
Enhanced L-β-Aminoisobutyric Acid Is Involved in the Pathophysiology of Effectiveness for Treatment-Resistant Schizophrenia and Adverse Reactions of Clozapine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Agents
2.2. Experimental Animals
2.3. Primary Cultured Astrocytes
2.3.1. Effects of L-BAIBA on GABAB-R and III-mGluR
2.3.2. Determination of Intracellular L-BAIBA Levels in the Astrocytes
2.3.3. Acute Effects of Clozapine on Astroglial Release of L-BAIBA Evoked by Ripple-Burst Stimulation
2.4. Determination of the Levels of cAMP, L-BAIBA, and L-Glutamate
2.5. Preparation of the Microdialysis System
2.6. Data Analysis
3. Results
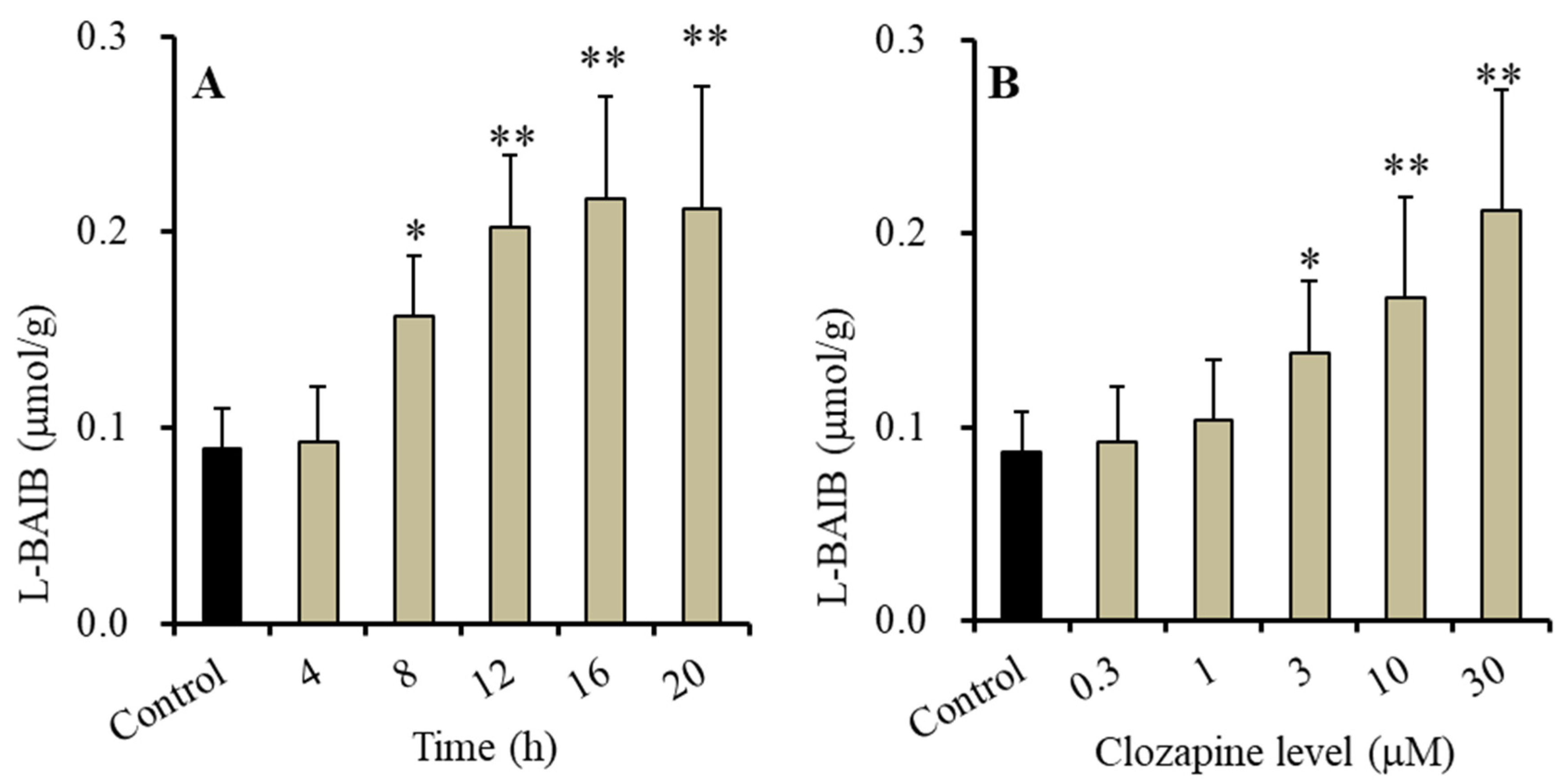
3.1. Time-Dependent and Concentration-Dependent Effects of Clozapine on Intracellular L-BAIBA Levels in Astrocytes
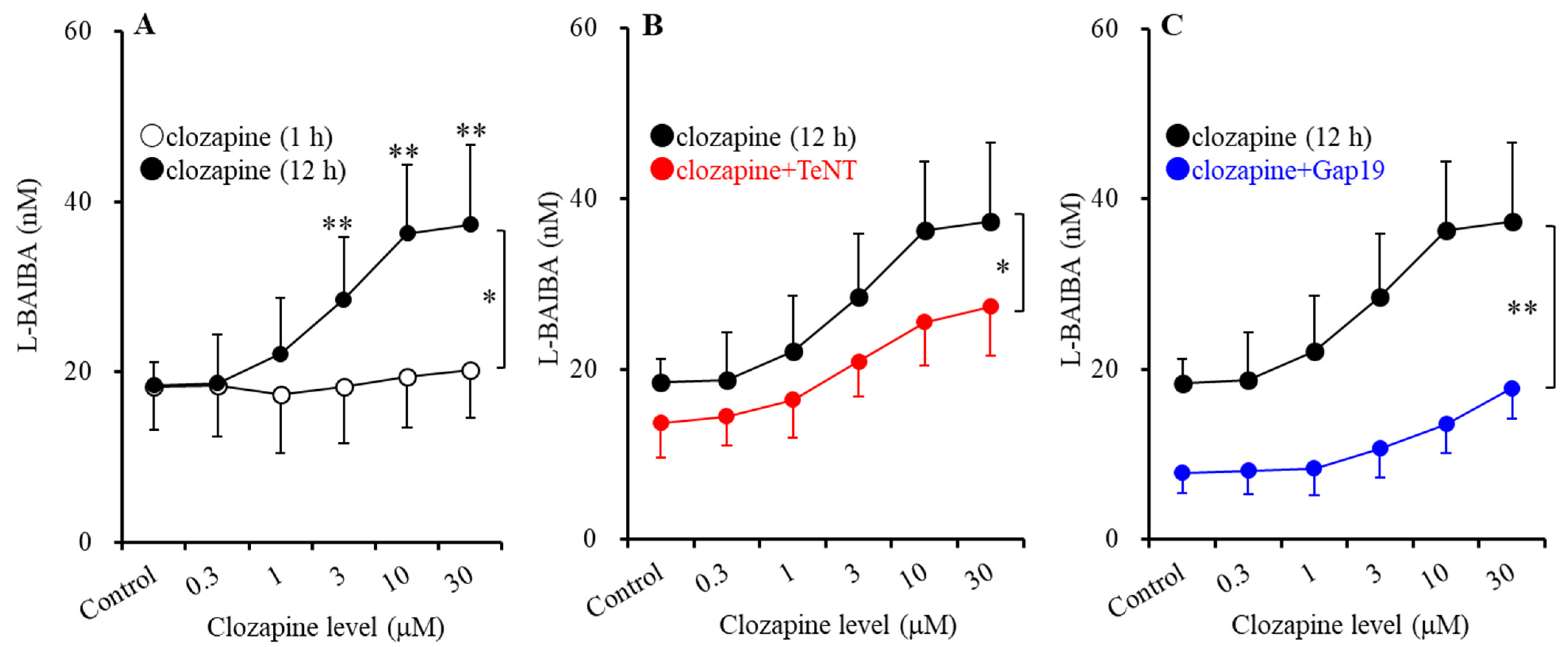
3.2. Time-Dependent and Concentration-Dependent Effects of Clozapine on Astroglial Ripple-Evoked L-BAIBA Release
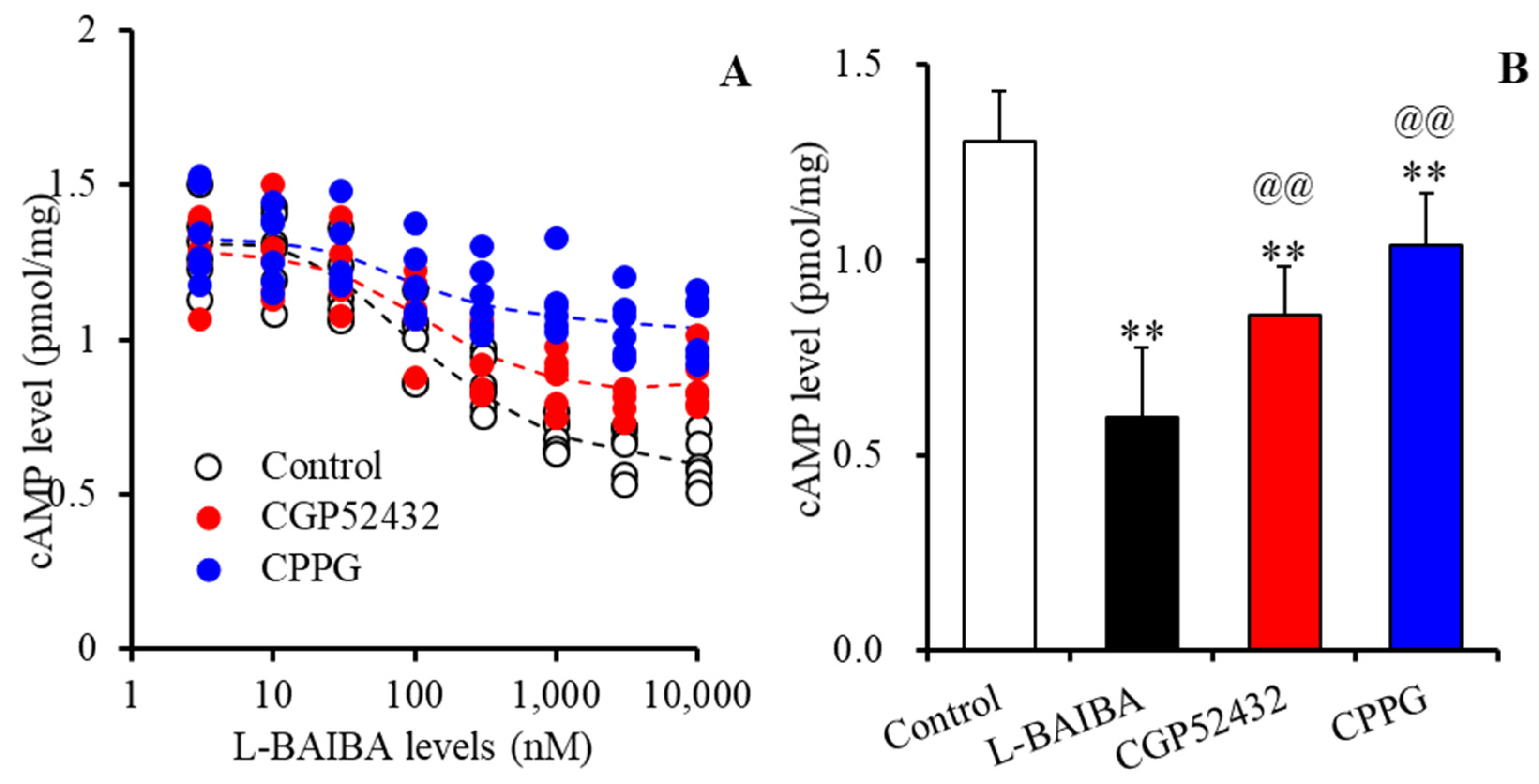
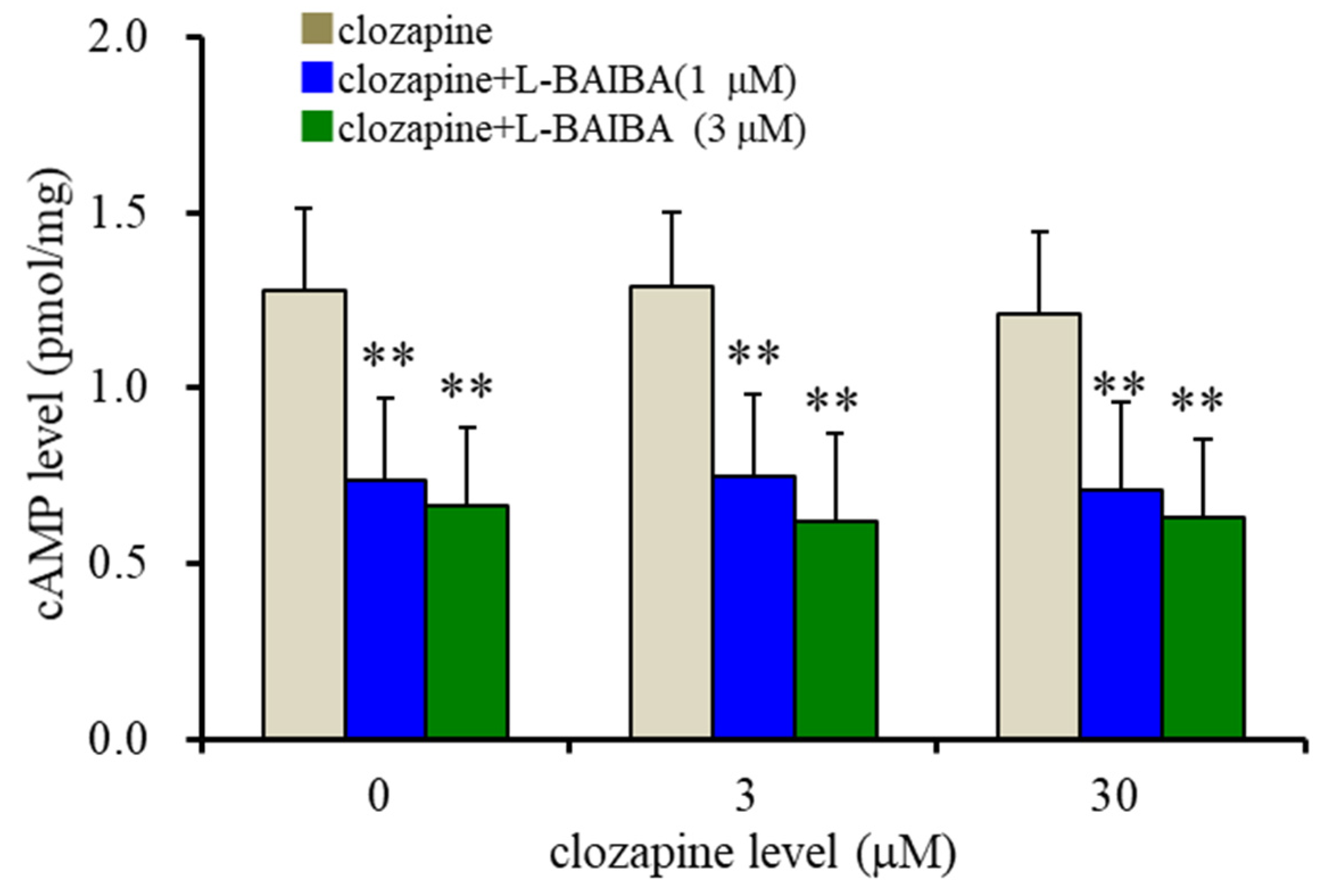
3.3. Concentration-Dependent Effects of L-BAIBA on GABAB-R and III-mGluR
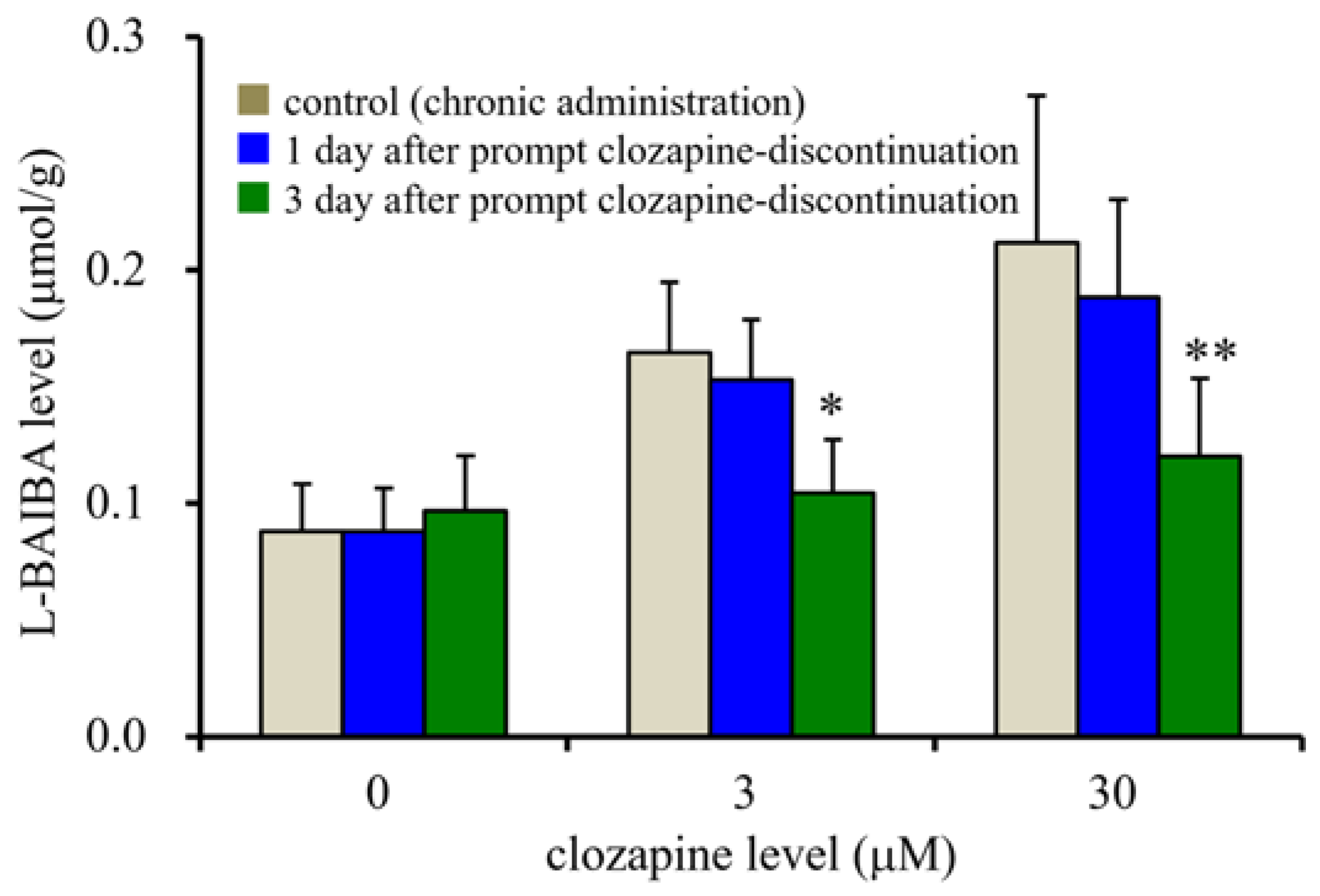
3.4. Effects of Prompt Clozapine Discontinuation on Intracellular L-BAIBA Levels in the Astrocytes
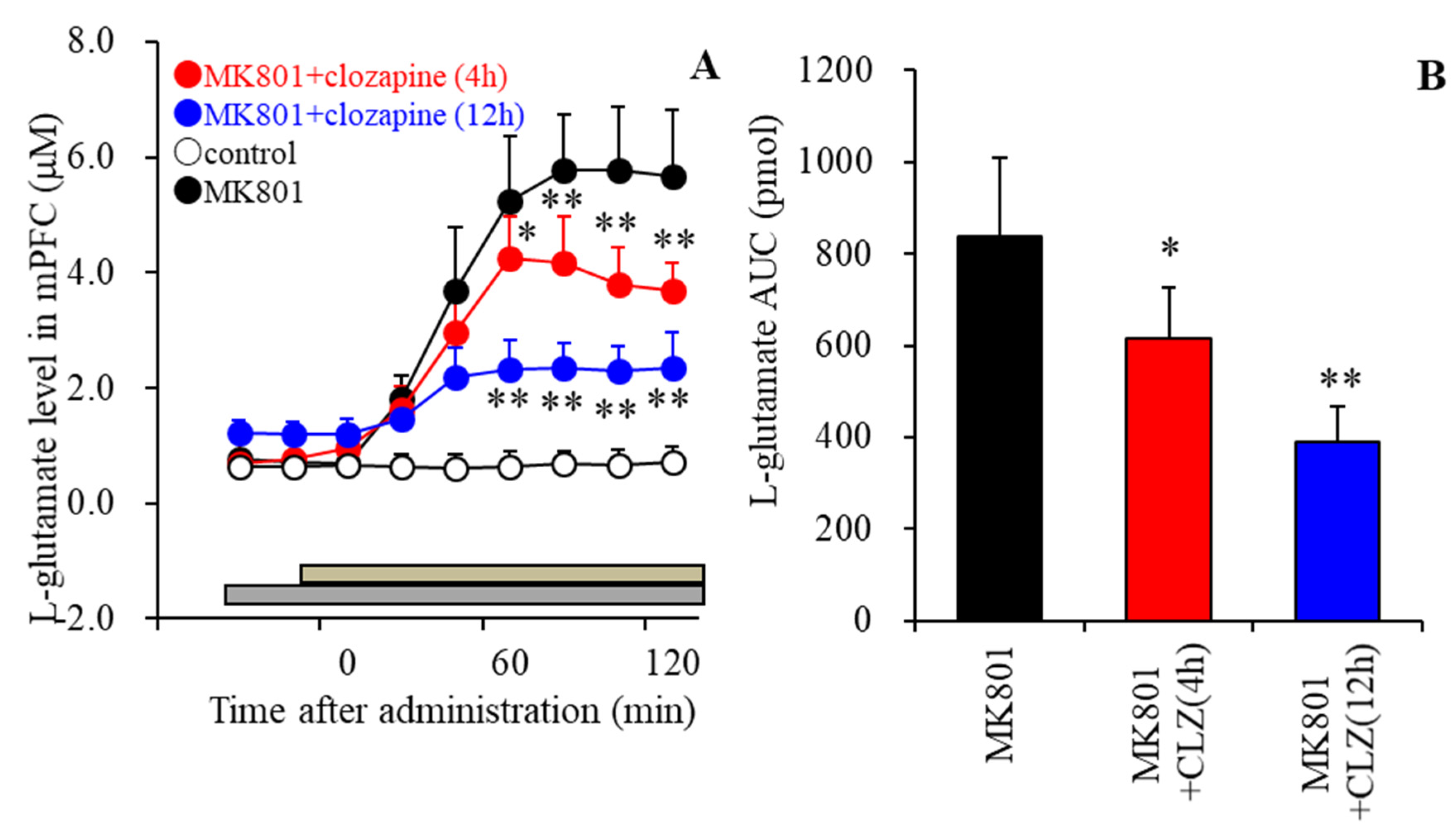
3.5. Effects of the Local Administration of Clozapine into the mPFC on MK801-Evoked L-Glutamate Release in the mPFC
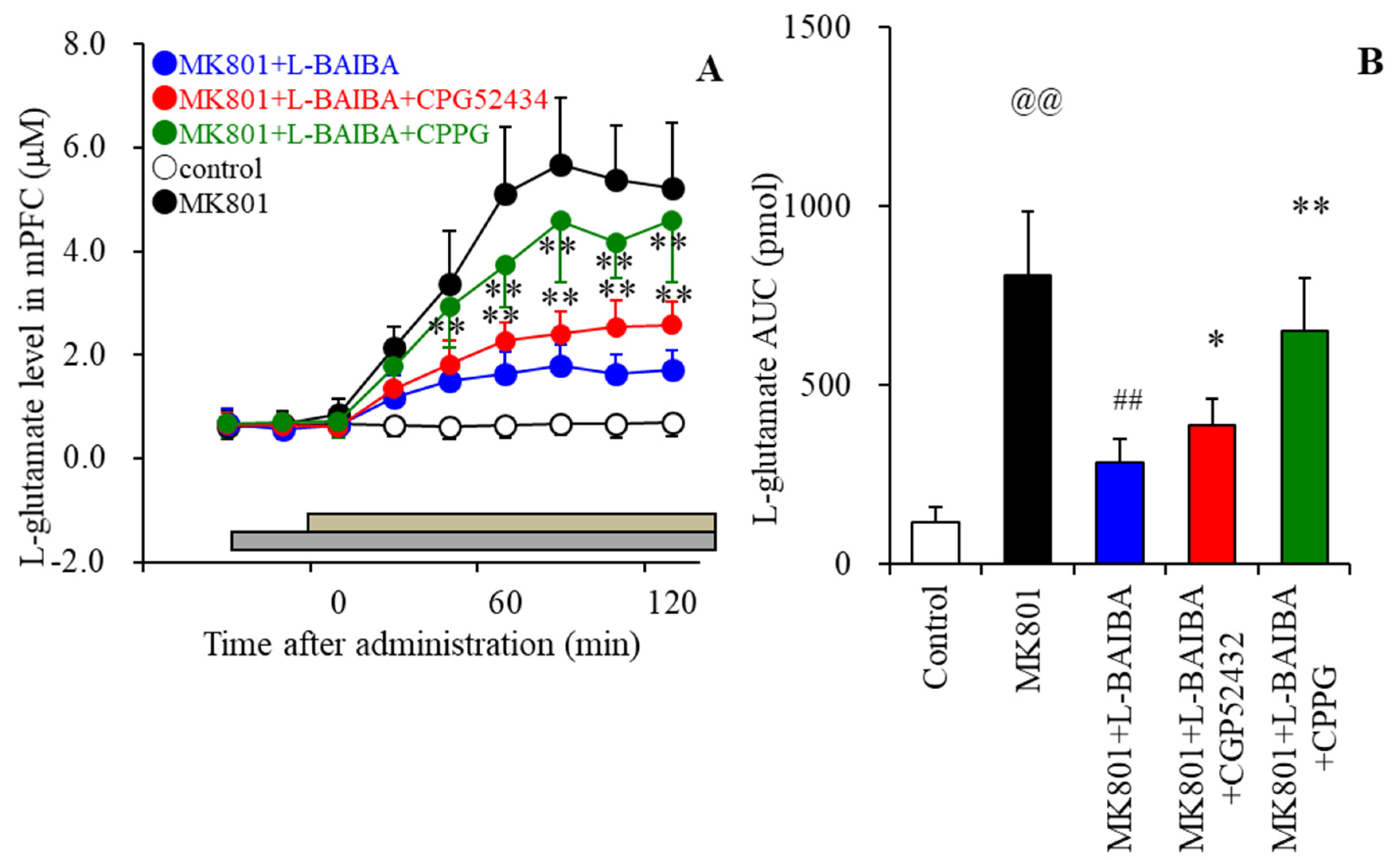
3.6. Effects of the Local Administration of L-BAIBA into the mPFC on MK801-Evoked L-Glutamate Release in the mPFC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siskind, D.; Orr, S.; Sinha, S.; Yu, O.; Brijball, B.; Warren, N.; MacCabe, J.H.; Smart, S.E.; Kisely, S. Rates of treatment-resistant schizophrenia from first-episode cohorts: Systematic review and meta-analysis. Br. J. Psychiatry 2022, 220, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Crippa, A.; Siafis, S.; Patel, M.X.; Orsini, N.; Davis, J.M. Dose-response meta-analysis of antipsychotic drugs for acute schizophrenia. Am. J. Psychiatry 2020, 177, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; McCutcheon, R.A.; Brugger, S.P.; Howes, O.D. Heterogeneity and efficacy of antipsychotic treatment for schizophrenia with or without treatment resistance: A meta-analysis. Neuropsychopharmacology 2020, 45, 622–631. [Google Scholar] [CrossRef]
- Okhuijsen-Pfeifer, C.; Huijsman, E.A.H.; Hasan, A.; Sommer, I.E.C.; Leucht, S.; Kahn, R.S.; Luykx, J.J. Clozapine as a first- or second-line treatment in schizophrenia: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2018, 138, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Tiihonen, J.; Mittendorfer-Rutz, E.; Majak, M.; Mehtala, J.; Hoti, F.; Jedenius, E.; Enkusson, D.; Leval, A.; Sermon, J.; Tanskanen, A.; et al. Real-world effectiveness of antipsychotic treatments in a nationwide cohort of 29,823 patients with schizophrenia. JAMA Psychiatry 2017, 74, 686–693. [Google Scholar] [CrossRef]
- Li, X.H.; Zhong, X.M.; Lu, L.; Zheng, W.; Wang, S.B.; Rao, W.W.; Wang, S.; Ng, C.H.; Ungvari, G.S.; Wang, G.; et al. The prevalence of agranulocytosis and related death in clozapine-treated patients: A comprehensive meta-analysis of observational studies. Psychol. Med. 2020, 50, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.; Ramineni, V.; Malacova, E.; Eriksson, L.; McMahon, K.; Moudgil, V.; Scott, J.; Siskind, D. Risk factors for clozapine-induced myocarditis and cardiomyopathy: A systematic review and meta-analysis. Acta Psychiatr. Scand. 2022, 145, 442–455. [Google Scholar] [CrossRef]
- Pillinger, T.; McCutcheon, R.A.; Vano, L.; Mizuno, Y.; Arumuham, A.; Hindley, G.; Beck, K.; Natesan, S.; Efthimiou, O.; Cipriani, A.; et al. Comparative effects of 18 antipsychotics on metabolic function in patients with schizophrenia, predictors of metabolic dysregulation, and association with psychopathology: A systematic review and network meta-analysis. Lancet Psychiatry 2020, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Blackman, G.; Oloyede, E. Clozapine discontinuation withdrawal symptoms in schizophrenia. Ther. Adv. Psychopharmacol. 2021, 11, 20451253211032053. [Google Scholar] [CrossRef]
- Lander, M.; Bastiampillai, T.; Sareen, J. Review of withdrawal catatonia: What does this reveal about clozapine? Transl. Psychiatry 2018, 8, 139. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A working hypothesis regarding identical pathomechanisms between clinical efficacy and adverse reaction of clozapine via the activation of connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Bymaster, F.P.; Meltzer, H.Y.; Deutch, A.Y.; Duncan, G.E.; Marx, C.E.; Aprille, J.R.; Dwyer, D.S.; Li, X.M.; Mahadik, S.P.; et al. Antipsychotic drugs: Comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol. Rev. 2008, 60, 358–403. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Sapkota, K. The origin of nmda receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.A.; Quincy Erickson-Oberg, M.; Logan, R.W.; Freyberg, Z. Relevance of interactions between dopamine and glutamate neurotransmission in schizophrenia. Mol. Psychiatry 2022, 27, 3583–3591. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Shatalina, E. Integrating the neurodevelopmental and dopamine hypotheses of schizophrenia and the role of cortical excitation-inhibition balance. Biol. Psychiatry 2022, 92, 501–513. [Google Scholar] [CrossRef]
- Javitt, D.C. Cognitive impairment associated with schizophrenia: From pathophysiology to treatment. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 119–141. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial d-serine and l-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine normalizes a glutamatergic transmission abnormality induced by an impaired nmda receptor in the thalamocortical pathway via the activation of a group iii metabotropic glutamate receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of astroglial connexin is involved in concentration-dependent double-edged sword clinical action of clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of atypical antipsychotics, clozapine, quetiapine and brexpiprazole on astroglial transmission associated with connexin43. Int. J. Mol. Sci. 2021, 22, 5623. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. Therapeutic potential and limitation of serotonin type 7 receptor modulation. Int. J. Mol. Sci. 2023, 24, 2070. [Google Scholar] [CrossRef] [PubMed]
- Okubo, R.; Hasegawa, T.; Fukuyama, K.; Shiroyama, T.; Okada, M. Current limitations and candidate potential of 5-ht7 receptor antagonism in psychiatric pharmacotherapy. Front. Psychiatry 2021, 12, 623684. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Motomura, E.; Okada, M. Opposing effects of clozapine and brexpiprazole on beta-aminoisobutyric acid: Pathophysiology of antipsychotics-induced weight gain. Schizophrenia 2023, 9, 8. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. A candidate gliotransmitter, l-β-aminoisobutyrate, contributes to weight gain and metabolic complication induced by atypical antipsychotics. Nutrients 2023, 15, 1621. [Google Scholar] [CrossRef] [PubMed]
- Crumpler, H.R.; Dent, C.E.; Harris, H.; Westall, R.G. Beta-aminoisobutyric acid (alpha-methyl-beta-alanine); a new amino-acid obtained from human urine. Nature 1951, 167, 307–308. [Google Scholar] [CrossRef]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. Beta-aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Jung, T.W.; Hwang, H.J.; Hong, H.C.; Yoo, H.J.; Baik, S.H.; Choi, K.M. Baiba attenuates insulin resistance and inflammation induced by palmitate or a high fat diet via an ampk-ppardelta-dependent pathway in mice. Diabetologia 2015, 58, 2096–2105. [Google Scholar] [CrossRef]
- Shi, C.X.; Zhao, M.X.; Shu, X.D.; Xiong, X.Q.; Wang, J.J.; Gao, X.Y.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. Beta-aminoisobutyric acid attenuates hepatic endoplasmic reticulum stress and glucose/lipid metabolic disturbance in mice with type 2 diabetes. Sci. Rep. 2016, 6, 21924. [Google Scholar] [CrossRef]
- Lopez, M. Hypothalamic ampk as a possible target for energy balance-related diseases. Trends Pharmacol. Sci. 2022, 43, 546–556. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef]
- Siskind, D.J.; Leung, J.; Russell, A.W.; Wysoczanski, D.; Kisely, S. Metformin for clozapine associated obesity: A systematic review and meta-analysis. PLoS ONE 2016, 11, e0156208. [Google Scholar] [CrossRef]
- Zheng, W.; Li, X.B.; Tang, Y.L.; Xiang, Y.Q.; Wang, C.Y.; de Leon, J. Metformin for weight gain and metabolic abnormalities associated with antipsychotic treatment: Meta-analysis of randomized placebo-controlled trials. J. Clin. Psychopharmacol. 2015, 35, 499–509. [Google Scholar] [CrossRef] [PubMed]
- de Silva, V.A.; Suraweera, C.; Ratnatunga, S.S.; Dayabandara, M.; Wanniarachchi, N.; Hanwella, R. Metformin in prevention and treatment of antipsychotic induced weight gain: A systematic review and meta-analysis. BMC Psychiatry 2016, 16, 341. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, Q.E.; Cai, D.B.; Yang, X.H.; Ungvari, G.S.; Ng, C.H.; Wu, R.R.; Xiang, Y.T. Combination of metformin and lifestyle intervention for antipsychotic-related weight gain: A meta-analysis of randomized controlled trials. Pharmacopsychiatry 2019, 52, 24–31. [Google Scholar] [CrossRef]
- Jiang, W.L.; Cai, D.B.; Yin, F.; Zhang, L.; Zhao, X.W.; He, J.; Ng, C.H.; Ungvari, G.S.; Sim, K.; Hu, M.L.; et al. Adjunctive metformin for antipsychotic-induced dyslipidemia: A meta-analysis of randomized, double-blind, placebo-controlled trials. Transl. Psychiatry 2020, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Decrock, E.; De Bock, M.; Wang, N.; Gadicherla, A.K.; Bol, M.; Delvaeye, T.; Vandenabeele, P.; Vinken, M.; Bultynck, G.; Krysko, D.V.; et al. Ip3, a small molecule with a powerful message. Biochim. Biophys. Acta 2013, 1833, 1772–1786. [Google Scholar] [CrossRef]
- Carli, M.; Kolachalam, S.; Longoni, B.; Pintaudi, A.; Baldini, M.; Aringhieri, S.; Fasciani, I.; Annibale, P.; Maggio, R.; Scarselli, M. Atypical antipsychotics and metabolic syndrome: From molecular mechanisms to clinical differences. Pharmaceuticals 2021, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Motomura, E. Dose-dependent biphasic action of quetiapine on ampk signalling via 5-ht7 receptor: Exploring pathophysiology of clinical and adverse effects of quetiapine. Int. J. Mol. Sci. 2022, 23, 9103. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Shiroyama, T.; Okada, M. Impact of 5-ht7 receptor inverse agonism of lurasidone on monoaminergic tripartite synaptic transmission and pathophysiology of lower risk of weight gain. Biomed. Pharmacother. 2022, 148, 112750. [Google Scholar] [CrossRef]
- Fukuyama, K.; Motomura, E.; Okada, M. Brexpiprazole reduces 5-ht7 receptor function on astroglial transmission systems. Int. J. Mol. Sci. 2022, 23, 6571. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Effects of an atypical antipsychotic, zotepine, on astroglial l-glutamate release through hemichannels: Exploring the mechanism of mood-stabilising antipsychotic actions and antipsychotic-induced convulsion. Pharmaceuticals 2021, 14, 1116. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.F.; Huang, A.S.; Snowman, A.M.; Teuscher, C.; Snyder, S.H. From the cover: Antipsychotic drug-induced weight gain mediated by histamine h1 receptor-linked activation of hypothalamic amp-kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 3456–3459. [Google Scholar] [CrossRef]
- He, M.; Deng, C.; Huang, X.F. The role of hypothalamic h1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs 2013, 27, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Siafis, S.; Hamza, T.; Schneider-Thoma, J.; Davis, J.M.; Salanti, G.; Leucht, S. Antipsychotic-induced weight gain: Dose-response meta-analysis of randomized controlled trials. Schizophr. Bull. 2022, 48, 643–654. [Google Scholar] [CrossRef]
- Okada, M.; Yoshida, S.; Zhu, G.; Hirose, S.; Kaneko, S. Biphasic actions of topiramate on monoamine exocytosis associated with both soluble n-ethylmaleimide-sensitive factor attachment protein receptors and ca(2+)-induced ca(2+)-releasing systems. Neuroscience 2005, 134, 233–246. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum-mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef]
- Fukuyama, K.; Tanahashi, S.; Nakagawa, M.; Yamamura, S.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Levetiracetam inhibits neurotransmitter release associated with cicr. Neurosci. Lett. 2012, 518, 69–74. [Google Scholar] [CrossRef]
- Horikoshi, T.; Asanuma, A.; Yanagisawa, K.; Anzai, K.; Goto, S. Taurine and beta-alanine act on both gaba and glycine receptors in xenopus oocyte injected with mouse brain messenger rna. Brain Res. 1988, 464, 97–105. [Google Scholar] [PubMed]
- Schmieden, V.; Betz, H. Pharmacology of the inhibitory glycine receptor: Agonist and antagonist actions of amino acids and piperidine carboxylic acid compounds. Mol. Pharmacol. 1995, 48, 919–927. [Google Scholar]
- Nair, P.C.; McKinnon, R.A.; Miners, J.O.; Bastiampillai, T. Binding of clozapine to the gabab receptor: Clinical and structural insights. Mol. Psychiatry 2020, 25, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Hirjak, D.; Northoff, G.; Taylor, S.F.; Wolf, R.C. Gabab receptor, clozapine, and catatonia-a complex triad. Mol. Psychiatry 2021, 26, 2683–2684. [Google Scholar] [CrossRef]
- Wu, Y.; Blichowski, M.; Daskalakis, Z.J.; Wu, Z.; Liu, C.C.; Cortez, M.A.; Snead, O.C., 3rd. Evidence that clozapine directly interacts on the gabab receptor. Neuroreport 2011, 22, 637–641. [Google Scholar] [CrossRef]
- Miyazawa, A.; Kanahara, N.; Shiko, Y.; Ozawa, Y.; Kawasaki, Y.; Komatsu, H.; Masumo, Y.; Nakata, Y.; Iyo, M. The cortical silent period in schizophrenia: A systematic review and meta-analysis focusing on disease stage and antipsychotic medication. J. Psychopharmacol. 2022, 36, 479–488. [Google Scholar] [CrossRef]
- Roth BL, D.J. Psychoactive Drug Screening Program (PDSP); University of North Carolina: Charlotte, NC, USA, 2017. [Google Scholar]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/glutamate antiporter and aripiprazole compensate nmda antagonist-induced dysfunction of thalamocortical l-glutamatergic transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone sub-chronically activates serotonergic transmission via desensitization of 5-ht1a and 5-ht7 receptors in dorsal raphe nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits nmda receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-ht7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of carbamazepine, lacosamide and zonisamide on gliotransmitter release associated with activated astroglial hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef]
- Lilley, E.; Stanford, S.C.; Kendall, D.E.; Alexander, S.P.H.; Cirino, G.; Docherty, J.R.; George, C.H.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Arrive 2.0 and the british journal of pharmacology: Updated guidance for 2020. Br. J. Pharmacol. 2020, 177, 3611–3616. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Hoshikawa, M.; Dai, K.; Saito, H.; Suzuki, N.; Niwa, O.; Okada, M. Ono-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br. J. Pharmacol. 2013, 168, 1088–1100. [Google Scholar] [CrossRef]
- Gjoni, T.; Urwyler, S. Changes in the properties of allosteric and orthosteric gabab receptor ligands after a continuous, desensitizing agonist pretreatment. Eur. J. Pharmacol. 2009, 603, 37–41. [Google Scholar] [CrossRef]
- Vernon, A.C.; Smith, E.J.; Stevanato, L.; Modo, M. Selective activation of metabotropic glutamate receptor 7 induces inhibition of cellular proliferation and promotes astrocyte differentiation of ventral mesencephalon human neural stem/progenitor cells. Neurochem. Int. 2011, 59, 421–431. [Google Scholar] [CrossRef]
- Azevedo, C.; Saiardi, A. Extraction and analysis of soluble inositol polyphosphates from yeast. Nat. Protoc. 2006, 1, 2416–2422. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. High frequency oscillations play important roles in development of epileptogenesis/ictogenesis via activation of astroglial signallings. Biomed. Pharmacother. 2022, 149, 112846. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okada, M. Brivaracetam and levetiracetam suppress astroglial l-glutamate release through hemichannel via inhibition of synaptic vesicle protein. Int. J. Mol. Sci. 2022, 23, 4473. [Google Scholar] [CrossRef]
- Latchoumane, C.V.; Ngo, H.V.; Born, J.; Shin, H.S. Thalamic spindles promote memory formation during sleep through triple phase-locking of cortical, thalamic, and hippocampal rhythms. Neuron 2017, 95, 424–435.e6. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C.; Bergemann, N.; Clement, H.W.; Conca, A.; Deckert, J.; Domschke, K.; Eckermann, G.; Egberts, K.; Gerlach, M.; Greiner, C.; et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: Update 2017. Pharmacopsychiatry 2018, 51, 9–62. [Google Scholar] [PubMed]
- Schoretsanitis, G.; Paulzen, M.; Unterecker, S.; Schwarz, M.; Conca, A.; Zernig, G.; Grunder, G.; Haen, E.; Baumann, P.; Bergemann, N.; et al. TDM in psychiatry and neurology: A comprehensive summary of the consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology, update 2017; a tool for clinicians. World J. Biol. Psychiatry 2018, 19, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Okubo, R.; Fukuyama, K. Vortioxetine subchronically activates serotonergic transmission via desensitization of serotonin 5-ht1a receptor with 5-ht3 receptor inhibition in rats. Int. J. Mol. Sci. 2019, 20, 6235. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Okada, M. Age-dependent and sleep/seizure-induced pathomechanisms of autosomal dominant sleep-related hypermotor epilepsy. Int. J. Mol. Sci. 2020, 21, 8142. [Google Scholar] [CrossRef]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine combines astroglial system xc− activation with glutamate/nmda receptor inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by nmda receptor antagonism via activation of system xc. Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Frau, R.; Orru, M.; Piras, A.P.; Fa, M.; Tuveri, A.; Puligheddu, M.; Gessa, G.L.; Castelli, M.P.; Mereu, G.; et al. Activation of gaba(b) receptors reverses spontaneous gating deficits in juvenile dba/2j mice. Psychopharmacology 2007, 194, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Nishikimi, M.; Inoue, M.; Muragaki, Y.; Ooshima, A. Specific expression of alanine-glyoxylate aminotransferase 2 in the epithelial cells of henle’s loop. Nephron 1999, 83, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, R.J.; Green, A.; Smith, R. Excessive excretion of beta-alanine and of 3-hydroxypropionic, r- and s-3-aminoisobutyric, r- and s-3-hydroxyisobutyric and s-2-(hydroxymethyl)butyric acids probably due to a defect in the metabolism of the corresponding malonic semialdehydes. J. Inherit. Metab. Dis. 1985, 8, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.R.; Struys, E.; Kok, R.M.; Roe, D.S.; Harris, R.A.; Jakobs, C. Methylmalonic semialdehyde dehydrogenase deficiency: Psychomotor delay and methylmalonic aciduria without metabolic decompensation. Mol. Genet. Metab. 1998, 65, 35–43. [Google Scholar] [CrossRef]
- Kakimoto, Y.; Kanazawa, A.; Taniguchi, K.; Sano, I. Beta-aminoisobutyrate-alpha-ketoglutarate transaminase in relation to beta-aminoisobutyric aciduria. Biochim. Biophys. Acta 1968, 156, 374–380. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Vellucci, L.; Barone, A.; Manchia, M.; De Luca, V.; Iasevoli, F.; Correll, C.U. Clozapine’s multiple cellular mechanisms: What do we know after more than fifty years? A systematic review and critical assessment of translational mechanisms relevant for innovative strategies in treatment-resistant schizophrenia. Pharmacol. Ther. 2022, 236, 108236. [Google Scholar] [CrossRef]
- Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate strategies for development of a rapid-acting antidepressant class that does not result in neuropsychiatric adverse effects: Prevention of ketamine-induced neuropsychiatric adverse reactions. Int. J. Mol. Sci. 2020, 21, 7951. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K. Interaction between mesocortical and mesothalamic catecholaminergic transmissions associated with nmda receptor in the locus coeruleus. Biomolecules 2020, 10, 990. [Google Scholar] [CrossRef]
- Cruz, K.G.; Leow, Y.N.; Le, N.M.; Adam, E.; Huda, R.; Sur, M. Cortical-subcortical interactions in goal-directed behavior. Physiol. Rev. 2023, 103, 347–389. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Nakano, T.; Shiroyama, T.; Okada, M. Chronic administrations of guanfacine on mesocortical catecholaminergic and thalamocortical glutamatergic transmissions. Int. J. Mol. Sci. 2021, 22, 4122. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with s284l-mutant alpha4 subunit of nicotinic ach receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Matsumoto, R.; Yamamoto, Y.; Fukuyama, K. Effects of subchronic administrations of vortioxetine, lurasidone, and escitalopram on thalamocortical glutamatergic transmission associated with serotonin 5-ht7 receptor. Int. J. Mol. Sci. 2021, 22, 1351. [Google Scholar] [CrossRef] [PubMed]
- Roenker, N.L.; Gudelsky, G.A.; Ahlbrand, R.; Horn, P.S.; Richtand, N.M. Evidence for involvement of nitric oxide and gaba(b) receptors in mk-801- stimulated release of glutamate in rat prefrontal cortex. Neuropharmacology 2012, 63, 575–581. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Haidary, H.A.; Padhy, R.K. Clozapine. In Statpearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Bilbily, J.; McCollum, B.; de Leon, J. Catatonia secondary to sudden clozapine withdrawal: A case with three repeated episodes and a literature review. Case Rep. Psychiatry 2017, 2017, 2402731. [Google Scholar] [CrossRef]
- Ito, Y.; Murata, M.; Taku, O.; Fukuyama, K.; Motomura, E.; Dohi, K.; Okada, M. Developed catatonia with rhabdomyolysis and exacerbated cardiac failure upon switching from clozapine to olanzapine owing to cardiomyopathy during clozapine medication—A case report. Asian J. Psychiatr. 2023, 80, 103376. [Google Scholar] [CrossRef]
- Ingole, A.; Bastiampillai, T.; Tibrewal, P. Clozapine withdrawal catatonia, psychosis and associated neuroleptic malignant syndrome. Asian J. Psychiatr. 2017, 30, 96–97. [Google Scholar] [CrossRef]
- O’Connor, W.T.; O’Shea, S.D. Clozapine and gaba transmission in schizophrenia disease models: Establishing principles to guide treatments. Pharmacol. Ther. 2015, 150, 47–80. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukuyama, K.; Motomura, E.; Okada, M. Enhanced L-β-Aminoisobutyric Acid Is Involved in the Pathophysiology of Effectiveness for Treatment-Resistant Schizophrenia and Adverse Reactions of Clozapine. Biomolecules 2023, 13, 862. https://doi.org/10.3390/biom13050862
Fukuyama K, Motomura E, Okada M. Enhanced L-β-Aminoisobutyric Acid Is Involved in the Pathophysiology of Effectiveness for Treatment-Resistant Schizophrenia and Adverse Reactions of Clozapine. Biomolecules. 2023; 13(5):862. https://doi.org/10.3390/biom13050862
Chicago/Turabian StyleFukuyama, Kouji, Eishi Motomura, and Motohiro Okada. 2023. "Enhanced L-β-Aminoisobutyric Acid Is Involved in the Pathophysiology of Effectiveness for Treatment-Resistant Schizophrenia and Adverse Reactions of Clozapine" Biomolecules 13, no. 5: 862. https://doi.org/10.3390/biom13050862