
Effect of Cucumber Continuous Monocropping on Traditional Chinese Medicine Residue through Analysis of Physicochemical Characteristics and Microbial Diversity
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Physicochemical Indices of the Substrates
2.3. DNA Extraction, PCR Amplification and Sequencing
2.4. Bioinformatics and Statistical Analysis
2.5. Species Diversity Analysis
2.6. Correlation Analysis between Physicochemical Properties and Microbial Communities
2.7. Analysis of Key Microbiota
3. Results
3.1. The Physicochemical Properties Changed Due to Continuous Monocropping
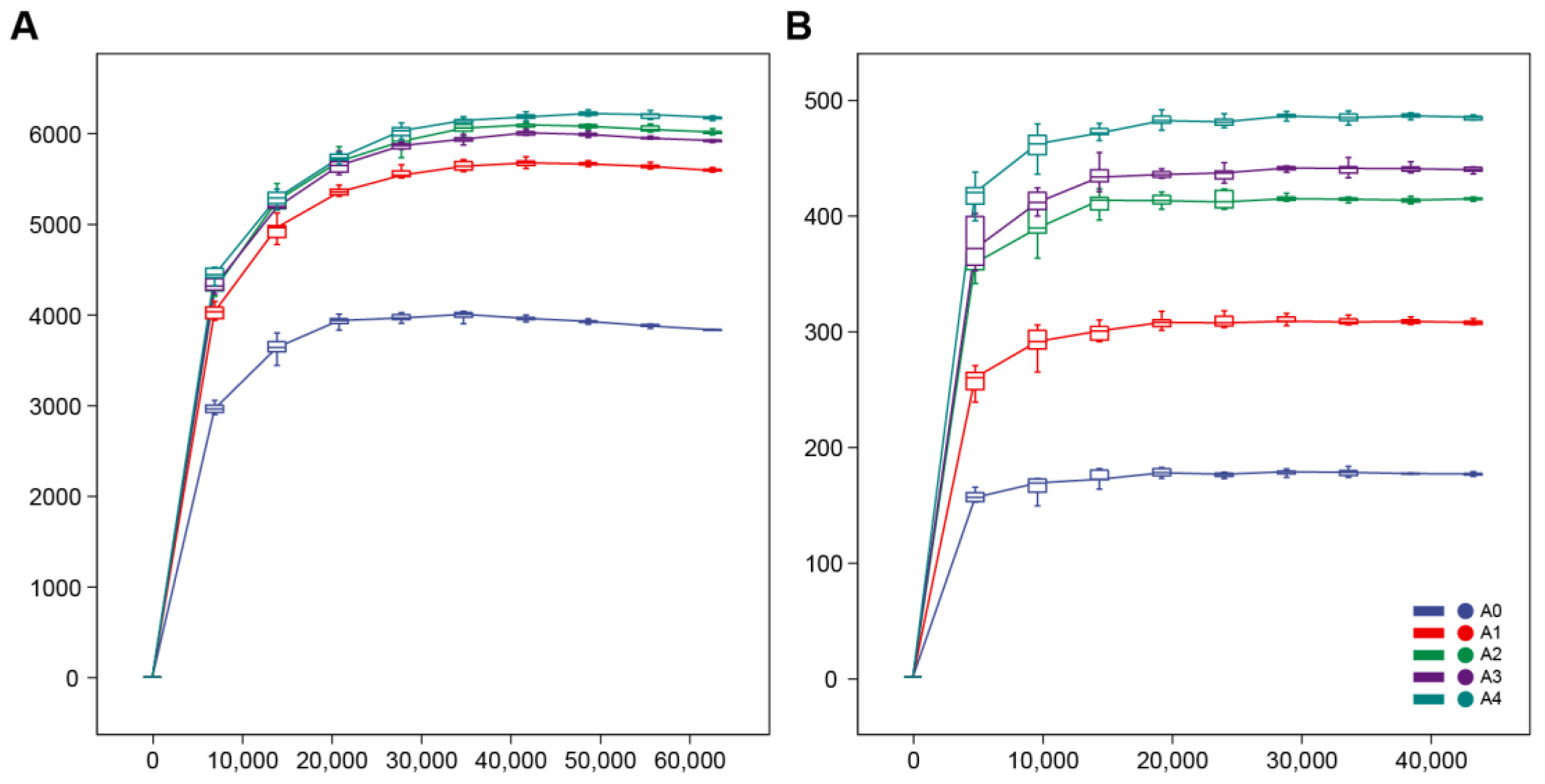
3.2. Assessment of the Sequencing Quality
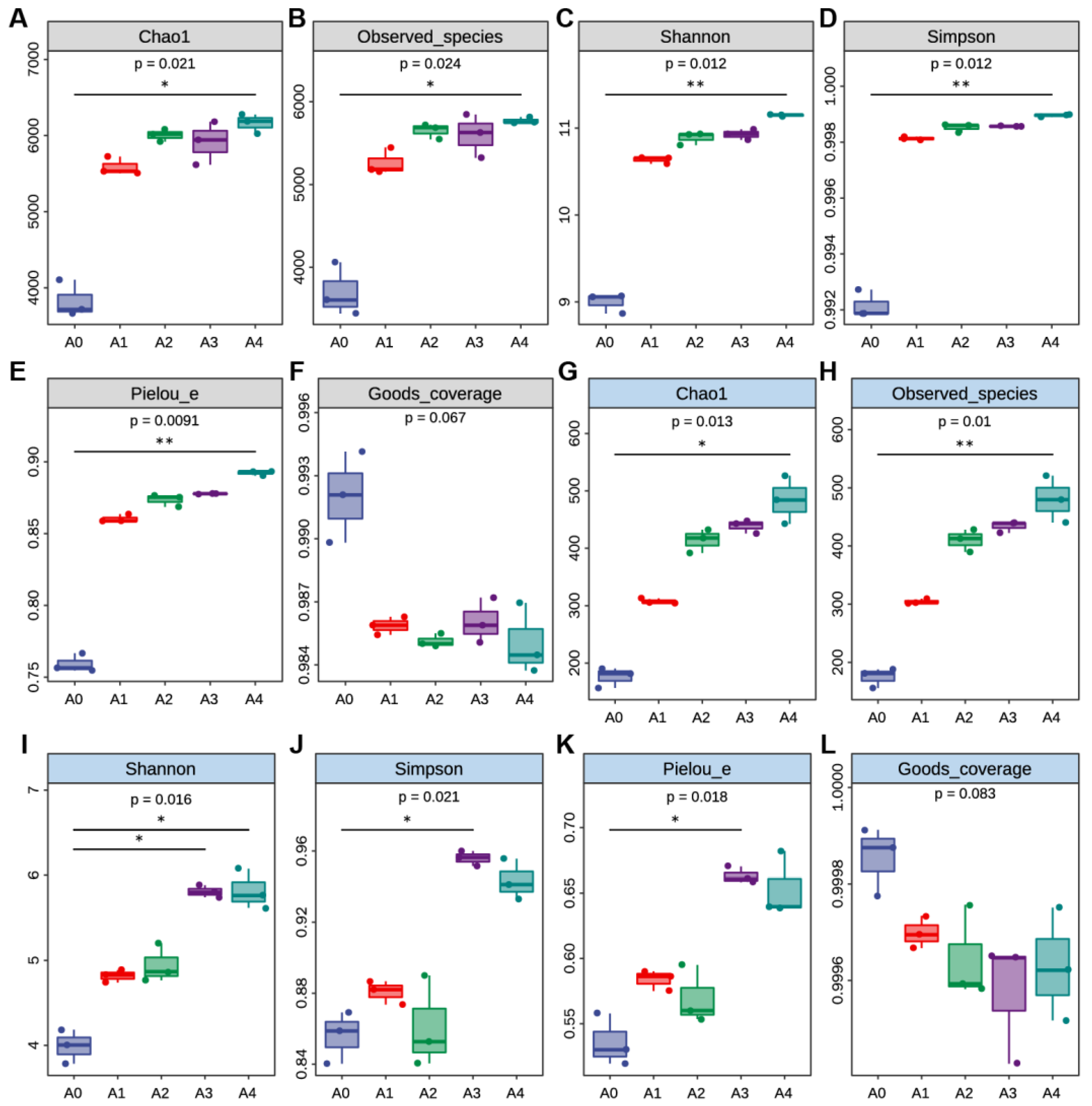
3.3. The Microbial α-Diversity Increased with Cucumber Continuous Cropping
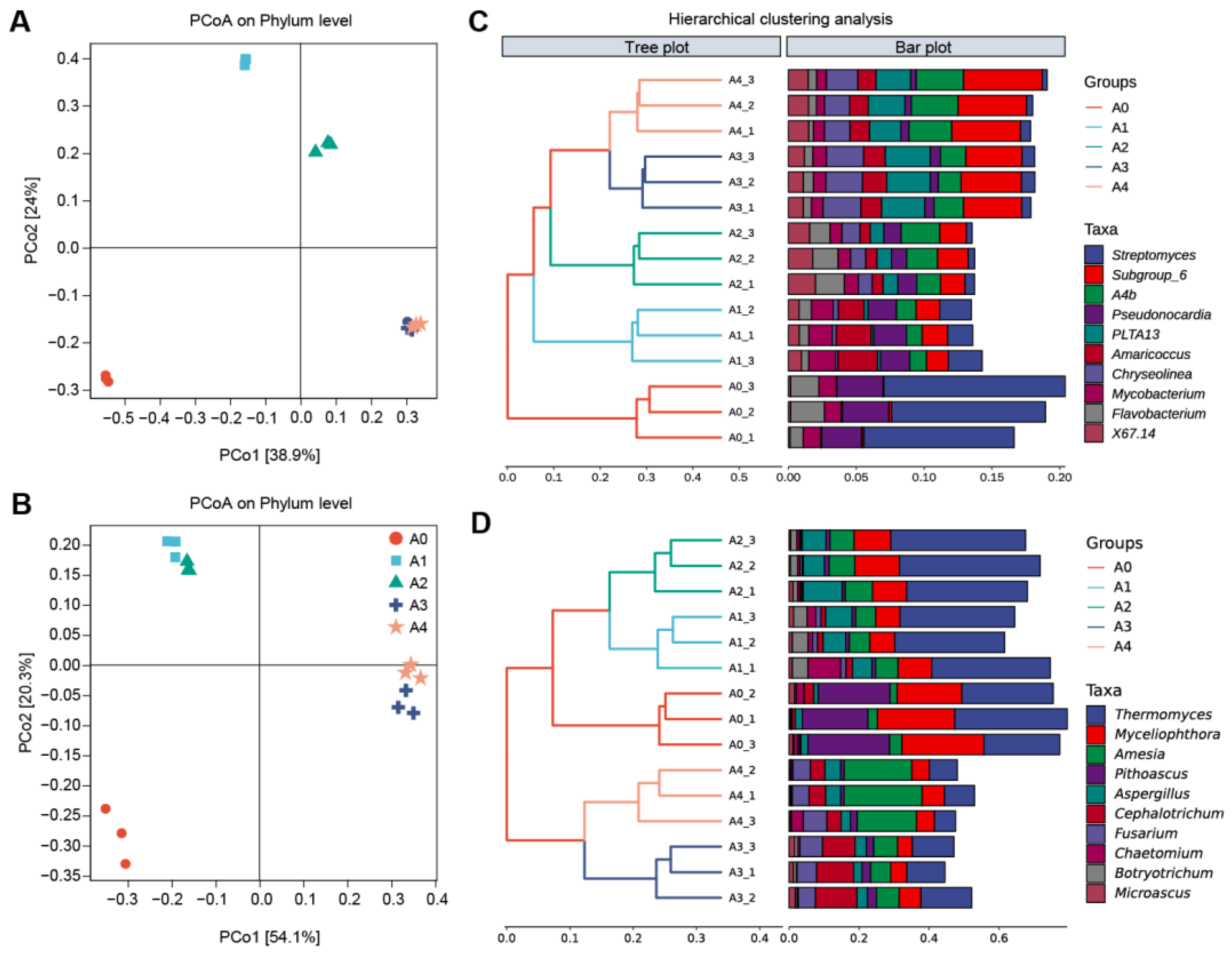
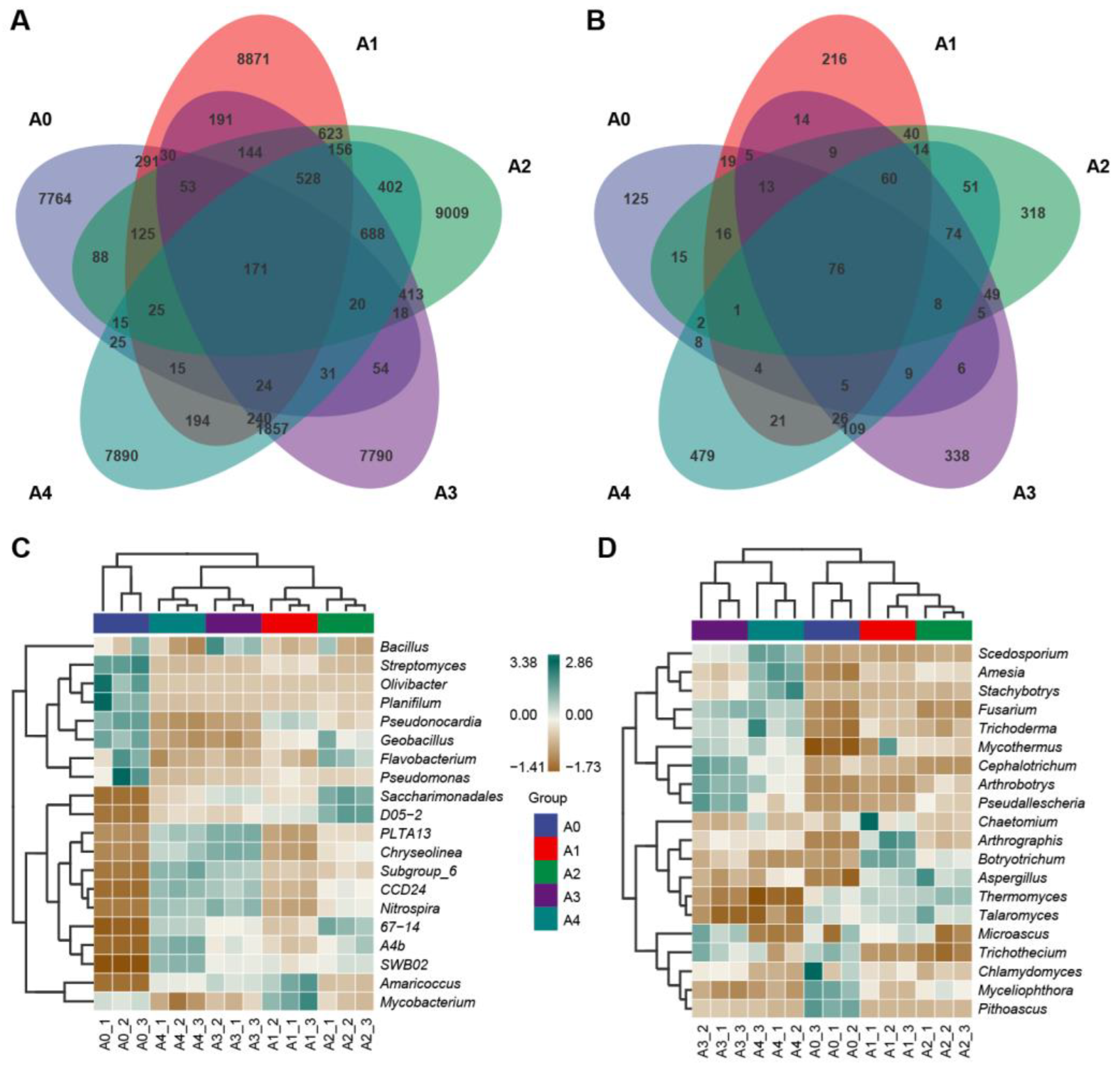
3.4. Analysis of Dissimilarities in the Composition of Microbial Communities
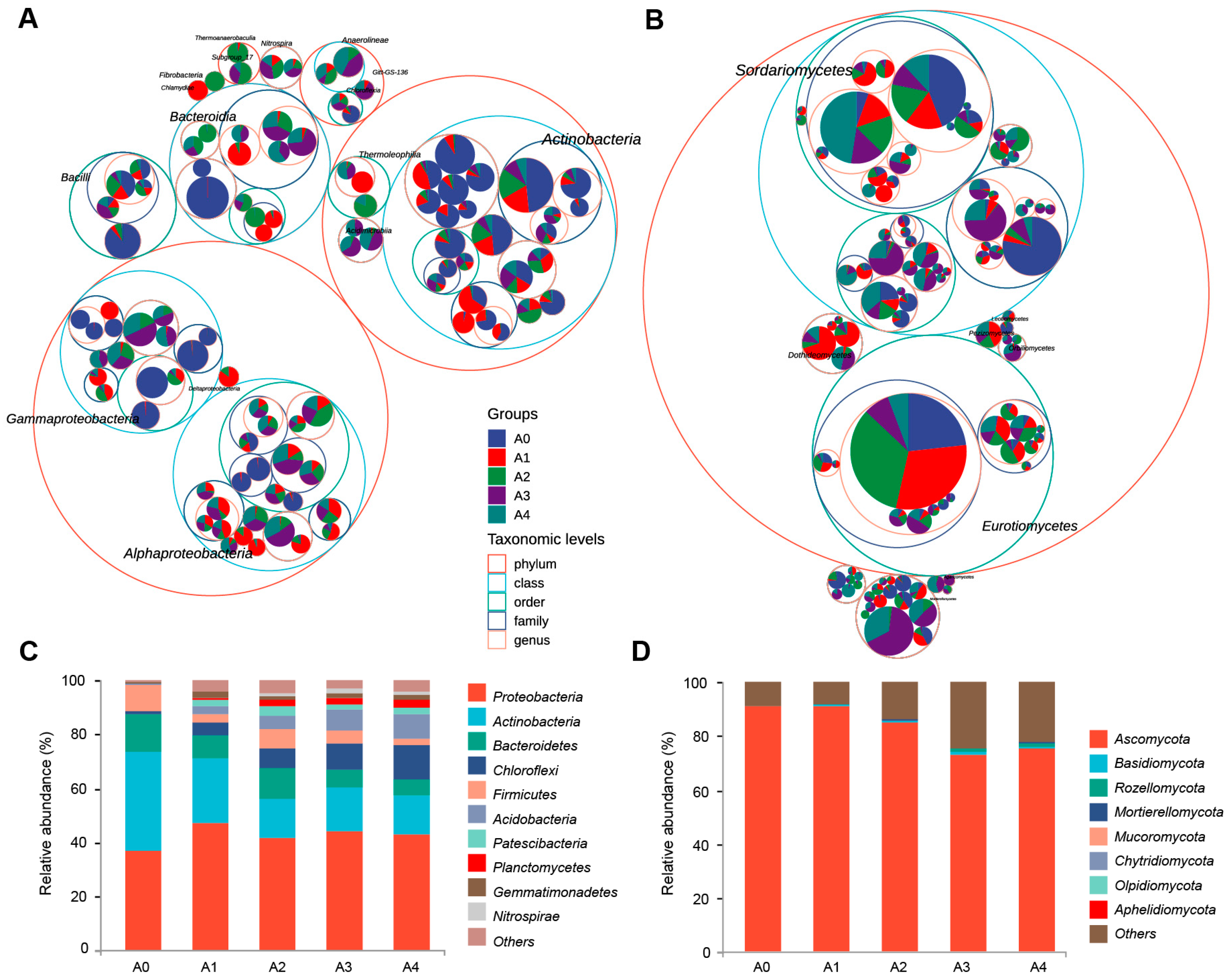
3.5. Composition Analysis of the Bacterial and Fungal Communities
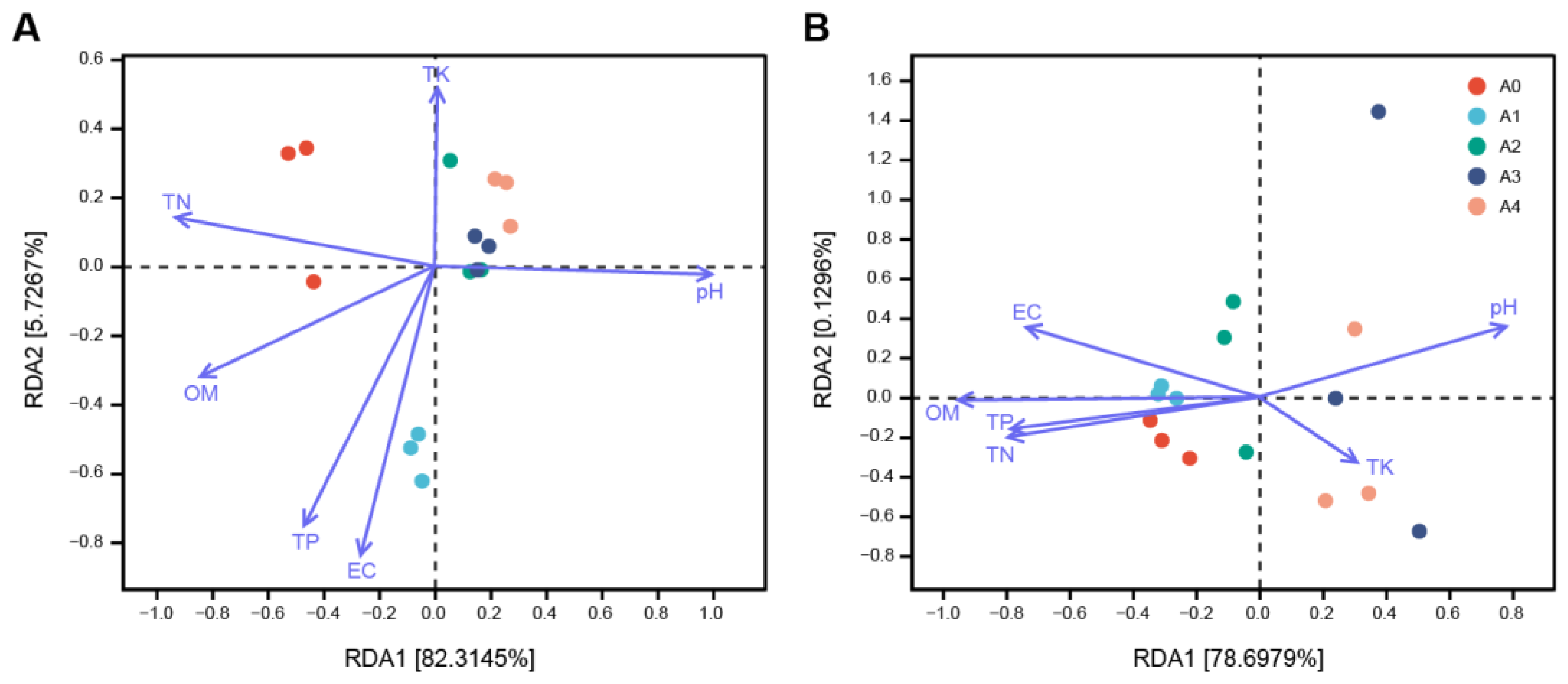
3.6. Correlation Analysis of Physicochemical Properties with Microbial Diversity
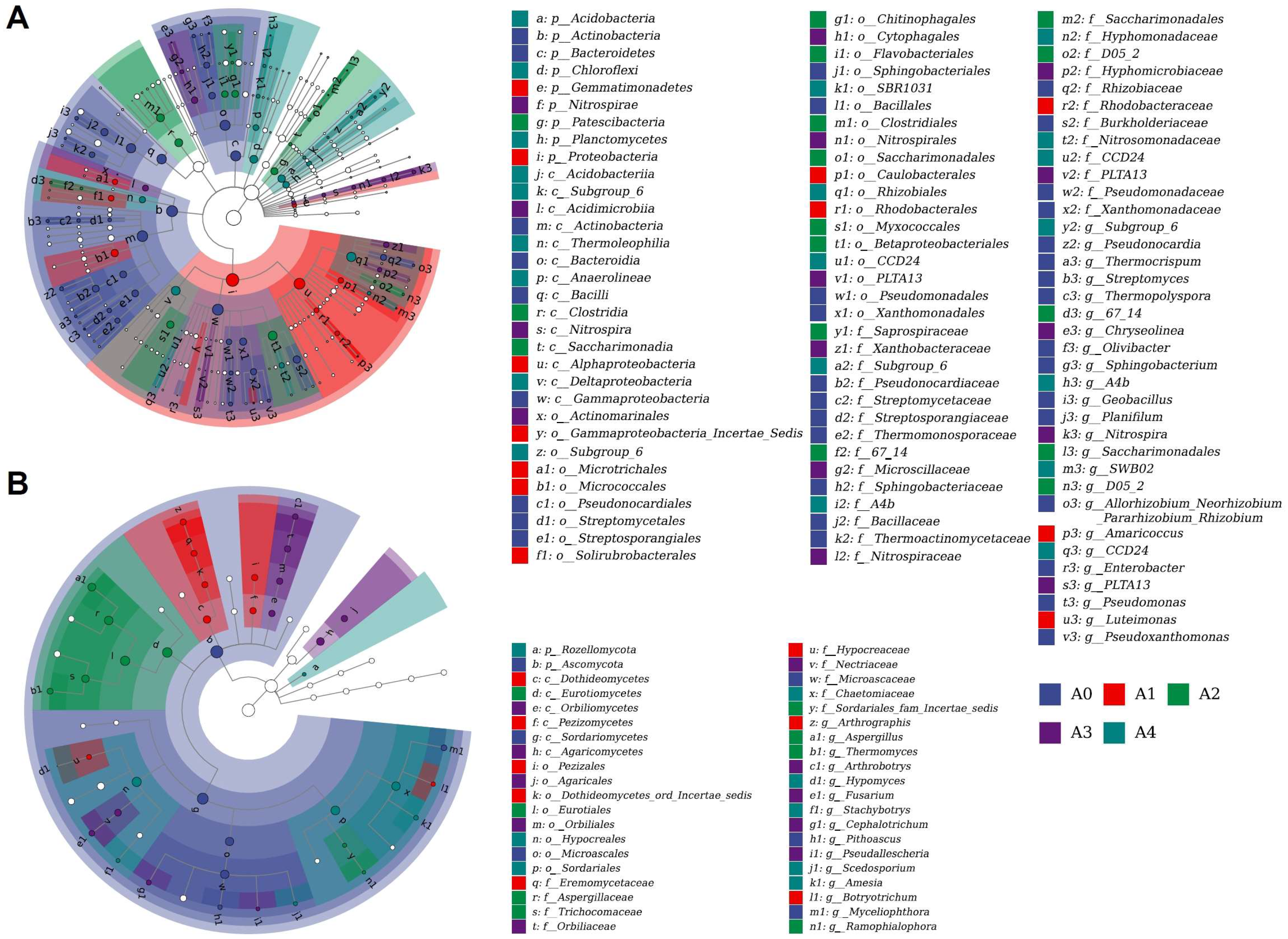
3.7. Analysis of Biomarkers in Substrate Samples
4. Discussion
4.1. Effect of TCM on the Physicochemical Properties of Substrates
4.2. Effect of TCM on the Diversity of the Bacterial and Fungal Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, B.; Wang, X.; Yang, S.; Chen, H.; Zhao, Y. Addition Pinus massoniana fallen wood improved the growth of Plagiomnium acutum in a substrate cultivation. Sci. Rep. 2022, 12, 17755. [Google Scholar] [CrossRef]
- Ulaganathan, K.; Dong, C.-J.; Li, Q.; Wang, L.-L.; Shang, Q.-M. Dynamic changes in bacterial communities in the recirculating nutrient solution of cucumber plug seedlings cultivated in an ebb-and-flow subirrigation system. PLoS ONE 2020, 15, e0232446. [Google Scholar] [CrossRef]
- Meng, L.; Fu, Y.; Li, D.; Sun, X.; Chen, Y.; Li, X.; Xu, S.; Li, X.; Li, C.; Song, B.; et al. Effects of corn stalk cultivation substrate on the growth of the slippery mushroom (Pholiota microspora). RSC Adv. 2019, 9, 5347–5353. [Google Scholar] [CrossRef]
- Siddiqui, Z.; Hagare, D.; Liu, M.-H.; Panatta, O.; Hussain, T.; Memon, S.; Noorani, A.; Chen, Z.-H. A Food Waste-Derived Organic Liquid Fertiliser for Sustainable Hydroponic Cultivation of Lettuce, Cucumber and Cherry Tomato. Foods 2023, 12, 719. [Google Scholar] [CrossRef]
- Conversa, G.; Bonasia, A.; Lazzizera, C.; La Rotonda, P.; Elia, A. Reduction of Nitrate Content in Baby-Leaf Lettuce and Cichorium endivia Through the Soilless Cultivation System, Electrical Conductivity and Management of Nutrient Solution. Front. Plant Sci. 2021, 12, 645671. [Google Scholar] [CrossRef]
- Tzortzakis, N.; Nicola, S.; Savvas, D.; Voogt, W. Editorial: Soilless Cultivation Through an Intensive Crop Production Scheme. Management Strategies, Challenges and Future Directions. Front. Plant Sci. 2020, 11, 529970. [Google Scholar] [CrossRef]
- Renna, M.; D’Imperio, M.; Maggi, S.; Serio, F. Soilless biofortification, bioaccessibility, and bioavailability: Signposts on the path to personalized nutrition. Front. Nutr. 2022, 9, 966018. [Google Scholar] [CrossRef]
- Xu, B.; Yang, X.; Li, Y.; Yang, K.; Xiong, Y.; Yuan, N. Pyrite-Based Autotrophic Denitrifying Microorganisms Derived from Paddy Soils: Effects of Organic Co-Substrate Addition. Int. J. Environ. Res. Public Health 2022, 19, 11763. [Google Scholar] [CrossRef]
- Zhou, F.; Wu, X.; Gao, Y.; Fan, S.; Zhou, H.; Zhang, X. Diversity Shifts in the Root Microbiome of Cucumber under Different Plant Cultivation Substrates. Front. Microbiol. 2022, 13, 878409. [Google Scholar] [CrossRef]
- Rabbani, M.; Kazemi, F. Water need and water use efficiency of two plant species in soil-containing and soilless substrates under green roof conditions. J. Environ. Manag. 2022, 302, 113950. [Google Scholar] [CrossRef]
- Suwannarach, N.; Kumla, J.; Zhao, Y.; Kakumyan, P. Impact of Cultivation Substrate and Microbial Community on Improving Mushroom Productivity: A Review. Biology 2022, 11, 569. [Google Scholar] [CrossRef]
- Anzalone, A.; Mosca, A.; Dimaria, G.; Nicotra, D.; Tessitori, M.; Privitera, G.F.; Pulvirenti, A.; Leonardi, C.; Catara, V. Soil and Soilless Tomato Cultivation Promote Different Microbial Communities That Provide New Models for Future Crop Interventions. Int. J. Mol. Sci. 2022, 23, 8820. [Google Scholar] [CrossRef]
- Burza, W.; Zuzga, S.; Yin, Z.; Malepszy, S. Cucumber (Cucumis sativus L.). In Agrobacterium Protocols; Wang, K., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 427–438. [Google Scholar]
- Zhang, Y.; Chen, H.; Zhu, W.; Li, C. Comparison of the carotenoid compositions and protection of in-season and anti-season tomato extracts againstd-galactose-induced cognition deficits and oxidative damage in mice. Int. J. Food Sci. Nutr. 2016, 67, 983–994. [Google Scholar] [CrossRef]
- Larkin, R.P. Characterization of soil microbial communities under different potato cropping systems by microbial population dynamics, substrate utilization, and fatty acid profiles. Soil Biol. Biochem. 2003, 35, 1451–1466. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Wang, B.; Wang, X.; Franks, A.E.; Teng, Y.; Li, Z.; Luo, Y. Changes in the abundance and structure of bacterial communities under long-term fertilization treatments in a peanut monocropping system. Plant Soil 2015, 395, 415–427. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, D.; Li, Y.; Li, H.; Lou, J.; Li, X.; Wei, M. Changes in rhizospheric microbiome structure and soil metabolic function in response to continuous cucumber cultivation. FEMS Microbiol. Ecol. 2022, 98, fiac129. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, D.; Li, X.; Li, X.; Lou, J.; Wei, M. Effect of vegetable residues incorporation on soil fertility, rhizosphere microbial community structure, and plant growth of continuously cropped cucumber in a solar greenhouse. Ann. Microbiol. 2022, 72, 32. [Google Scholar] [CrossRef]
- Ding, H.; Ali, A.; Cheng, Z. Effect of green garlic/cucumber crop rotation for 3 years on the dynamics of soil properties and cucumber yield in Chinese anthrosol. J. Sci. Food Agric. 2019, 100, 362–370. [Google Scholar] [CrossRef]
- Xu, S.-Y.; Wei, J.-K.; Xue, F.-Y.; Li, W.-C.; Guan, T.-K.; Hu, B.-Y.; Chen, Q.-J.; Han, Y.-Y.; Liu, C.-J.; Zhang, G.-Q. Microbial inoculation influences microbial communities and physicochemical properties during lettuce seedling using composted spent mushroom substrate. Appl. Soil Ecol. 2022, 174, 104418. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, L.; Zhou, X.; Tian, T.; Xu, S.; Li, D.; Li, C.; Li, Y. Optimizing Straw-Rotting Cultivation for Sustainable Edible Mushroom Production: Composting Spent Mushroom Substrate with Straw Additions. J. Fungi 2023, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Vandecasteele, B.; Amery, F.; Ommeslag, S.; Vanhoutte, K.; Visser, R.; Robbens, J.; De Tender, C.; Debode, J. Chemically versus thermally processed brown shrimp shells or Chinese mitten crab as a source of chitin, nutrients or salts and as microbial stimulant in soilless strawberry cultivation. Sci. Total Environ. 2021, 771, 145263. [Google Scholar] [CrossRef]
- Zheng, W.; Ma, Y.; Wang, X.; Wang, X.; Li, J.; Tian, Y.; Zhang, X. Producing high-quality cultivation substrates for cucumber production by in-situ composting of corn straw blocks amended with biochar and earthworm casts. Waste Manag. 2022, 139, 179–189. [Google Scholar] [CrossRef]
- Huang, C.; Li, Z.-X.; Wu, Y.; Huang, Z.-Y.; Hu, Y.; Gao, J. Treatment and bioresources utilization of traditional Chinese medicinal herb residues: Recent technological advances and industrial prospect. J. Environ. Manag. 2021, 299, 113607. [Google Scholar] [CrossRef]
- Ma, J.; Chen, Y.; Zhao, Y.; Chen, D.; Wang, H. Effects of traditional Chinese medicine residue on plant growth and soil properties: A case study with maize (Zea mays L.). Environ. Sci. Pollut. Res. 2019, 26, 32880–32890. [Google Scholar] [CrossRef]
- Liu, S.; Yang, G.; Wu, F.; Ge, Y.; Liu, F.; Pu, C.; Wang, Z.; Shen, Y.; Zhou, X.; Luo, Y.; et al. Traditional Chinese medicine residues promote the growth and quality of Salvia miltiorrhiza Bunge by improving soil health under continuous monoculture. Front. Plant Sci. 2023, 14, 1112382. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Yao, W.; Wang, F.; Li, X.; Wang, W.; Li, J.; Gao, Z.; Qiu, L.; You, R.; et al. Compound kushen injection relieves tumor-associated macrophage-mediated immunosuppression through TNFR1 and sensitizes hepatocellular carcinoma to sorafenib. J. Immunother. Cancer 2020, 8, e000317. [Google Scholar] [CrossRef]
- Zhao, Z.; Fan, H.; Higgins, T.; Qi, J.; Haines, D.; Trivett, A.; Oppenheim, J.J.; Wei, H.; Li, J.; Lin, H.; et al. Fufang Kushen injection inhibits sarcoma growth and tumor-induced hyperalgesia via TRPV1 signaling pathways. Cancer Lett. 2014, 355, 232–241. [Google Scholar] [CrossRef]
- Wang, W.; You, R.-l.; Qin, W.-j.; Hai, L.-n.; Fang, M.-j.; Huang, G.-h.; Kang, R.-x.; Li, M.-h.; Qiao, Y.-f.; Li, J.-w.; et al. Anti-tumor activities of active ingredients in Compound Kushen Injection. Acta Pharmacol. Sin. 2015, 36, 676–679. [Google Scholar] [CrossRef]
- Lei, H.; Niu, T.; Song, H.; Bai, B.; Han, P.; Wang, Z.; Liu, A. Comparative transcriptome profiling reveals differentially expressed genes involved in flavonoid biosynthesis between biennial and triennial Sophora flavescens. Ind. Crops Prod. 2021, 161, 113217. [Google Scholar] [CrossRef]
- Ciavatta, C.; Govi, M.; Antisari, L.V.; Sequi, P. Determination of organic carbon in aqueous extracts of soils and fertilizers. Commun. Soil Sci. Plant Anal. 2008, 22, 795–807. [Google Scholar] [CrossRef]
- Sun, K.; Fu, L.; Song, Y.; Yuan, L.; Zhang, H.; Wen, D.; Yang, N.; Wang, X.; Yue, Y.; Li, X.; et al. Effects of continuous cucumber cropping on crop quality and soil fungal community. Environ. Monit. Assess. 2021, 193, 436. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, H.M. The determination of total phosphorus in soils by acid digestion. Chem. Anal. Environ. Res. 1987, 18, 92–96. [Google Scholar]
- Guo, Y.-X.; Chen, Q.-J.; Qin, Y.; Yang, Y.-R.; Yang, Q.-Z.; Wang, Y.-X.; Cheng, Z.-A.; Cao, N.; Zhang, G.-Q. Succession of the microbial communities and function prediction during short-term peach sawdust-based composting. Bioresour. Technol. 2021, 332, 125079. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Good, I.J. The Population Frequencies of Species and the Estimation of Population Parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Nabout, J.C.; Walters, K.E.; Martiny, J.B.H. Alpha-, beta-, and gamma-diversity of bacteria varies across habitats. PLoS ONE 2020, 15, e0233872. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, J.; Liang, H.; Ye, L.; Lan, L.; Lu, F.; Wang, Q.; Lei, T.; Yang, X.; Cui, P.; et al. Differences in Alpha Diversity of Gut Microbiota in Neurological Diseases. Front. Neurosci. 2022, 16, 879318. [Google Scholar] [CrossRef] [PubMed]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- Zeeshan Ul Haq, M.; Yu, J.; Yao, G.; Yang, H.; Iqbal, H.A.; Tahir, H.; Cui, H.; Liu, Y.; Wu, Y. A Systematic Review on the Continuous Cropping Obstacles and Control Strategies in Medicinal Plants. Int. J. Mol. Sci. 2023, 24, 12470. [Google Scholar] [CrossRef] [PubMed]
- van Es, H.M.; Karlen, D.L. Reanalysis Validates Soil Health Indicator Sensitivity and Correlation with Long-term Crop Yields. Soil Sci. Soc. Am. J. 2019, 83, 721–732. [Google Scholar] [CrossRef]
- Drew, G.C.; Stevens, E.J.; King, K.C. Microbial evolution and transitions along the parasite–mutualist continuum. Nat. Rev. Microbiol. 2021, 19, 623–638. [Google Scholar] [CrossRef]
- Olanrewaju, O.S.; Babalola, O.O. The rhizosphere microbial complex in plant health: A review of interaction dynamics. J. Integr. Agric. 2022, 21, 2168–2182. [Google Scholar] [CrossRef]
- Lei, H.; Liu, A.; Hou, Q.; Zhao, Q.; Guo, J.; Wang, Z. Diversity patterns of soil microbial communities in the Sophora flavescens rhizosphere in response to continuous monocropping. BMC Microbiol. 2020, 20, 272. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, R.; Luu, V.T.; Weinhold, A.; Goldberg, J.; Oh, Y.; Baldwin, I.T. Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. Proc. Natl. Acad. Sci. USA 2015, 112, E5013–E5020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, X.; Liu, T.; Wang, H.; Yang, Y.; Chen, X.; Zhu, S. Analysis of bacterial and fungal communities in continuous-cropping ramie (Boehmeria nivea L. Gaud) fields in different areas in China. Sci. Rep. 2020, 10, 3264. [Google Scholar] [CrossRef] [PubMed]
- Buijs, Y.; Bech, P.K.; Vazquez-Albacete, D.; Bentzon-Tilia, M.; Sonnenschein, E.C.; Gram, L.; Zhang, S.-D. Marine Proteobacteria as a source of natural products: Advances in molecular tools and strategies. Nat. Prod. Rep. 2019, 36, 1333–1350. [Google Scholar] [CrossRef]
- Daims, H.; Lücker, S.; Wagner, M. A New Perspective on Microbes Formerly Known as Nitrite-Oxidizing Bacteria. Trends Microbiol. 2016, 24, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Barrera, V.A.; Martin, M.E.; Aulicino, M.; Martínez, S.; Chiessa, G.; Saparrat, M.C.N.; Gasoni, A.L. Carbon-substrate utilization profiles by Cladorrhinum (Ascomycota). Rev. Argent. Microbiol. 2019, 51, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Challacombe, J.F.; Hesse, C.N.; Bramer, L.M.; McCue, L.A.; Lipton, M.; Purvine, S.; Nicora, C.; Gallegos-Graves, L.V.; Porras-Alfaro, A.; Kuske, C.R. Genomes and secretomes of Ascomycota fungi reveal diverse functions in plant biomass decomposition and pathogenesis. BMC Genom. 2019, 20, 976. [Google Scholar] [CrossRef] [PubMed]
- Bastida, F.; Torres, I.F.; Moreno, J.L.; Baldrian, P.; Ondoño, S.; Ruiz-Navarro, A.; Hernández, T.; Richnow, H.H.; Starke, R.; García, C.; et al. The active microbial diversity drives ecosystem multifunctionality and is physiologically related to carbon availability in Mediterranean semi-arid soils. Mol. Ecol. 2016, 25, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Dannert, C. Biocatalytic portfolio of Basidiomycota. Curr. Opin. Chem. Biol. 2016, 31, 40–49. [Google Scholar] [CrossRef]
- Yan, H.; Ge, C.; Zhou, J.; Li, J. Diversity of soil fungi in the vineyards of Changli region in China. Can. J. Microbiol. 2022, 68, 341–352. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, J.; Yu, Z.; Yao, Q.; Li, Y.; Liang, A.; Zhang, W.; Mi, G.; Jin, J.; Liu, X.; et al. Long-term continuous cropping of soybean is comparable to crop rotation in mediating microbial abundance, diversity and community composition. Soil Tillage Res. 2020, 197, 104503. [Google Scholar] [CrossRef]
- Fernandez-Gnecco, G.; Smalla, K.; Maccario, L.; Sørensen, S.J.; Barbieri, P.; Consolo, V.F.; Covacevich, F.; Babin, D. Microbial community analysis of soils under different soybean cropping regimes in the Argentinean south-eastern Humid Pampas. FEMS Microbiol. Ecol. 2021, 97, fiab007. [Google Scholar] [CrossRef]
- Nevarez, L.; Vasseur, V.; Le Madec, A.; Le Bras, M.A.; Coroller, L.; Leguérinel, I.; Barbier, G. Physiological traits of Penicillium glabrum strain LCP 08.5568, a filamentous fungus isolated from bottled aromatised mineral water. Int. J. Food Microbiol. 2009, 130, 166–171. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, F. Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol. Ecol. 2012, 80, 469–478. [Google Scholar] [CrossRef]
- Xiao, X.; Cheng, Z.; Lv, J.; Xie, J.; Ma, N.; Yu, J. A green garlic (Allium sativum L.) based intercropping system reduces the strain of continuous monocropping in cucumber (Cucumis sativus L.) by adjusting the micro-ecological environment of soil. PeerJ 2019, 7, e7267. [Google Scholar] [CrossRef]
- Li, B.; Zhao, L.; Liu, D.; Zhang, Y.; Wang, W.; Miao, Y.; Han, L. Bacillus subtilis Promotes Cucumber Growth and Quality under Higher Nutrient Solution by Altering the Rhizospheric Microbial Community. Plants 2023, 12, 298. [Google Scholar] [CrossRef]
- Ali, A.; Ghani, M.I.; Haiyan, D.; Iqbal, M.; Cheng, Z.; Cai, Z. Garlic Substrate Induces Cucumber Growth Development and Decreases Fusarium Wilt through Regulation of Soil Microbial Community Structure and Diversity in Replanted Disturbed Soil. Int. J. Mol. Sci. 2020, 21, 6008. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | EC | pH | OM | TN (g/kg) | TP (g/kg) | TK (g/kg) |
|---|---|---|---|---|---|---|
| A0 | 1.75 ± 0.002 c | 6.93 ± 0.06 d | 0.38 ± 0.01 a | 17.3 ± 0.07 a | 10.02 ± 0.32 b | 9.80 ± 0.07 a |
| A1 | 2.64 ± 0.035 a | 7.65 ± 0.02 c | 0.35 ± 0.01 b | 16.23 ± 0.28 b | 11.14 ± 0.43 a | 9.08 ± 0.88 a |
| A2 | 2.23 ± 0.047 b | 8.05 ± 0.03 ab | 0.34 ± 0.003 c | 16.18 ± 0.43 b | 9.73 ± 0.51 bc | 9.48 ± 0.32 a |
| A3 | 1.73 ± 0.057 c | 7.97 ± 0.07 b | 0.31 ± 0.01 d | 15.93 ± 0.04 bc | 9.18 ± 0.61 bc | 10.04 ± 0.77 a |
| A4 | 1.03 ± 0.001 d | 8.12 ± 0.05 a | 0.25 ± 0.01 e | 15.66 ± 0.3 c | 8.88 ± 0.33 c | 9.64 ± 0.25 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Q.; Dong, J.; Yan, Z.; Xu, L.; Liu, A. Effect of Cucumber Continuous Monocropping on Traditional Chinese Medicine Residue through Analysis of Physicochemical Characteristics and Microbial Diversity. Agronomy 2024, 14, 709. https://doi.org/10.3390/agronomy14040709
Zhao Q, Dong J, Yan Z, Xu L, Liu A. Effect of Cucumber Continuous Monocropping on Traditional Chinese Medicine Residue through Analysis of Physicochemical Characteristics and Microbial Diversity. Agronomy. 2024; 14(4):709. https://doi.org/10.3390/agronomy14040709
Chicago/Turabian StyleZhao, Qingsong, Jingjing Dong, Zhiyong Yan, Ling Xu, and Ake Liu. 2024. "Effect of Cucumber Continuous Monocropping on Traditional Chinese Medicine Residue through Analysis of Physicochemical Characteristics and Microbial Diversity" Agronomy 14, no. 4: 709. https://doi.org/10.3390/agronomy14040709