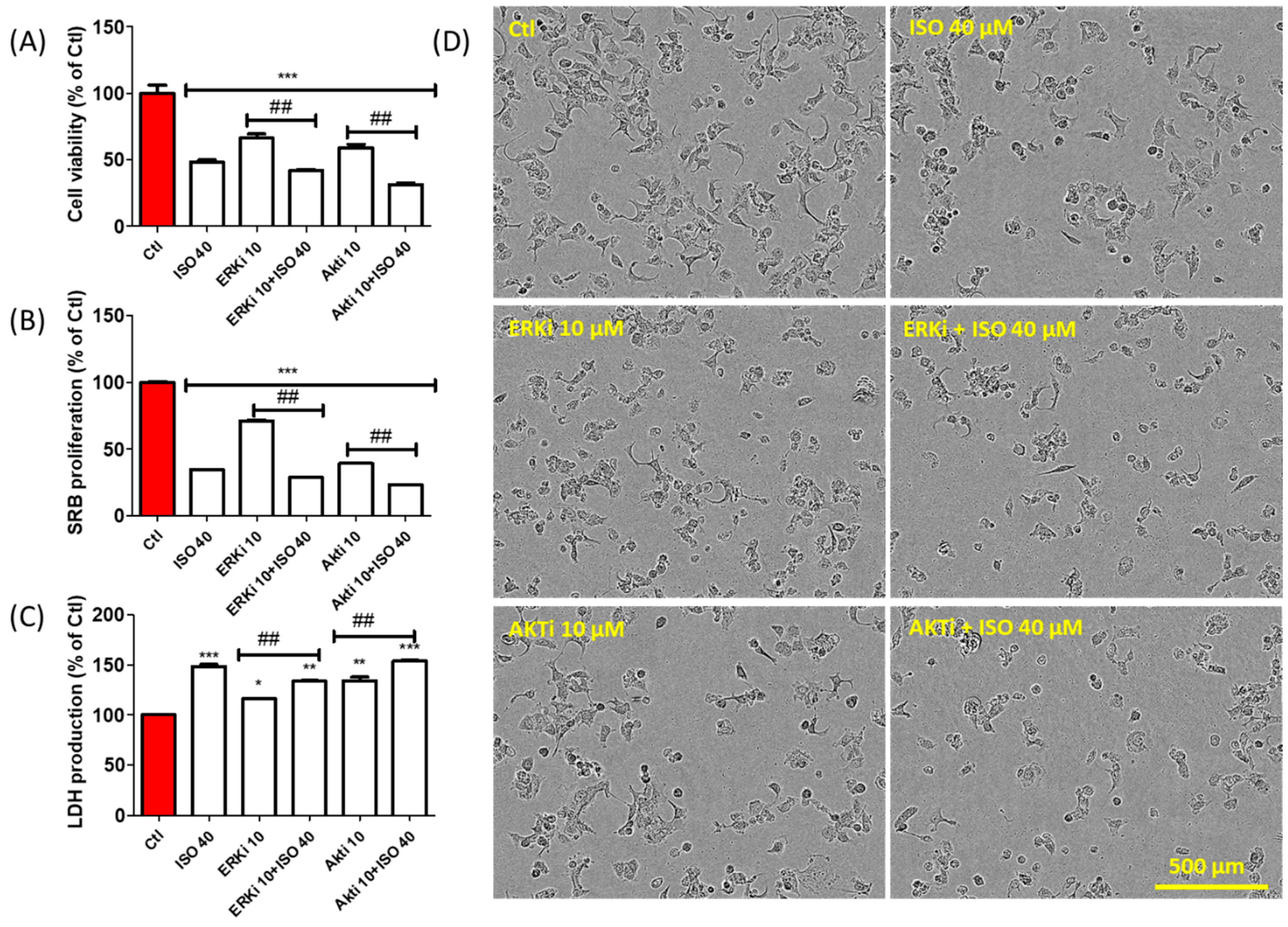
3.8. ISO-Mediated Anti-Proliferation Was further Increased by Co-Treatment with ERK and AKT Inhibitor
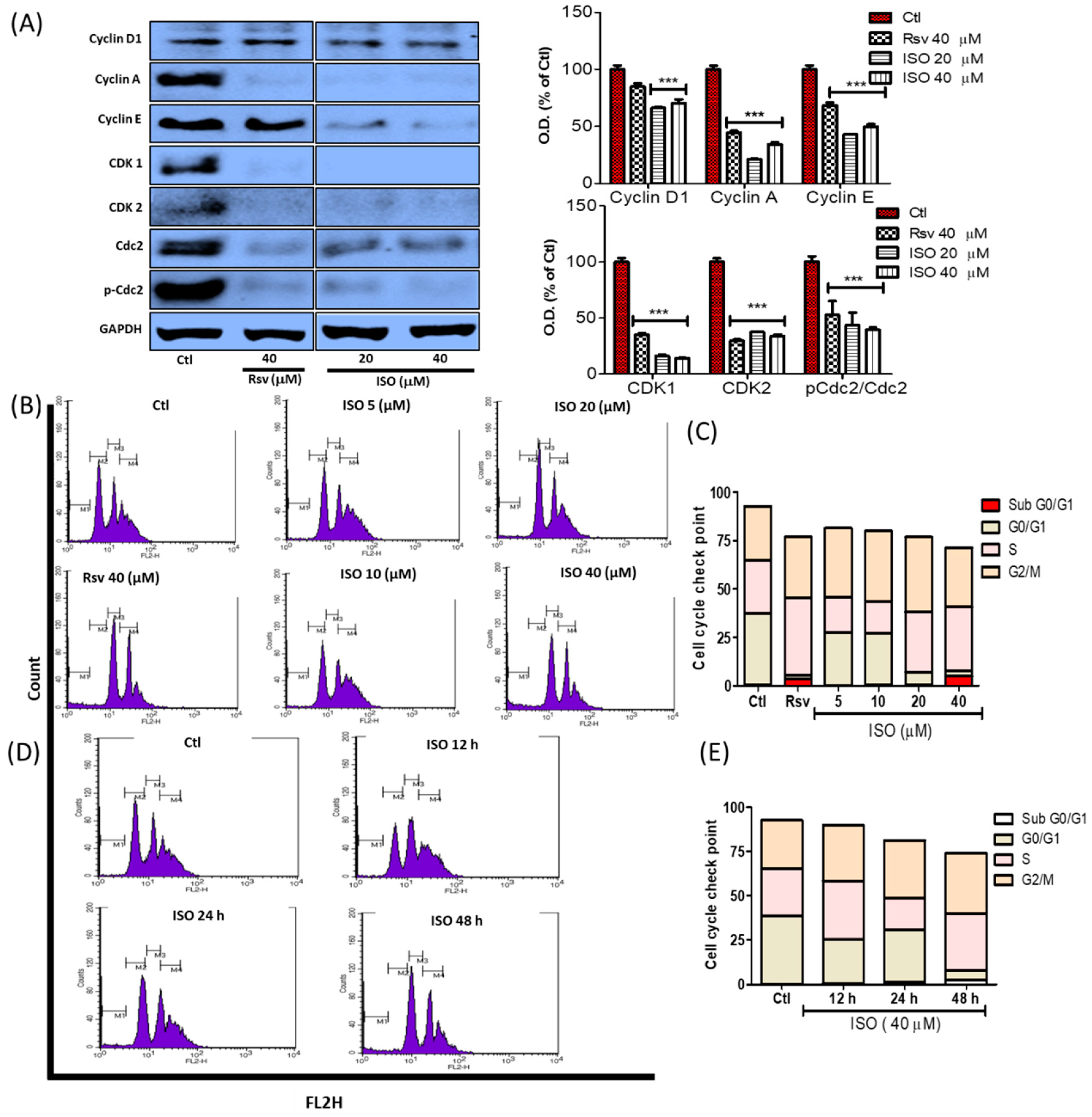
3.9. ISO Regulates G0/G1 and G2/M Phase Arrest in MCF7 Cells
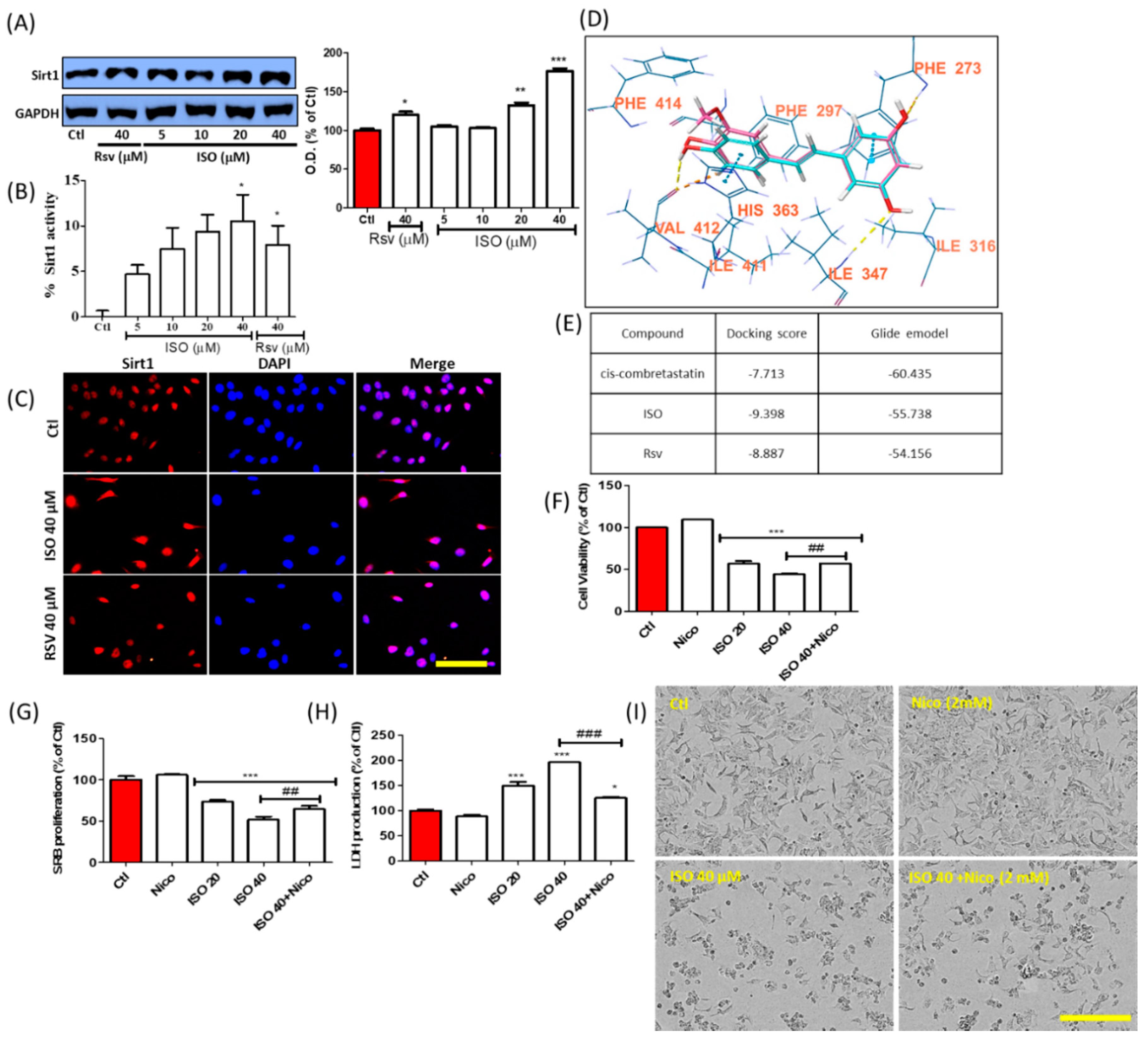
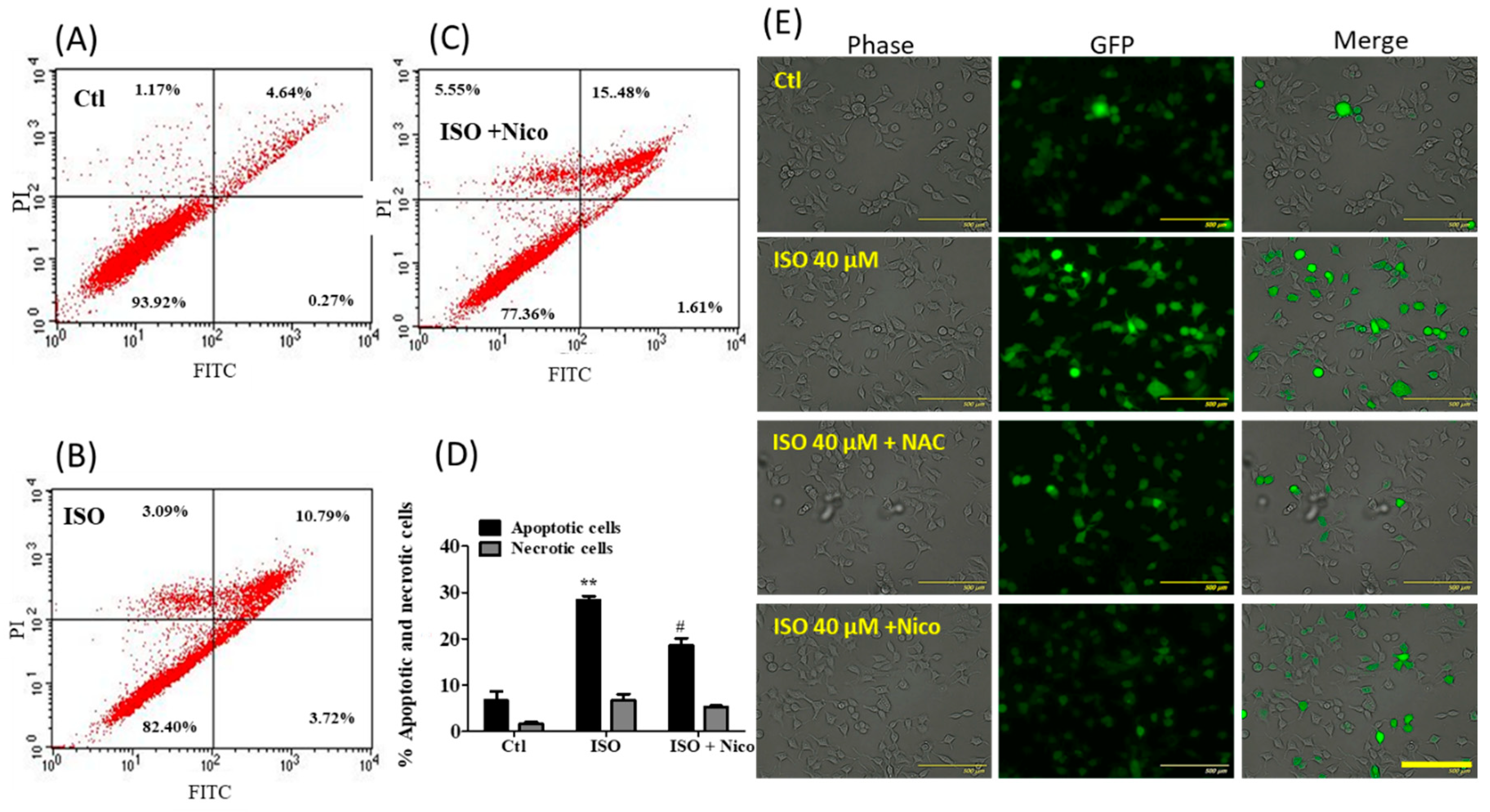
3.10. ISO-Mediated Anti-Cancer Effect Was Recovered by Sirt1 Inhibitor Nicotinamide
3.11. ISO Induced MDA-MB-231 Cell Death and Growth Arrest
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, H., Jr.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12, 1179554918790563. [Google Scholar] [CrossRef] [PubMed]
- Tariq, K.; Rana, F. TNBC vs. Non-TNBC: A Five-Year Retrospective Review of Differences in Mean Age, Family History, Smoking History and Stage at Diagnosis at an Inner City University Program. World J. Oncol. 2013, 4, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sledge, G.W.; Mamounas, E.P.; Hortobagyi, G.N.; Burstein, H.J.; Goodwin, P.J.; Wolff, A.C. Past, present, and future challenges in breast cancer treatment. J. Clin. Oncol. 2014, 32, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.K.; George, J.; Ahmad, N. Resveratrol-based combinatorial strategies for cancer management. Ann. N. Y. Acad. Sci. 2013, 1290, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [Green Version]
- Hengst, J.A.; Yun, J.K. Sphingosine kinase: A key to solving the ‘French Paradox’? Br. J. Pharmacol. 2012, 166, 1603–1604. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.G.; Gray, A.I.; Pyne, S.; Pyne, N.J. Resveratrol dimers are novel sphingosine kinase 1 inhibitors and affect sphingosine kinase 1 expression and cancer cell growth and survival. Br. J. Pharmacol. 2012, 166, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; He, Y.Y. Targeting the AMP-Activated Protein Kinase for Cancer Prevention and Therapy. Front. Oncol. 2013, 3, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horgan, X.J.; Tatum, H.; Brannan, E.; Paull, D.H.; Rhodes, L.V. Resveratrol analogues surprisingly effective against triplenegative breast cancer, independent of ERalpha. Oncol. Rep. 2019, 41, 3517–3526. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.; Fu, H.; Zheng, Z.; Liu, T.; Ji, S.; Li, G. Resveratrol inhibits proliferation and migration through SIRT1 mediated posttranslational modification of PI3K/AKT signaling in hepatocellular carcinoma cells. Mol. Med. Rep. 2017, 16, 8037–8044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.W.; Kang, H.J.; Kim, H.J.; Kong, Y.; Brown, M.L.; Bae, I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget 2013, 4, 984–994. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Nagahashi, M.; Rashid, O.M.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in the tumor microenvironment and its clinical implications. Tumour Biol. 2017, 39, 1010428317699133. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Wang, J.; Guan, H.; Wu, J.; Xu, R.; Wang, M.; Rong, X.; Huang, K.; Huang, J.; Liao, Q.; et al. SphK1 confers resistance to apoptosis in gastric cancer cells by downregulating Bim via stimulating Akt/FoxO3a signaling. Oncol. Rep. 2014, 32, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Hannun, Y.A.; Proia, R.L.; Brenner, D.A. Roles of AKT and sphingosine kinase in the antiapoptotic effects of bile duct ligation in mouse liver. Hepatology 2005, 42, 1320–1328. [Google Scholar] [CrossRef]
- Pyne, S.; Edwards, J.; Ohotski, J.; Pyne, N.J. Sphingosine 1-phosphate receptors and sphingosine kinase 1: Novel biomarkers for clinical prognosis in breast, prostate, and hematological cancers. Front. Oncol. 2012, 2, 168. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, J.; Nagahashi, M.; Takabe, K.; Wakai, T. Clinical Impact of Sphingosine-1-Phosphate in Breast Cancer. Mediat. Inflamm. 2017, 2017, 2076239. [Google Scholar] [CrossRef] [Green Version]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 2011, 11, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-phosphate transporters as targets for cancer therapy. BioMed Res. Int. 2014, 2014, 651727. [Google Scholar] [CrossRef] [PubMed]
- Xun, C.; Chen, M.B.; Qi, L.; Zhang, T.-N.; Peng, X.; Ning, L.; Chen, Z.-X.; Wang, L.-W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, L.; Qi, Y.; Chen, J.; Kaczorowski, D.; Di, W.; Wang, W.; Xia, P. Sphingosine kinase (SphK) 1 and SphK2 play equivalent roles in mediating insulin’s mitogenic action. Mol. Endocrinol. 2014, 28, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hengst, J.A.; Dick, T.E.; Sharma, A.; Doi, K.; Hegde, S.; Tan, S.F.; Geffert, L.M.; Fox, T.E.; Sharma, A.K.; Desai, D.; et al. SKI-178: A Multitargeted Inhibitor of Sphingosine Kinase and Microtubule Dynamics Demonstrating Therapeutic Efficacy in Acute Myeloid Leukemia Models. Cancer Transl. Med. 2017, 3, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakchaisri, K.; Kim, S.O.; Hwang, J.; Soung, N.K.; Lee, K.H.; Choi, T.W.; Lee, Y.; Park, C.M.; Thimmegowda, N.R.; Lee, P.Y.; et al. Anticancer activity of a novel small molecule tubulin inhibitor STK899704. PLoS ONE 2017, 12, e0173311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.; Gopalakrishnan, V.; Hegde, M.; Kumar, S.; Karki, S.S.; Raghavan, S.C.; Choudhary, B. A Novel Resveratrol Based Tubulin Inhibitor Induces Mitotic Arrest and Activates Apoptosis in Cancer Cells. Sci. Rep. 2016, 6, 34653. [Google Scholar] [CrossRef] [Green Version]
- Sergides, C.; Chirila, M.; Silvestro, L.; Pitta, D.; Pittas, A. Bioavailability and safety study of resveratrol 500 mg tablets in healthy male and female volunteers. Exp. Ther. Med. 2016, 11, 164–170. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, E.; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef]
- Johnson, J.J.; Nihal, M.; Siddiqui, I.A.; Scarlett, C.O.; Bailey, H.H.; Mukhtar, H.; Ahmad, N. Enhancing the bioavailability of resveratrol by combining it with piperine. Mol. Nutr. Food Res. 2011, 55, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhu, H.L. Resveratrol and its analogues: Promising antitumor agents. Anticancer Agents Med. Chem. 2011, 11, 479–490. [Google Scholar]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marin, M.I.; Guerrero, R.F.; Garcia-Parrilla, M.C.; Puertas, B.; Richard, T.; Rodriguez-Werner, M.A.; Winterhalter, P.; Monti, J.P.; Cantos-Villar, E. Isorhapontigenin: A novel bioactive stilbene from wine grapes. Food Chem. 2012, 135, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.L.; Lin, M.; Liu, G.T. Antioxidative activity of natural isorhapontigenin. Jpn. J. Pharmacol. 2001, 87, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, H.; Tewtrakul, S.; Morikawa, T.; Yoshikawa, M. Anti-allergic activity of stilbenes from Korean rhubarb (Rheum undulatum L.): Structure requirements for inhibition of antigen-induced degranulation and their effects on the release of TNF-alpha and IL-4 in RBL-2H3 cells. Bioorg. Med. Chem. 2004, 12, 4871–4876. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.L.; Yang, F.; Zhu, M.; Zhou, D.; Lin, M.; Lee, S.M.; Wang, Y.T.; Du, G.H. In vitro anti-influenza viral activities of stilbenoids from the lianas of Gnetum pendulum. Planta Med. 2010, 76, 1874–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.M. Cardioprotective effect of resveratrol analogue isorhapontigenin versus omega-3 fatty acids in isoproterenol-induced myocardial infarction in rats. J. Physiol. Biochem. 2016, 72, 469–484. [Google Scholar] [CrossRef]
- Yeo, S.C.M.; Fenwick, P.S.; Barnes, P.J.; Lin, H.S.; Donnelly, L.E. Isorhapontigenin, a bioavailable dietary polyphenol, suppresses airway epithelial cell inflammation through a corticosteroid-independent mechanism. Br. J. Pharmacol. 2017, 174, 2043–2059. [Google Scholar] [CrossRef]
- Dai, Y.; Yeo, S.C.M.; Barnes, P.J.; Donnelly, L.E.; Loo, L.C.; Lin, H.S. Pre-clinical Pharmacokinetic and Metabolomic Analyses of Isorhapontigenin, a Dietary Resveratrol Derivative. Front. Pharmacol. 2018, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Yu, Y.; Hou, Q.; Zheng, X.; Zhang, M.; Zhang, D.; Li, J.; Wu, X.R.; Huang, C. The Chinese herb isolate isorhapontigenin induces apoptosis in human cancer cells by down-regulating overexpression of antiapoptotic protein XIAP. J. Biol. Chem. 2012, 287, 35234–35243. [Google Scholar] [CrossRef] [Green Version]
- Mohammadizadeh, F.; Hani, M.; Ranaee, M.; Bagheri, M. Role of cyclin D1 in breast carcinoma. J. Res. Med. Sci. 2013, 18, 1021–1025. [Google Scholar]
- Zhu, Y.; Murphy, A.; Kluz, T.; Li, B.; Costa, M.; Huang, C.; Sun, H. Abstract 5857: Isorhapontigenin inhibits triple-negative breast cancer via activating NRF2-mediated pathway. Cancer Res. 2018, 78, 5857. [Google Scholar]
- Mirmalek, S.A.; Azizi, M.A.; Jangholi, E.; Yadollah-Damavandi, S.; Javidi, M.A.; Parsa, Y.; Parsa, T.; Salimi-Tabatabaee, S.A.; Ghasemzadeh Kolagar, H.; Alizadeh-Navaei, R. Cytotoxic and apoptogenic effect of hypericin, the bioactive component of Hypericum perforatum on the MCF-7 human breast cancer cell line. Cancer Cell Int. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.K.; Rajanikant, G.K. Computational repositioning and experimental validation of approved drugs for HIF-prolyl hydroxylase inhibition. J. Chem. Inf. Model. 2013, 53, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.K.; Rajanikant, G.K. Identification of novel potential HIF-prolyl hydroxylase inhibitors by in silico screening. Mol. Divers. 2012, 16, 193–202. [Google Scholar] [CrossRef]
- Teli, M.K.; Krishnamurthy, R.G. A combination of 3D-QSAR modeling and molecular docking approach for the discovery of potential HIF prolyl hydroxylase inhibitors. Med. Chem. 2013, 9, 360–370. [Google Scholar] [CrossRef]
- Parasuraman, S.; Raveendran, R.; Vijayakumar, B.; Velmurugan, D.; Balamurugan, S. Molecular docking and ex vivo pharmacological evaluation of constituents of the leaves of Cleistanthus collinus (Roxb.) (Euphorbiaceae). Indian J. Pharmacol. 2012, 44, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.H.; Kim, S.Y. Resveratrol-Enriched Rice Attenuates UVB-ROS-Induced Skin Aging via Downregulation of Inflammatory Cascades. Oxid. Med. Cell. Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef]
- Du, Q.; Hu, B.; An, H.M.; Shen, K.P.; Xu, L.; Deng, S.; Wei, M.M. Synergistic anticancer effects of curcumin and resveratrol in Hepa1-6 hepatocellular carcinoma cells. Oncol. Rep. 2013, 29, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Medina-Aguilar, R.; Marchat, L.A.; Arechaga Ocampo, E.; Gariglio, P.; Garcia Mena, J.; Villegas Sepulveda, N.; Martinez Castillo, M.; Lopez-Camarillo, C. Resveratrol inhibits cell cycle progression by targeting Aurora kinase A and Polo-like kinase 1 in breast cancer cells. Oncol. Rep. 2016, 35, 3696–3704. [Google Scholar] [CrossRef] [Green Version]
- Sprouse, A.A.; Herbert, B.S. Resveratrol augments paclitaxel treatment in MDA-MB-231 and paclitaxel-resistant MDA-MB-231 breast cancer cells. Anticancer Res. 2014, 34, 5363–5374. [Google Scholar] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Subedi, L.; Kwon, O.W.; Pak, C.; Lee, G.; Lee, K.; Kim, H.; Kim, S.Y. N,N-disubstituted azines attenuate LPS-mediated neuroinflammation in microglia and neuronal apoptosis via inhibiting MAPK signaling pathways. BMC Neurosci. 2017, 18, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P1) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Shen, W.; Zhao, Y.; Chen, H.; Zhang, T.; Wu, S.; Liu, P. M3, a natural lignan xyloside, exhibits potent anticancer activity in HCT116 cells. Oncol. Lett. 2019, 17, 2117–2122. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, J.; Hou, L.; Cui, X. The effect of beta-elemene on alpha-tubulin polymerization in human hepatoma HepG2 cells. Chin. J. Cancer Res. 2013, 25, 770–776. [Google Scholar] [CrossRef]
- Kamal, A.; Ashraf, M.; Basha, S.T.; Ali Hussaini, S.M.; Singh, S.; Vishnuvardhan, M.V.; Kiran, B.; Sridhar, B. Design, synthesis and antiproliferative activity of the new conjugates of E7010 and resveratrol as tubulin polymerization inhibitors. Org. Biomol. Chem. 2016, 14, 1382–1394. [Google Scholar] [CrossRef]
- Aka, J.A.; Lin, S.X. Comparison of functional proteomic analyses of human breast cancer cell lines T47D and MCF7. PLoS ONE 2012, 7, e31532. [Google Scholar] [CrossRef]
- Chao, S.C.; Chen, Y.J.; Huang, K.H.; Kuo, K.L.; Yang, T.H.; Huang, K.Y.; Wang, C.C.; Tang, C.H.; Yang, R.S.; Liu, S.H. Induction of sirtuin-1 signaling by resveratrol induces human chondrosarcoma cell apoptosis and exhibits antitumor activity. Sci. Rep. 2017, 7, 3180. [Google Scholar] [CrossRef]
- Tian, H.; Yu, Z. Resveratrol induces apoptosis of leukemia cell line K562 by modulation of sphingosine kinase-1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 2755–2762. [Google Scholar]
- Chimento, A.; De Amicis, F.; Sirianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to Improve Oral Bioavailability and Beneficial Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, S.N.A.; Kumar, G.B.; Revankar, H.M.; Qin, H.L. Development of combretastatins as potent tubulin polymerization inhibitors. Bioorg. Chem. 2017, 72, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Schneider, Y.; Chabert, P.; Stutzmann, J.; Coelho, D.; Fougerousse, A.; Gosse, F.; Launay, J.F.; Brouillard, R.; Raul, F. Resveratrol analog (Z)-3,5,4’-trimethoxystilbene is a potent anti-mitotic drug inhibiting tubulin polymerization. Int. J. Cancer 2003, 107, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Madadi, N.R.; Zong, H.; Ketkar, A.; Zheng, C.; Penthala, N.R.; Janganati, V.; Bommagani, S.; Eoff, R.L.; Guzman, M.L.; Crooks, P.A. Synthesis and evaluation of a series of resveratrol analogues as potent anti-cancer agents that target tubulin. MedChemComm 2015, 6, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Baek, D.J.; MacRitchie, N.; Anthony, N.G.; Mackay, S.P.; Pyne, S.; Pyne, N.J.; Bittman, R. Structure-activity relationships and molecular modeling of sphingosine kinase inhibitors. J. Med. Chem. 2013, 56, 9310–9327. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, N.; Omori, Y.; Tanaka, K.; Ito, H.; Takagi, A.; Kojima, T.; Nakatochi, M.; Ogiso, H.; Kawamoto, Y.; Nakamura, M.; et al. Increased SPHK2 Transcription of Human Colon Cancer Cells in Serum-Depleted Culture: The Involvement of CREB Transcription Factor. J. Cell. Biochem. 2015, 116, 2227–2238. [Google Scholar] [CrossRef]
- Sareen, D.; Darjatmoko, S.R.; Albert, D.M.; Polans, A.S. Mitochondria, calcium, and calpain are key mediators of resveratrol-induced apoptosis in breast cancer. Mol. Pharmacol. 2007, 72, 1466–1475. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yu, G.; Wang, G.; Liu, H.; Wu, X.; Wang, Q.; Liu, M.; Liao, K.; Wu, M.; Cheng, X.; et al. An NQO1-initiated and p53-independent apoptotic pathway determines the anti-tumor effect of tanshinone IIA against non-small cell lung cancer. PLoS ONE 2012, 7, e42138. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Modulating both tumor cell death and innate immunity is essential for improving radiation therapy effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Gestaut, M.M.; Antoon, J.W.; Burow, M.E.; Beckman, B.S. Inhibition of sphingosine kinase-2 ablates androgen resistant prostate cancer proliferation and survival. Pharmacol. Rep. 2014, 66, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Loo, S.Y.; Huang, B.; Wong, L.; Tan, S.S.; Tan, T.Z.; Lee, S.C.; Thiery, J.P.; Lim, Y.C.; Yong, W.P.; et al. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget 2014, 5, 5920–5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.H.; Tao, Y.F.; Xu, L.X.; Zhao, H.; Li, X.L.; Fang, F.; Wu, Y.; Lu, J.; Li, Y.H.; Du, W.W.; et al. A novel sphingosine kinase 1 inhibitor (SKI-5C) induces cell death of Wilms’ tumor cells in vitro and in vivo. Am. J. Transl. Res. 2016, 8, 4548–4563. [Google Scholar] [PubMed]
- Hamada, M.; Kameyama, H.; Iwai, S.; Yura, Y. Induction of autophagy by sphingosine kinase 1 inhibitor PF-543 in head and neck squamous cell carcinoma cells. Cell Death Discov. 2017, 3, 17047. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.Q.; Irwan, A.W.; Goh, H.H.; Howe, H.S.; Yu, D.T.; Valle-Onate, R.; McInnes, I.B.; Melendez, A.J.; Leung, B.P. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J. Immunol. 2008, 181, 8010–8017. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Thangada, S.; Dasgupta, O.; Khanna, K.M.; Yamase, H.T.; Kashgarian, M.; Hla, T.; Shapiro, L.H.; Ferrer, F.A. Cell-intrinsic sphingosine kinase 2 promotes macrophage polarization and renal inflammation in response to unilateral ureteral obstruction. PLoS ONE 2018, 13, e0194053. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Van Der Kooi, K.; Messina, J.L.; Smalley, K.S. Measurement of constitutive MAPK and PI3K/AKT signaling activity in human cancer cell lines. Methods Enzymol. 2010, 484, 549–567. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Irshad, S.; Yu, W.; Belakavadi, M.; Chekmareva, M.; Ittmann, M.M.; Abate-Shen, C.; Fondell, J.D. ERK and AKT signaling drive MED1 overexpression in prostate cancer in association with elevated proliferation and tumorigenicity. Mol. Cancer Res. 2013, 11, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Adlung, L.; Kar, S.; Wagner, M.C.; She, B.; Chakraborty, S.; Bao, J.; Lattermann, S.; Boerries, M.; Busch, H.; Wuchter, P.; et al. Protein abundance of AKT and ERK pathway components governs cell type-specific regulation of proliferation. Mol. Syst. Biol. 2017, 13, 904. [Google Scholar] [CrossRef]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Han, L.; Ding, N.; Mu, Y.; Guan, P.; Hu, C.; Huang, X. Bafilomycin C1 induces G0/G1 cell-cycle arrest and mitochondrial-mediated apoptosis in human hepatocellular cancer SMMC7721 cells. J. Antibiot. 2018, 71, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Peng, W.; Sun, Q.Q.; Li, Y.H.; Chen, B.; Yu, L.T.; Xu, Y.Z.; Wang, S.Y.; Zhao, Y.L. #2714, a novel active inhibitor with potent G2/M phase arrest and antitumor efficacy in preclinical models. Cell Death Discov. 2018, 4, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blajeski, A.L.; Phan, V.A.; Kottke, T.J.; Kaufmann, S.H. G1 and G2 cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Investig. 2002, 110, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Franzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef]
- Faggioli, L.; Costanzo, C.; Merola, M.; Bianchini, E.; Furia, A.; Carsana, A.; Palmieri, M. Nuclear factor kappa B (NF-kappa B), nuclear factor interleukin-6 (NFIL-6 or C/EBP beta) and nuclear factor interleukin-6 beta (NFIL6-beta or C/EBP delta) are not sufficient to activate the endogenous interleukin-6 gene in the human breast carcinoma cell line MCF-7. Comparative analysis with MDA-MB-231 cells, an interleukin-6-expressing human breast carcinoma cell line. Eur. J. Biochem. 1996, 239, 624–631. [Google Scholar] [CrossRef]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subedi, L.; Teli, M.K.; Lee, J.H.; Gaire, B.P.; Kim, M.-h.; Kim, S.Y. A Stilbenoid Isorhapontigenin as a Potential Anti-Cancer Agent against Breast Cancer through Inhibiting Sphingosine Kinases/Tubulin Stabilization. Cancers 2019, 11, 1947. https://doi.org/10.3390/cancers11121947
Subedi L, Teli MK, Lee JH, Gaire BP, Kim M-h, Kim SY. A Stilbenoid Isorhapontigenin as a Potential Anti-Cancer Agent against Breast Cancer through Inhibiting Sphingosine Kinases/Tubulin Stabilization. Cancers. 2019; 11(12):1947. https://doi.org/10.3390/cancers11121947
Chicago/Turabian StyleSubedi, Lalita, Mahesh Kumar Teli, Jae Hyuk Lee, Bhakta Prasad Gaire, Mi-hyun Kim, and Sun Yeou Kim. 2019. "A Stilbenoid Isorhapontigenin as a Potential Anti-Cancer Agent against Breast Cancer through Inhibiting Sphingosine Kinases/Tubulin Stabilization" Cancers 11, no. 12: 1947. https://doi.org/10.3390/cancers11121947