
An Integrated Approach Including CRISPR/Cas9-Mediated Nanopore Sequencing, Mate Pair Sequencing, and Cytogenomic Methods to Characterize Complex Structural Rearrangements in Acute Myeloid Leukemia


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Data and Diagnosis of Acute Myeloid Leukemia/Myelodysplastic Syndrome
2.2. Cytogenetics Data: Conventional Chromosome Analysis, FISH, and SNP Microarray
2.3. Mate Pair Sequencing
2.4. CRISPR/Cas9-Mediated Nanopore Sequencing
2.5. Data Comparison among MPseq, Nanopore Sequencing, and SNP Microarray
2.6. Gene Mutation Panel by Next-Generation Sequencing (NGS)
3. Results
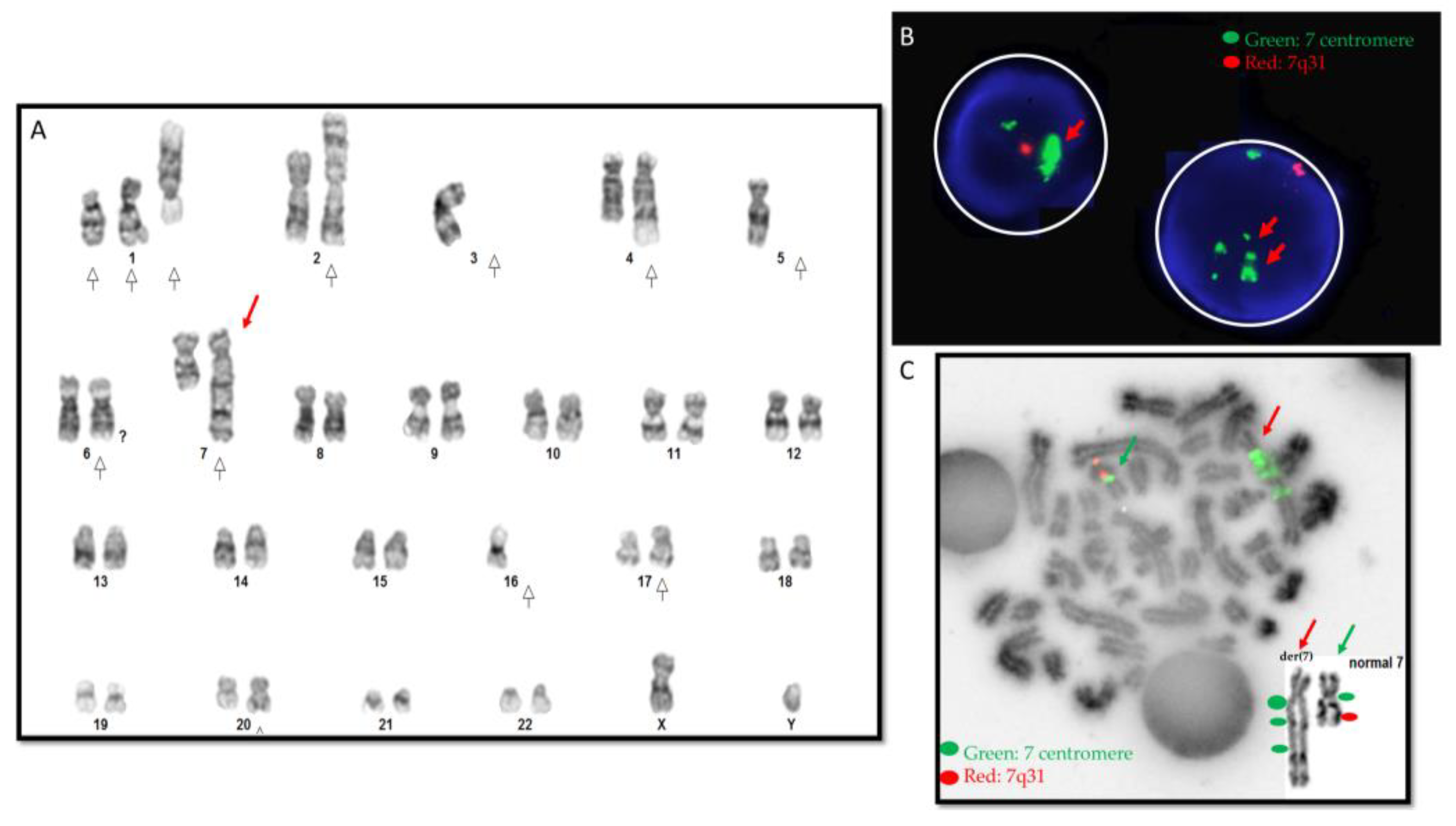
3.1. Cytogenetic Results
3.2. Mate Pair Sequencing
3.3. SVs by Nanopore Sequencing
3.3.1. SVs Based on CRISPR/Cas9 crRNA Sequences
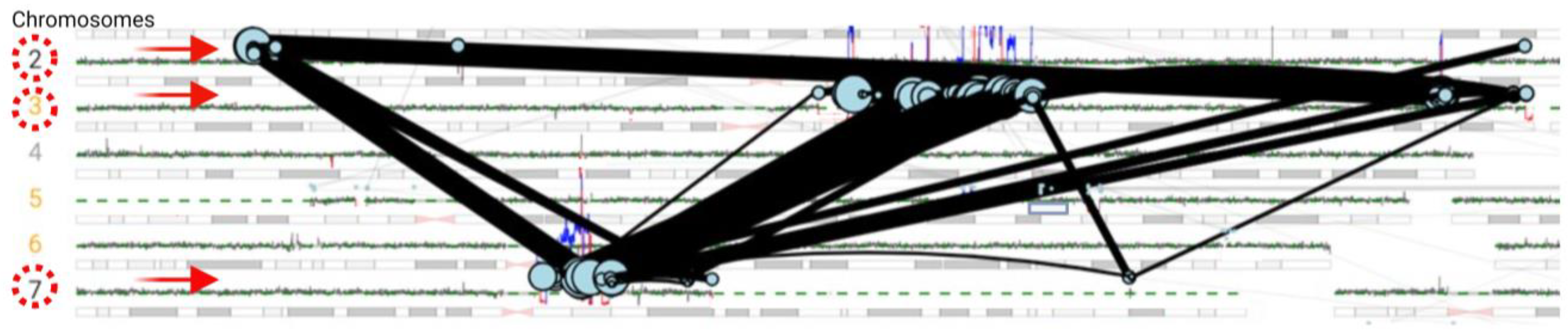
3.3.2. SVs Involving Chromosomes 2, 3, and 7
3.4. Comparison between MPseq and Nanopore Sequencing
3.5. Copy Number Variant Analysis
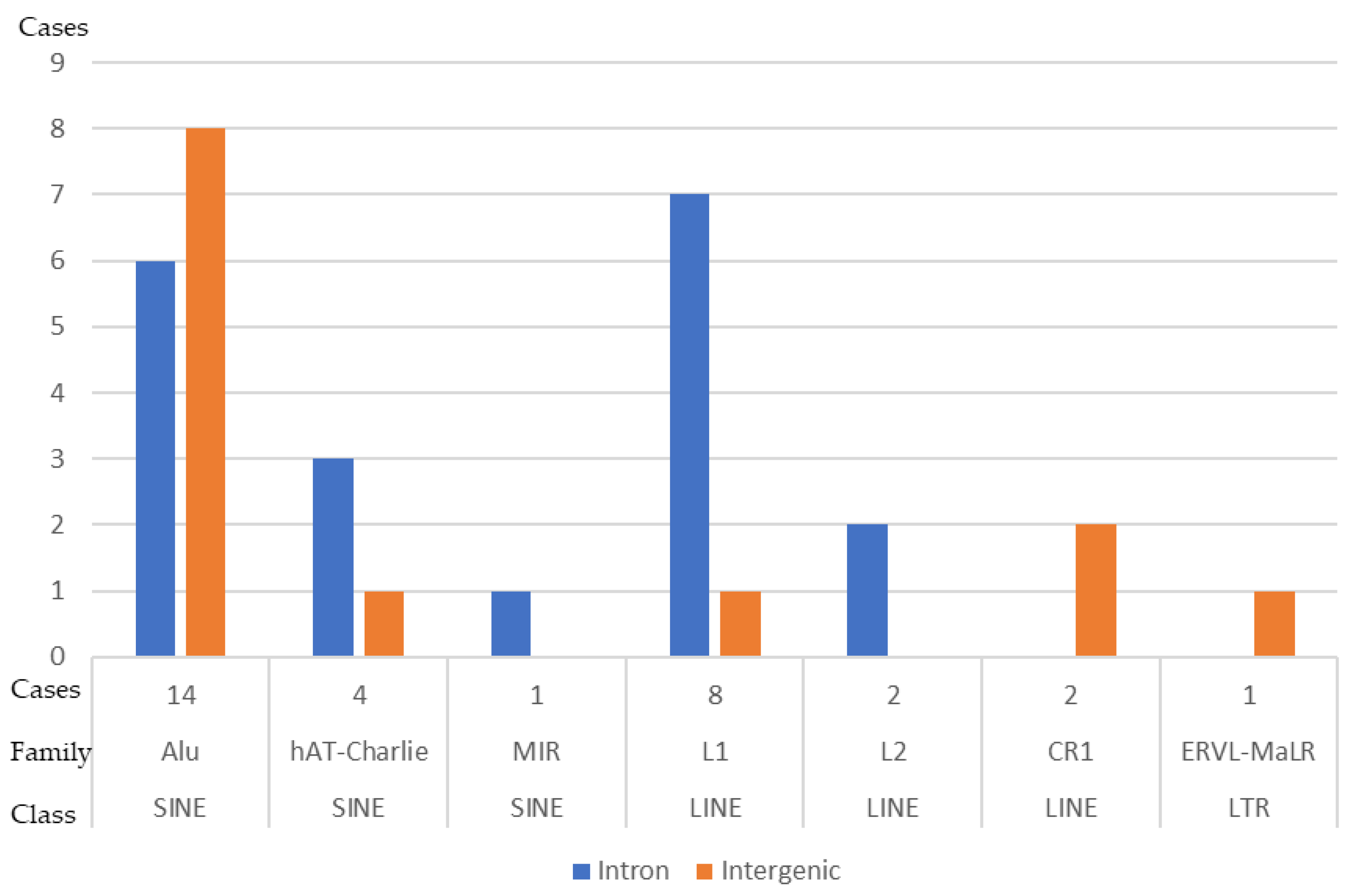
3.6. DNA Sequences Flanking the SV Breakpoints
3.7. NGS Gene Mutation Panel
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| alt-EJ | Alternative end joining |
| AML | Acute myeloid leukemia |
| ALL | Acute lymphoblastic leukemia |
| Cas9 | CRISPR-associated Protein 9 |
| CIN | Chromosome instability |
| CNevents | Copy number events |
| CNs | Copy numbers |
| CNV | Copy number variants |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DSBs | Double-strand breaks |
| DNA | Deoxyribonucleic acid |
| FAB | French–American–British |
| FISH | Fluorescence in situ hybridization |
| FoSTeS | Fork stalling and template switching |
| guideRNA | Guide ribonucleic acid |
| IDT | Integrated DNA Technologies |
| IGV | Integrative Genomics Viewer |
| LINE | Long interspersed nuclear elements |
| MDS | Myelodysplastic syndrome |
| MMBIR | Microhomology-mediated break-induced relocation |
| MPseq | Mate pair sequencing |
| NGS | Next-generation sequencing |
| NHEJ | Non-homologous end joining |
| PCR | Polymerase chain reaction |
| RNA | Ribonucleic acid |
| SOB | Shortness of breath |
| SNP | Single-nucleotide polymorphism |
| SHH-MB | Sonic Hedgehog-induced medulloblastoma |
| SVs | Structural variants |
| UCSC | University of California Santa Cruz |
| VIA | Variant Intelligence Applications™ |
| WGS | Whole-genome sequencing |
References
- Holland, A.J.; Cleveland, D.W. Chromoanagenesis and cancer: Mechanisms and consequences of localized, complex chromosomal rearrangements. Nat. Med. 2012, 18, 1630–1638. [Google Scholar] [CrossRef]
- Pellestor, F. Chromoanagenesis: Cataclysms behind complex chromosomal rearrangements. Mol. Cytogenet. 2019, 12, 6. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Maher, C.A.; Wilson, R.K. Chromothripsis and human disease: Piecing together the shattering process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for inference of chromothripsis in cancer genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Masset, H.; Hestand, M.S.; Van Esch, H.; Kleinfinger, P.; Plaisancie, J.; Afenjar, A.; Molignier, R.; Schluth-Bolard, C.; Sanlaville, D.; Vermeesch, J.R. A Distinct Class of Chromoanagenesis Events Characterized by Focal Copy Number Gains. Hum. Mutat. 2016, 37, 661–668. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Le Guen, T.; Lopez, B.S.; Guirouilh-Barbat, J. Genomic rearrangements induced by unscheduled DNA double strand breaks in somatic mammalian cells. FEBS J. 2017, 284, 2324–2344. [Google Scholar] [CrossRef]
- Liu, P.; Erez, A.; Nagamani, S.C.; Dhar, S.U.; Kolodziejska, K.E.; Dharmadhikari, A.V.; Cooper, M.L.; Wiszniewska, J.; Zhang, F.; Withers, M.A.; et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2011, 146, 889–903. [Google Scholar] [CrossRef]
- Lee, J.A.; Carvalho, C.M.; Lupski, J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007, 131, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. The DNA damage response during DNA replication. Curr. Opin. Cell. Biol. 2005, 17, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Natarajan, A.T.; Hande, M.P. Chromosomal instability--mechanisms and consequences. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 176–184. [Google Scholar] [CrossRef]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Hoogstraat, M.; Paling, O.; Tavakoli-Yaraki, M.; Renkens, I.; Vermaat, J.S.; van Roosmalen, M.J.; van Lieshout, S.; Nijman, I.J.; Roessingh, W.; et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. 2011, 12, R103. [Google Scholar] [CrossRef]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef]
- Wu, C.; Wyatt, A.W.; McPherson, A.; Lin, D.; McConeghy, B.J.; Mo, F.; Shukin, R.; Lapuk, A.V.; Jones, S.J.; Zhao, Y.; et al. Poly-gene fusion transcripts and chromothripsis in prostate cancer. Genes Chromosomes Cancer 2012, 51, 1144–1153. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Magrangeas, F.; Avet-Loiseau, H.; Munshi, N.C.; Minvielle, S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood 2011, 118, 675–678. [Google Scholar] [CrossRef]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; van Sluis, P.; Valentijn, L.J.; van der Ploeg, I.; Hamdi, M.; van Nes, J.; Westerman, B.A.; van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef]
- Wahl, G.M. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989, 49, 1333–1340. [Google Scholar]
- Kou, F.; Wu, L.; Ren, X.; Yang, L. Chromosome Abnormalities: New Insights into Their Clinical Significance in Cancer. Mol. Ther. Oncolytics 2020, 17, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell. Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 2008, 14, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.A. Chromosome abnormalities in human cancer and leukemia. Mutat. Res. 1991, 247, 231–240. [Google Scholar] [CrossRef]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rora, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef]
- Gao, J.; Chen, Y.H.; Mina, A.; Altman, J.K.; Kim, K.Y.; Zhang, Y.; Lu, X.; Jennings, L.; Sukhanova, M. Unique morphologic and genetic characteristics of acute myeloid leukemia with chromothripsis: A clinicopathologic study from a single institution. Hum. Pathol. 2020, 98, 22–31. [Google Scholar] [CrossRef]
- MacKinnon, R.N. Analysis of Chromothripsis by Combined FISH and Microarray Analysis. Methods Mol. Biol. 2018, 1769, 53–77. [Google Scholar]
- Boyd, R.J.; Murry, J.B.; Morsberger, L.A.; Klausner, M.; Chen, S.; Gocke, C.D.; McCallion, A.S.; Zou, Y.S. Ring Chromosomes in Hematological Malignancies Are Associated with TP53 Gene Mutations and Characteristic Copy Number Variants. Cancers 2023, 15, 5439. [Google Scholar] [CrossRef]
- Bailey, J.A.; Yavor, A.M.; Massa, H.F.; Trask, B.J.; Eichler, E.E. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res. 2001, 11, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Chen, Y.; Eyre-Walker, A. The large-scale distribution of somatic mutations in cancer genomes. Hum. Mutat. 2012, 33, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Hills, M.; Jeyapalan, J.N.; Foxon, J.L.; Royle, N.J. Mutation mechanisms that underlie turnover of a human telomere-adjacent segmental duplication containing an unstable minisatellite. Genomics 2007, 89, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Hastings, P.J.; Lupski, J.R.; Rosenberg, S.M.; Ira, G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009, 10, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.O.; Gurdasani, D.; Mentzer, A.J.; Porter, T.; Sandhu, M.S. Long reads: Their purpose and place. Hum. Mol. Genet. 2018, 27, R234–R241. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Pitel, B.A.; Zuckerman, E.Z.; Baughn, L.B. Mate Pair Sequencing: Next-Generation Sequencing for Structural Variant Detection. Methods Mol. Biol. 2023, 2621, 127–149. [Google Scholar]
- Smadbeck, J.; Peterson, J.F.; Pearce, K.E.; Pitel, B.A.; Figueroa, A.L.; Timm, M.; Jevremovic, D.; Shi, M.; Stewart, A.K.; Braggio, E.; et al. Mate pair sequencing outperforms fluorescence in situ hybridization in the genomic characterization of multiple myeloma. Blood Cancer J. 2019, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, J.B.; Johnson, S.H.; Smoley, S.A.; Gaitatzes, A.; Drucker, T.M.; Zenka, R.M.; Kosari, F.; Murphy, S.J.; Hoppman, N.; Aypar, U.; et al. Copy number variant analysis using genome-wide mate-pair sequencing. Genes Chromosomes Cancer 2018, 57, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.H.; Smadbeck, J.B.; Smoley, S.A.; Gaitatzes, A.; Murphy, S.J.; Harris, F.R.; Drucker, T.M.; Zenka, R.M.; Pitel, B.A.; Rowsey, R.A.; et al. SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq). Cancer Genet. 2018, 221, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Drucker, T.M.; Johnson, S.H.; Murphy, S.J.; Cradic, K.W.; Therneau, T.M.; Vasmatzis, G. BIMA V3: An aligner customized for mate pair library sequencing. Bioinformatics 2014, 30, 1627–1629. [Google Scholar] [CrossRef]
- Jiang, L.; Pallavajjala, A.; Huang, J.; Haley, L.; Morsberger, L.; Stinnett, V.; Hardy, M.; Park, R.; Ament, C.; Finch, A.; et al. Clinical Utility of Targeted Next-Generation Sequencing Assay to Detect Copy Number Variants Associated with Myelodysplastic Syndrome in Myeloid Malignancies. J. Mol. Diagn. 2021, 23, 467–483. [Google Scholar] [CrossRef]
- Pallavajjala, A.; Haley, L.; Stinnett, V.; Adams, E.; Pallavajjala, R.; Huang, J.; Morsberger, L.A.; Hardy, M.; Long, P.; Gocke, C.D.; et al. Utility of targeted next-generation sequencing assay to detect 1p/19q co-deletion in formalin-fixed paraffin-embedded glioma specimens. Hum. Pathol. 2022, 126, 63–76. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 1999, 9, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F. The origin of interspersed repeats in the human genome. Curr. Opin. Genet. Dev. 1996, 6, 743–748. [Google Scholar] [CrossRef]
- Jurka, J. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 2000, 16, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Chen, C.W.; Sundaramurthy, V.; Slabicki, M.; Hao, D.; Watson, C.J.; Tovy, A.; Reyes, J.M.; Dakhova, O.; Crovetti, B.R.; et al. Systematic Profiling of DNMT3A Variants Reveals Protein Instability Mediated by the DCAF8 E3 Ubiquitin Ligase Adaptor. Cancer Discov. 2022, 12, 220–235. [Google Scholar] [CrossRef]
- Mackinnon, R.N.; Campbell, L.J. Chromothripsis under the microscope: A cytogenetic perspective of two cases of AML with catastrophic chromosome rearrangement. Cancer Genet. 2013, 206, 238–251. [Google Scholar] [CrossRef]
- Mackinnon, R.N.; Wall, M.; Zordan, A.; Nutalapati, S.; Mercer, B.; Peverall, J.; Campbell, L.J. Genome organization and the role of centromeres in evolution of the erythroleukaemia cell line HEL. Evol. Med. Public Health 2013, 2013, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Garsed, D.W.; Marshall, O.J.; Corbin, V.D.; Hsu, A.; Di Stefano, L.; Schroder, J.; Li, J.; Feng, Z.P.; Kim, B.W.; Kowarsky, M.; et al. The architecture and evolution of cancer neochromosomes. Cancer Cell 2014, 26, 653–667. [Google Scholar] [CrossRef]
- Macchia, G.; Nord, K.H.; Zoli, M.; Purgato, S.; D’Addabbo, P.; Whelan, C.W.; Carbone, L.; Perini, G.; Mertens, F.; Rocchi, M.; et al. Ring chromosomes, breakpoint clusters, and neocentromeres in sarcomas. Genes Chromosomes Cancer 2015, 54, 156–167. [Google Scholar] [CrossRef]
- Yadav, V.; Sun, S.; Coelho, M.A.; Heitman, J. Centromere scission drives chromosome shuffling and reproductive isolation. Proc. Natl. Acad. Sci. USA 2020, 117, 7917–7928. [Google Scholar] [CrossRef]
- Singh, Z.N.; Richards, S.; El Chaer, F.; Duong, V.H.; Gudipati, M.A.; Waters, E.O.; Koon, S.; Webley, M.; Pitel, B.; Hoppman, N.L.; et al. Cryptic ETV6-PDGFRB fusion in a highly complex rearrangement of chromosomes 1, 5, and 12 due to a chromothripsis-like event in a myelodysplastic syndrome/myeloproliferative neoplasm. Leuk. Lymphoma 2019, 60, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Dolnik, A.; Blätte, T.J.; Teleanu, V.; Ernst, A.; Thol, F.; Heuser, M.; Ganser, A.; Döhner, H.; Döhner, K.; et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica 2018, 103, e17–e20. [Google Scholar] [CrossRef] [PubMed]
- Gudipati, M.A.; Waters, E.; Greene, C.; Goel, N.; Hoppman, N.L.; Pitel, B.A.; Webley, M.R.; Zou, Y. Stable transmission of complex chromosomal rearrangements involving chromosome 1q derived from constitutional chromoanagenesis. Mol. Cytogenet. 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Hirai, H.; Yoshida, M.C.; Takaku, F. Evolution, expression, and chromosomal location of a novel receptor tyrosine kinase gene, eph. Mol. Cell. Biol. 1988, 8, 3770–3776. [Google Scholar] [PubMed]
- Kiyokawa, E.; Takai, S.; Tanaka, M.; Iwase, T.; Suzuki, M.; Xiang, Y.Y.; Naito, Y.; Yamada, K.; Sugimura, H.; Kino, I. Overexpression of ERK, an EPH family receptor protein tyrosine kinase, in various human tumors. Cancer Res. 1994, 54, 3645–3650. [Google Scholar] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Azim, A.C.; Knoll, J.H.; Marfatia, S.M.; Peel, D.J.; Bryant, P.J.; Chishti, A.H. DLG1: Chromosome location of the closest human homologue of the Drosophila discs large tumor suppressor gene. Genomics 1995, 30, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Iwao, K.; Miyoshi, Y.; Nakagawara, A.; Kofu, K.; Akiyama, T.; Arita, N.; Hayakawa, T.; Nakamura, Y. Identification of brain-specific splicing variants of the hDLG1 gene and altered splicing in neuroblastoma cell lines. J. Hum. Genet. 1998, 43, 123–127. [Google Scholar] [CrossRef]
- Bodmer, D.; Eleveld, M.; Kater-Baats, E.; Janssen, I.; Janssen, B.; Weterman, M.; Schoenmakers, E.; Nickerson, M.; Linehan, M.; Zbar, B.; et al. Disruption of a novel MFS transporter gene, DIRC2, by a familial renal cell carcinoma-associated t(2;3)(q35;q21). Hum. Mol. Genet. 2002, 11, 641–649. [Google Scholar] [CrossRef]
- George, C.M.; Alani, E. Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 297–313. [Google Scholar] [CrossRef]
- Weckselblatt, B.; Rudd, M.K. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015, 31, 587–599. [Google Scholar] [CrossRef]
- Feng, Q.; Moran, J.V.; Kazazian, H.H., Jr.; Boeke, J.D. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 1996, 87, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Mathias, S.L.; Scott, A.F.; Kazazian, H.H., Jr.; Boeke, J.D.; Gabriel, A. Reverse transcriptase encoded by a human transposable element. Science 1991, 254, 1808–1810. [Google Scholar] [CrossRef]
- Moran, J.V.; Holmes, S.E.; Naas, T.P.; DeBerardinis, R.J.; Boeke, J.D.; Kazazian, H.H., Jr. High frequency retrotransposition in cultured mammalian cells. Cell 1996, 87, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef]
- Kajikawa, M.; Okada, N. LINEs mobilize SINEs in the eel through a shared 3′ sequence. Cell 2002, 111, 433–444. [Google Scholar] [CrossRef]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef]
- Pascarella, G.; Hon, C.C.; Hashimoto, K.; Busch, A.; Luginbuhl, J.; Parr, C.; Hin Yip, W.; Abe, K.; Kratz, A.; Bonetti, A.; et al. Recombination of repeat elements generates somatic complexity in human genomes. Cell 2022, 185, 3025–3040.e6. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef]
- Lee, J.; Han, K.; Meyer, T.J.; Kim, H.S.; Batzer, M.A. Chromosomal inversions between human and chimpanzee lineages caused by retrotransposons. PLoS ONE 2008, 3, e4047. [Google Scholar] [CrossRef]
- Deininger, P.L.; Batzer, M.A. Alu repeats and human disease. Mol. Genet. Metab. 1999, 67, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Nazaryan-Petersen, L.; Bertelsen, B.; Bak, M.; Jonson, L.; Tommerup, N.; Hancks, D.C.; Tumer, Z. Germline Chromothripsis Driven by L1-Mediated Retrotransposition and Alu/Alu Homologous Recombination. Hum. Mutat. 2016, 37, 385–395. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A.; et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef]
- Miao, H.; Zhou, J.; Yang, Q.; Liang, F.; Wang, D.; Ma, N.; Gao, B.; Du, J.; Lin, G.; Wang, K.; et al. Long-read sequencing identified a causal structural variant in an exome-negative case and enabled preimplantation genetic diagnosis. Hereditas 2018, 155, 32. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Huddleston, J.; Chaisson, M.J.; Hill, C.M.; Kronenberg, Z.N.; Munson, K.M.; Malig, M.; Raja, A.; Fiddes, I.; Hillier, L.W.; et al. Long-read sequence assembly of the gorilla genome. Science 2016, 352, aae0344. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Vollger, M.R.; Logsdon, G.A.; Audano, P.A.; Sulovari, A.; Porubsky, D.; Peluso, P.; Wenger, A.M.; Concepcion, G.T.; Kronenberg, Z.N.; Munson, K.M.; et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet. 2020, 84, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Dong, K.; Wu, J.; Chen, Z.; Najm, F.J.; Zhang, Y.; Moore, M.M.; Hecht, V.; Shoresh, N.; Bernstein, B.E. Long-range phasing of dynamic, tissue-specific and allele-specific regulatory elements. Nat. Genet. 2022, 54, 1504–1513. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Chromo-some | Structural Variant (SV) Type | crRNA # | Copy Number Variant (CNV) | Genomic Location (Exon, Intron, Non-Gene Regions) |
|---|---|---|---|---|---|
| 1 | 3 | Simple | 1 | Gain | intron (ABI3BP) |
| 2 | 3 | Complex | 2 | Gain, amplification | intergenic region |
| 3 | 3 | Complex | 3 | Gain, amplification | intron (CBLB) |
| 4 | 3 | Complex | 4 | Loss, gains | exon (USF3), intergenic |
| 5 | 3 | Simple | 5 | Gain, loss | intron (ATP6V1A) |
| 6 | 3 | Simple | 6 | Loss, gain, amplification | intron (LSAMP) |
| 7 | 3 | Complex | 7 | Gains, loss | intron (LSAMP) |
| 8 | 3 | Complex | 8 | Normal | intergenic region |
| 9 | 3 | Complex | 9 | Normal | intron (NR_135547.1) |
| 10 | 3 | Complex | 11 | Gain | intron (CFAP91), intergenic |
| 11 | 3 | Complex | 12 | Gains, loss | intron (CASR), intergenic |
| 12 | 3 | Complex | 13 | Gain | intron (SLC49A4), exon and intron (SEMA5B), intergenic |
| 13 | 3 | Complex | 14 | Gain, amplification, normal | intron (ALG1L) |
| 14 | 3 | Complex | 15 | Gain, amplification | intron (ALD1H1) |
| 15 | 3 | Simple | 16 | Normal | intron (ALD1H1) |
| 16 | 3 | Simple | 17 | Normal | intron (CHST13) |
| 17 | 3 | Complex | 18 | Normal | exon and intron (CHST13) |
| 18 | 3 | Simple | 19 | Gain | intron (CHCHD6) |
| 19 | 3 | Complex | 20 | Gain | intron (PODXL2), intron (MGLL) |
| 20 | 7 | Complex | 21 | Gain, amplification, normal | intergenic region |
| 21 | 7 | Complex | 23 | Loss, gains | intergenic region |
| 22 | 7 | Complex | 24 | Gain | intergenic region |
| 23 | 7 | Simple | 25 | Gain | intergenic region |
| 24 | 7 | Complex | 26 | Gain | intron (AUTS2) |
| 25 | 7 | Complex | 27 | Gain | intron (AUTS2) |
| 26 | 7 | Simple | 28/29 | Gain, loss | intron (AUTS2) |
| 27 | 7 | Complex | 30 | Loss, normal | intron (GALNT17) |
| Chromosome Bands | Genomic Regions | SNP Microarray | Mate Pair Sequencing | CRISPR/Cas9 Nanopore Sequencing | |
|---|---|---|---|---|---|
| Structural Variant (SV) | Copy Number Variant (CNV) | ||||
| Chromoanagenesis | |||||
| 3q12.2–q13.31 | chr3:100712059-116712193 | 2 losses, 3 gains | 13 breakpoints | 27 breakpoints | 6 losses, 9 gains, 3 amplification |
| 3q13.32–q22.1 | chr3:118281297-129837325 | 7 gains | 47 breakpoints | 50 breakpoints | 2 losses, 13 gains, 6 amplification |
| 7p12.2–q21.11 | chr7:62012221-71918506 | 2 losses, 4 gains | 40 breakpoints | 31 breakpoints | 7 losses, 11 gains, 2 amplification |
| Other genomic regions that involved in chromosomes 2, 3, and 7 | |||||
| 2p23.3–p16.3 | chr2:24066341-52227175 | 2 losses, 1 gain | 17 breakpoints | 4 breakpoints | 1 gain, 1 amplification |
| 3q27.2–q29 | chr3:185359470-196944509 | 1 loss, 1 gain | 19 breakpoints | none | none |
| Others | various genomic regions | none | 2 breakpoints | 9 breakpoints | 1 gain |
| Total | 4 losses, 13 gains | 138 breakpoints (69 SVs) | 121 breakpoints (41 SVs) | 62 CNVs (15 losses, 35 gains, 12 amplifications) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, M.; Gomes, M.A.; Stinnett, V.; Morsberger, L.; Hoppman, N.L.; Pearce, K.E.; Smith, K.; Phan, B.; Jiang, L.; Zou, Y.S. An Integrated Approach Including CRISPR/Cas9-Mediated Nanopore Sequencing, Mate Pair Sequencing, and Cytogenomic Methods to Characterize Complex Structural Rearrangements in Acute Myeloid Leukemia. Biomedicines 2024, 12, 598. https://doi.org/10.3390/biomedicines12030598
Phan M, Gomes MA, Stinnett V, Morsberger L, Hoppman NL, Pearce KE, Smith K, Phan B, Jiang L, Zou YS. An Integrated Approach Including CRISPR/Cas9-Mediated Nanopore Sequencing, Mate Pair Sequencing, and Cytogenomic Methods to Characterize Complex Structural Rearrangements in Acute Myeloid Leukemia. Biomedicines. 2024; 12(3):598. https://doi.org/10.3390/biomedicines12030598
Chicago/Turabian StylePhan, Michael, Maria A. Gomes, Victoria Stinnett, Laura Morsberger, Nicole L. Hoppman, Kathryn E. Pearce, Kirstin Smith, Brian Phan, Liqun Jiang, and Ying S. Zou. 2024. "An Integrated Approach Including CRISPR/Cas9-Mediated Nanopore Sequencing, Mate Pair Sequencing, and Cytogenomic Methods to Characterize Complex Structural Rearrangements in Acute Myeloid Leukemia" Biomedicines 12, no. 3: 598. https://doi.org/10.3390/biomedicines12030598