 Yaling Liu
Yaling Liu Xinyu Tao
Xinyu Tao Jinhui Tao
Jinhui Tao- 1Department of Rheumatology and Immunology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
- 2Department of Clinical Medicine “5 + 3” Integration, The First Clinical College, Anhui Medical University, Hefei, China
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by multiple organ dysfunction resulting from the production of multiple autoantibodies and adaptive immune system abnormalities involving T and B lymphocytes. In recent years, inflammasomes have been recognized as an important component of innate immunity and have attracted increasing attention because of their pathogenic role in SLE. In short, inflammasomes regulate the abnormal differentiation of immune cells, modulate pathogenic autoantibodies, and participate in organ damage. However, due to the clinical heterogeneity of SLE, the pathogenic roles of inflammasomes are variable, and thus, the efficacy of inflammasome-targeting therapies is uncertain. To provide a foundation for the development of such therapeutic strategies, in this paper, we review the role of different inflammasomes in the pathogenesis of SLE and their correlation with clinical phenotypes and propose some corresponding treatment strategies.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the production of autoantibodies and multisystem damage. The main pathophysiological factors involve adaptive immune system abnormalities, manifested by an imbalance of T cells, excessive B-cell activation, and the production of pathogenic autoantibodies. At present, SLE treatment mainly involves hormonal and immunosuppressant therapies. The resulting immunosuppression is nonspecific, associated with multiple side effects, and its efficacy is uncertain. A better understanding of SLE pathophysiology would help us develop more targeted drug therapies.
Given that abnormal differentiation of T and B cells is critical to the pathogenesis of SLE, most drug development has targeted these cells, their cytokines, and key signal transduction pathways. However clinical translation in large-scale trials has often failed; the main reason for this seems to be that SLE is a clinically and pathologically heterogeneous disease (1). Therefore, a clearer understanding of SLE pathophysiology and more accurate patient phenotyping should result in a more accurate selection of appropriate targeted therapeutic drugs that achieve better clinical outcomes.
Although SLE involves abnormal adaptive immunity, the normal interactions between the innate and adaptive immune systems (2, 3) have attracted interest in how the former is involved in the pathogenesis of SLE. Inflammasomes are oligomeric complexes that make up a critical component of innate immunity as major driving forces for inducing inflammation via activation of pathogen-recognition receptors (PRRs). Activated inflammasomes promote the production and secretion of inflammatory cytokines such as IL-1 and IL-18 and play an important role in regulating immune response. Because inflammasomes affect the differentiation of T and B cells (4, 5), they also participate in SLE pathogenesis. In fact, it has been reported that inflammasome-dependent cytokines including IL-1 and IL-18 are involved in the pathogenesis of SLE (6). In patients with SLE, free IL-18 is significantly higher than in controls and is correlated with disease activity (7) related to active kidney disease (8); serum IL-18 can be used as a predictive biomarker for the long-term prognosis of renal function in patients with SLE (9).
SLE is characterized by the production of multiple autoantibodies and multiple organ damage. This is dominated by renal damage and vasculitis. Inhibition of caspase-1 or NLRP3-mediated inflammasome signaling in murine models has demonstrated that inflammasome pathways contribute to SLE pathogenesis by promoting autoantibody production, endothelial dysfunction, and nephritis (10–14). Abnormal activation of the inflammasome was shown to not only be involved in SLE pathogenesis but also be an important risk factor in some SLE patients. It has been reported that inflammasome polymorphisms are associated with susceptibility to SLE (15, 16). Therefore, targeted inflammasome therapy appears to offer a particularly attractive target.
Given the heterogeneity of SLE and a desire for targeted inflammasome inhibition, it will be necessary to select appropriate inflammasome inhibitors for specific SLE patients to achieve maximal efficacy. So far, many proteins that belong to the nucleus binding domain leucine-rich repeat (NLR) family such as NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4, and AIM2 (absent in melanoma-2) have been reported to initiate assembling the formation of an inflammasome and in recent years have attracted a lot of attention (17). Each inflammasome is activated in response to different stimuli These inflammasomes play different pathogenic roles. Understanding these roles and their correlation with clinical phenotypes underlies our ability to correctly select matching inhibitors for individual patients. In this review, we will summarize the role and clinical characteristics of the different inflammasomes and put forward a strategy for the targeted inhibition of inflammasomes in the treatment of SLE. We believe that this work will provide an important reference for clinical research and drug development.
Inflammasomes Are Involved in the Pathogenesis of SLE
Many studies have proposed a pathogenic role for inflammasomes in SLE. Upregulation of inflammasome gene expression and activity has been found in both human and murine lupus (14, 18–21). Due to the interaction between inflammasomes and acquired immunity (22), inflammasomes participate in the pathogenesis of SLE mainly by regulating the abnormal differentiation of immune cells, mediating the pathogenicity of a variety of pathogenic autoantibodies, affecting the pathogenicity of IFN-I, and abnormally activating various signaling pathways including those involved in inflammation. As such, inflammasomes have direct and indirect effects on immune function and thus play a critical role in the pathogenesis of SLE.
Inflammasomes Regulate the Abnormal Differentiation of Immune Cells
SLE patients have disordered Th1/Th2 and Treg/Th17 balance, which is related to disease activity (23). Inflammasomes help mediate T-cell differentiation. For example, various animal and human studies have shown that NLRP3, NLRP1, NLRC4, and AIM2 may regulate Th1, Tfh, and Th17 cell-mediated immune responses through different pathways, in different immune disorders (24–28). Although there has been no direct evidence showing that inflammasomes mediate T-cell differentiation in SLE, it has been shown that leptin promotes Th17 cell differentiation in lupus erythematosus mice by activating the NLRP3 inflammasome and thus participating in the pathogenesis of SLE (29).
In addition to disordered T-cell balance, abnormal B-cell activation is also critically important in the pathogenesis of SLE. Studies have shown that B cells constitutively express the NLRP3 inflammasome in the cytoplasm, which is activated by typical pathogen-associated molecular products (PAMPs) (30). The expression and activation of NLRP3 in B cells help to maintain B-cell homeostasis and humoral immune responses and are independent of IL-1β participation (31). Given the role of NLRP3 inflammasome in maintaining B-cell homeostasis, it has been suggested that the regulation of B-cell inflammasomes is also an important mechanism in SLE. In addition to NLRP3, it has been found that AIM2 is highly expressed in B cells from SLE patients, promoting B-cell differentiation by regulating the Bcl-6–Blimp-1 axis; this provides a novel target for SLE treatment (32).
Because the primary focus has long been set on the adaptive immune system, the role of neutrophils in SLE-related inflammation has been neglected. However, in recent years, it was discovered that neutrophil extracellular traps (NETs) are critical in driving autoimmune responses (33) and that they are involved in the pathogenesis of SLE (34, 35). In fact, a vicious SLE pathogenic cycle appears to be at play: NETs activate the NLRP3 inflammasome (20) while inflammasomes induce the formation of NETs (36, 37).
Inflammasomes Mediate the Pathogenicity of Autoantibodies
SLE is characterized by the production of numerous autoantibodies. An interaction between autoantibodies and inflammasomes is involved in the pathogenesis of SLE. As mentioned earlier, inflammasomes are involved in the production of interferon-induced autoantibodies (10), and in turn, autoantibodies produce pathogenicity through inflammasomes.
Production of anti-dsDNA antibodies, diagnosed via the antinuclear (ANA) test, is characteristic of SLE and is related to disease activity. In addition to forming immune complexes with dsDNA to produce pathogenicity, anti-dsDNA antibodies also activate the NLRP3 inflammasome and induce human monocytes to secrete IL-1β, triggering a downstream immune cascade and thus participating in the pathogenesis of SLE (18, 19). Anti-dsDNA also promotes lupus nephritis (LN) through the PKCδ–NLRC4 axis (38).
In addition to anti-dsDNA, U1-snRNP antibodies are also important in SLE. It has been found that this antibody activates the NLRP3 inflammasome in monocytes, thereby contributing to the pathogenesis of SLE (39, 40).
Inflammasomes Affect the Pathogenicity of the IFN-I Pathway
It is recognized that IFN-I plays an important role in SLE (41). Genetic variations of the IFN-I signaling pathway have been shown to increase the risk of SLE in humans and mouse models (42–44).
IFN-I regulates immune responses by activating inflammasomes. For example, in influenza A virus-infected cells, IFN-I signals activate the NLRP3 inflammasome through the TLR3 pathway (45). During streptococcus pneumonia infection, the IFN-I signaling pathway regulates the activation of the AIM2 inflammasome (46). Thus, in SLE induced by IFN-I, inflammasome activation may be an important component of its pathogenicity. Caspase-1 knockout (Casp1−/−) mice demonstrate strong protection against prion-induced autoantibody production and IFN-I responses, indicating that caspase-1 is likely to be necessary for IFN-I-induced lupus (10). IFN regulatory factor 1 induced by IFN-I was shown to upregulate inflammasome activity and participate in the pathogenesis of SLE (47).
How IFN-I regulates the inflammasome is incompletely understood and is dependent on the signaling pathway. For example, IFN-I activates the NLRP3 inflammasome through the RIG-I/TLR3 pathway (45), while the STAT1 transcription factor inhibits the activity of the NLRP1 and NLRP3 inflammasomes (48). In addition, the inflammasome itself also affects the expression of IFN-I, mainly playing a negative regulatory role. Studies have found that mice lacking inflammasome sensor AIM2, NLRP3, or adapter caspase-1 produce high levels of IFN-I cytokines, and the activated inflammasome negatively regulates IFN-I signal transduction through MyD88-IRF7 (49).
Therefore, the interaction between the inflammasome and IFN-I is complex, and unexpected results often appear in clinical studies. Clinical studies have shown that expression of the NLRP3/NLRP1 inflammasomes was significantly downregulated in PBMCs obtained from patients with SLE compared with healthy controls; further analysis showed that IFN-I levels were significantly inversely correlated with the expression of NLRP3/NLRP1 inflammasomes, suggesting a negative regulatory effect between IFN-I and the inflammasome in SLE (50).
Inflammasomes Interact With Various Signaling Pathways
The pathogenesis of SLE is extremely complex and involves many signaling pathways such as NF-KB (51–54), JAK/STAT (55–57), mTOR (58, 59), PI3K/AKT (55), RhoA-ROCK (60), and NETs (34, 35, 61). Direct or indirect evidence has shown that different inflammasomes and various pathways play important roles [(20, 38, 55, 56, 58, 60–99) Supplementary Table]. Although most of these findings have not been verified in SLE and the specific mechanisms underlying the interactions have not been made clear, we need to recognize the potential importance of their role. This also highlights the great complexity inherent in the involvement of the inflammasome in the pathogenesis of SLE. For example, NLRP3 (62, 63), NLRP1 (64), and AIM2 (65) inflammasomes are upregulated by NF-KB signaling, while the NF-KB pathway is upregulated by the NLRP3 and NLRP1 inflammasomes (66, 67); these interactions again show the complexity of these mechanisms.
Different Roles of Inflammasomes in the Pathogenesis of SLE
As stated, inflammasomes promote the pathogenesis of SLE through different mechanisms, among which NLRP3 has the most direct evidence. It has been shown that NLRP3 mediates the pathogenicity of autoantibodies and IFN-I, interacts with NF-KB and other pathways to regulate the differentiation of T and B lymphocytes, and participates in the pathogenesis of SLE (Figure 1). However, in its interaction with IFN-I, IFN-I exerts different effects on NLRP3 through different signaling pathways (45, 48), and NLRP3 inhibits IFN-I signal transduction (49). Therefore, it is not certain what role NLRP3 plays in SLE patients with highly abnormal IFN-I activation.
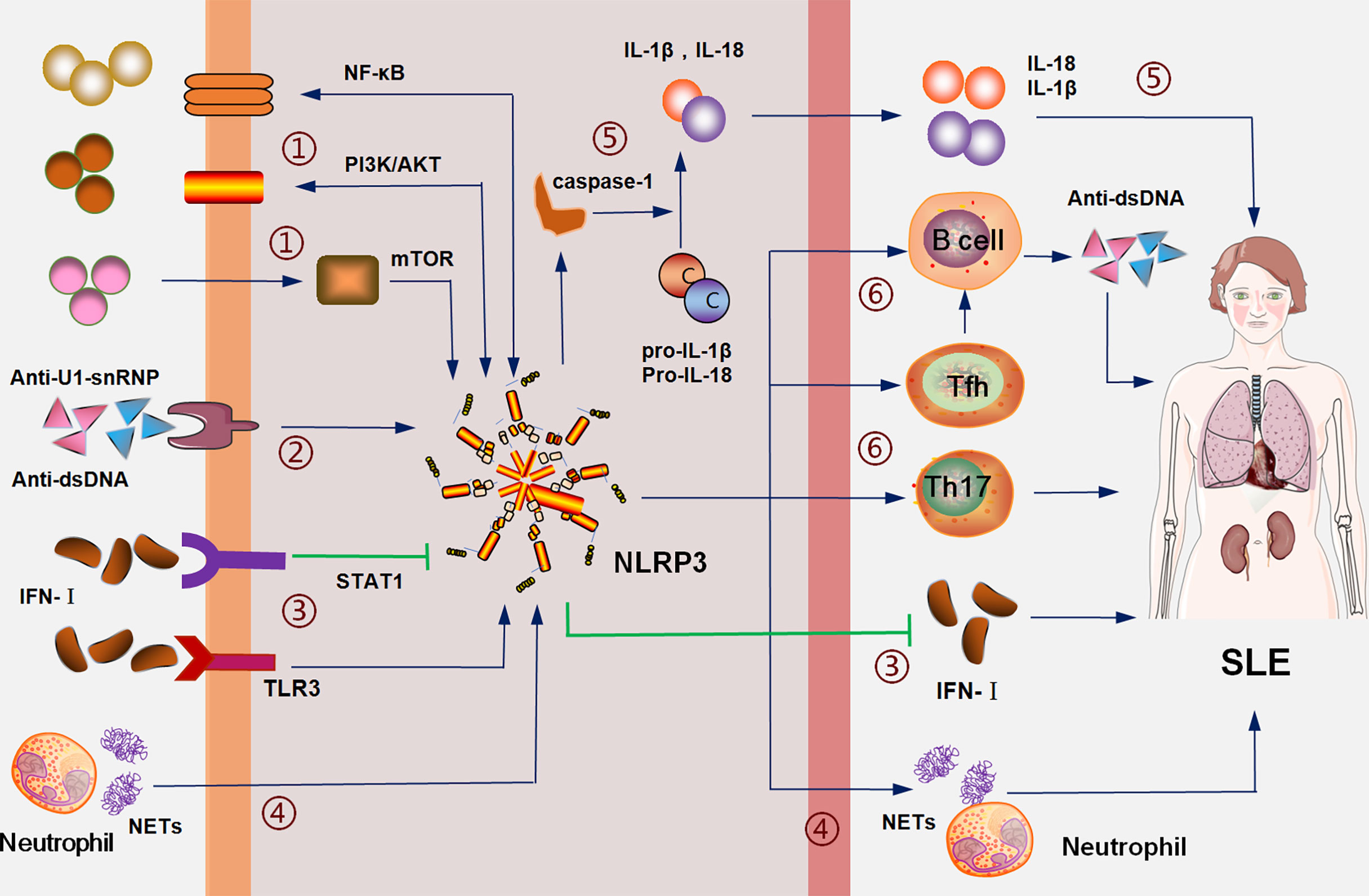
Figure 1 The NLRP3 inflammasome is involved in the pathogenesis of SLE: (1) interacts with SLE pathogenic signals, such as NF-KB, PI3K/AKT and mTOR; (2) mediates the inflammation of pathogenic antibodies including anti-dsDNA; (3) IFN-I activates NLRP3 inflammasome through the TLR3 pathway and inhibits NLRP3 inflammasome through the STAT1 pathway; NLRP3 inhibits the expression of IFN-I; (4) neutrophil NETs activate the NLRP3-associated inflammation, which, in turn, induces the formation of NETs; (5) the NLRP3 inflammasome promotes the maturation and secretion of IL-1β and IL-18 through caspase-1; (6) the NLRP3 inflammasome promotes the proliferation and activation of Th17, Tfh, and B cells to mediate the disorder of cellular immune balance and promote the production of antibodies.
In addition, AIM2 has a unique dual role in the pathogenesis of SLE. In terms of the IFN pathway, AIM2 inhibits DNA-induced IFN signals and plays a protective role in SLE. For example, in LN, IFN-I induced AIM2 negatively regulates the IFN-I response (100), suggesting that AIM2 may play a protective role in IFN-induced SLE. However, this effect is affected by p202. Studies have shown that increased expression of p202 inhibits AIM2, thus mediating the pathogenicity of IFN in SLE (101); murine studies have also confirmed that p202 inhibits the activation of the AIM2 inflammasome (102, 103).
The expression of AIM2 and p202 proteins differ by cell type and sex (104, 105); AIM2 is mainly expressed in male patients and innate immune cells, and the p202 protein is mainly expressed in female patients and adaptive immune cells. Therefore, in terms of the impact on IFN signals, AIM2 has stronger pathogenicity in male SLE patients and may be protective in female patients.
Unlike its effect on the IFN signaling pathway, AIM2 promotes SLE through other signaling pathways. AIM2 mediates DNA-induced macrophage functional maturation and SLE pathogenesis (106). AIM2 promotes the Th17 cell contributions to SLE (28) and inhibition of AIM2 expression significantly improves the SLE syndrome in mice (106).
In general, AIM2 promotes SLE. Bioinformatics analyses have found significant differences in the gene expression of the AIM2 inflammasome complex in SLE (107). AIM2 mRNA levels were found to be upregulated in the liver, PBMCs, and the spleen of SLE patients compared with healthy individuals (108). Using the AIM2 inhibitor class C1 decoy oligodeoxynucleotides (ODNs) in lupus-prone (NZB x NZW) F1 mice resulted in a delayed onset of glomerulonephritis and prolonged survival (109).
In addition to NLRP3 and AIM2, the NLRC4 and NLRP1 inflammasomes are also involved in the pathogenesis of SLE. For example, genetic evidence shows that NLRP1 gene polymorphisms are associated with SLE (15, 110) and NLRC4-mediated LN caused by anti-dsDNA antibodies (38). However, the few available relevant studies are unable to clearly explain their exact roles in SLE.
The Role of Inflammasomes in SLE Organ Damage
SLE is a heterogeneous autoimmune disease characterized by multiple organ damage. Various inflammasomes are involved in the pathogenesis of SLE through a variety of mechanisms, resulting in different pathophysiologies and clinical manifestations of SLE (Figure 2).
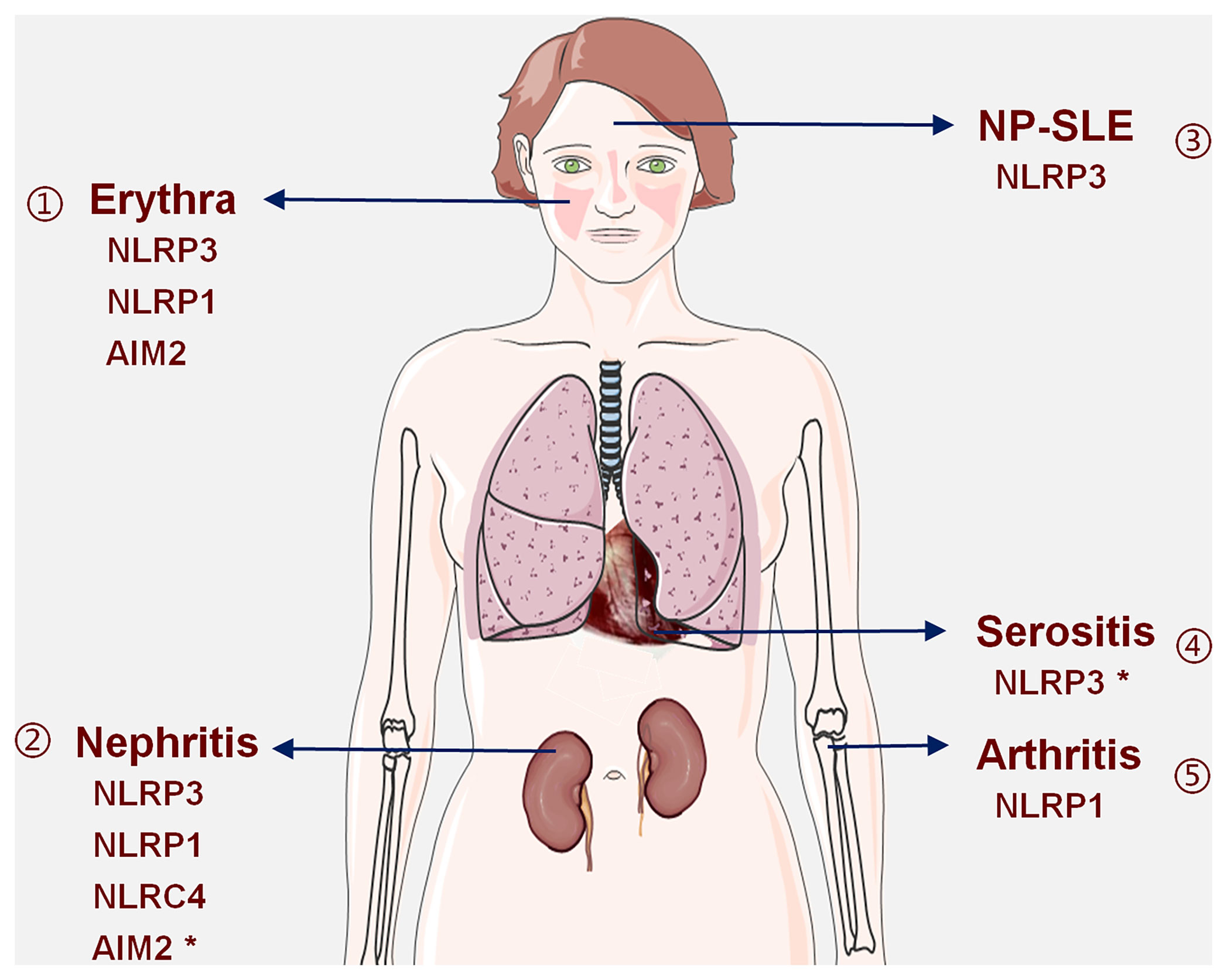
Figure 2 Inflammasomes are involved in SLE-related organ damage: available evidence shows that (1) NLRP3, NLRP1, and AIM2 inflammasomes are involved in erythrina; (2) NLRP3, NLRP1, NLRC4, and AIM2 inflammasomes are involved in LN, and the role of AIM2 inflammasome is complex and may have a protective effect; (3) the NLRP3 inflammasome is involved in the pathogenesis of NP-SLE; (4) the NLRP3 inflammasome may play a promotive or inhibitory role in serositis; (5) the NLRP1 inflammasome is involved in arthritis. *Indicates that this inflammasome has a dual role.
Kidney Damage and Inflammasomes
Kidney damage is the most common organ damage in SLE. As such, it is used as an indication of a successful disease model. Studies have shown that various inflammasomes are involved in this renal damage. For instance, animal experiments have confirmed that activation of the NLRC4 and NLRP3 inflammasomes promotes kidney damage (38, 111–114). Human experiments have also shown that activated NLRP3, NLRP1, and AIM2 inflammasomes are associated with the progression of LN, and that NLRP3 activation has a positive correlation with the activity index (AI) score in LN (115, 116). Moreover, gene expression studies have confirmed that NLRP3 and NLRP1 gene polymorphisms are associated with the development of LN (16, 110, 117) and that the acquired functional variant rs10754558 (NLRP3) is more common in patients with LN, strengthening the view that the NLRP3 inflammasome plays a key role not only in lupus but also in renal damage (16).
Among the many inflammasomes involved in the pathogenesis of LN, the role of AIM2 is particularly uncertain. Human and animal studies have suggested that the AIM2 inflammasome promotes the progression of LN (106). However, from the perspective of the influence of AIM2 on IFN-I signals, AIM2 may play a protective role in the pathogenesis of LN (100). As such, at this stage, it is difficult to explain the role of AIM2 in the pathogenesis of LN.
Erythema and Inflammasomes
SLE has a strong genetic background; two genetic association studies revealed that variations in IL-1β and NLRP1 are associated with SLE-related erythema in Brazilian cohorts (110, 118). IL-1β is a potent player in cutaneous inflammation and is central in the development of a Th17 micro-milieu in SLE. Studies have also confirmed that the Th17 micro-environment also regulates NLRP1-dependent caspase-5 activity in skin inflammation, suggesting that NLRP1 has an important relationship with skin inflammation (25).
Serositis and Inflammasomes
Currently, there is inadequate direct evidence to support that inflammasomes cause serositis in SLE. However, previous studies had shown that the 489c > T polymorphism of the P2RX7 gene is associated with the activation of the NLRP3 inflammasome and an increased release of the pro-inflammatory cytokines IL-1β and IL-18; this indirectly reflects that the NLRP3 inflammasome may be involved in the pathogenesis of SLE complicated with pericarditis (119). However, there have also been conflicting findings; the expression and function of P2X7R were shown to be decreased in SLE patients with lupus serositis (120). Therefore, it is uncertain whether serositis is related to the activation of the NLRP3 inflammasome.
Arthritis and Inflammasomes
Although the role of the inflammasome in the pathogenesis of arthritis has attracted increasing attention (121), their effect on SLE arthritis is not known. At present, it has only been confirmed that NLRP1 gene polymorphisms are associated with SLE arthritis at the genetic level (110).
Nervous System Damage and Inflammasomes
Neuropsychiatric symptoms of SLE (NP-SLE) are a common manifestation and include psychosis, epileptic seizures, and cognitive dysfunction in severe cases (122, 123). Gene-level studies have shown that NLRP3 gene variations are associated with susceptibility and neurological symptoms of SLE (117). Inhibition of the NLRP3 inflammasome by procyanidin B2 has been shown to be one of the neuropsychiatric symptoms found in the systemic autoimmune MRL-LPR mouse (124), suggesting that the damage to the nervous system may also be related to NLRP3 inflammasome.
Strategies for Selecting Inflammasome Inhibitors in SLE
Activated inflammasomes promote the production of IL-1 and IL-18, and other inflammatory cytokines, thereby playing an important role in regulating inflammation in SLE. In addition, inflammasomes also interact with many signal pathways related to the pathogenesis of SLE, regulate the differentiation of T, B, and other immune cells, and participate in the pathogenesis of SLE. Therefore, therapeutic inhibition of inflammasomes is a promising direction for precision therapy of SLE. However, there are no clinical, preclinical, or animal studies testing the treatment of SLE with inflammasome inhibitors. Due to the previously outlined disease heterogeneity, the specific inflammasome inhibitors will need to match the underlying pathophysiology. In clinical situations, we will need to comprehensively test which inflammasomes are involved and select the corresponding inhibitors to effect accurate treatment (Figure 3). Herein, we propose such a therapeutic approach utilizing the targeted inhibition of inflammasomes.
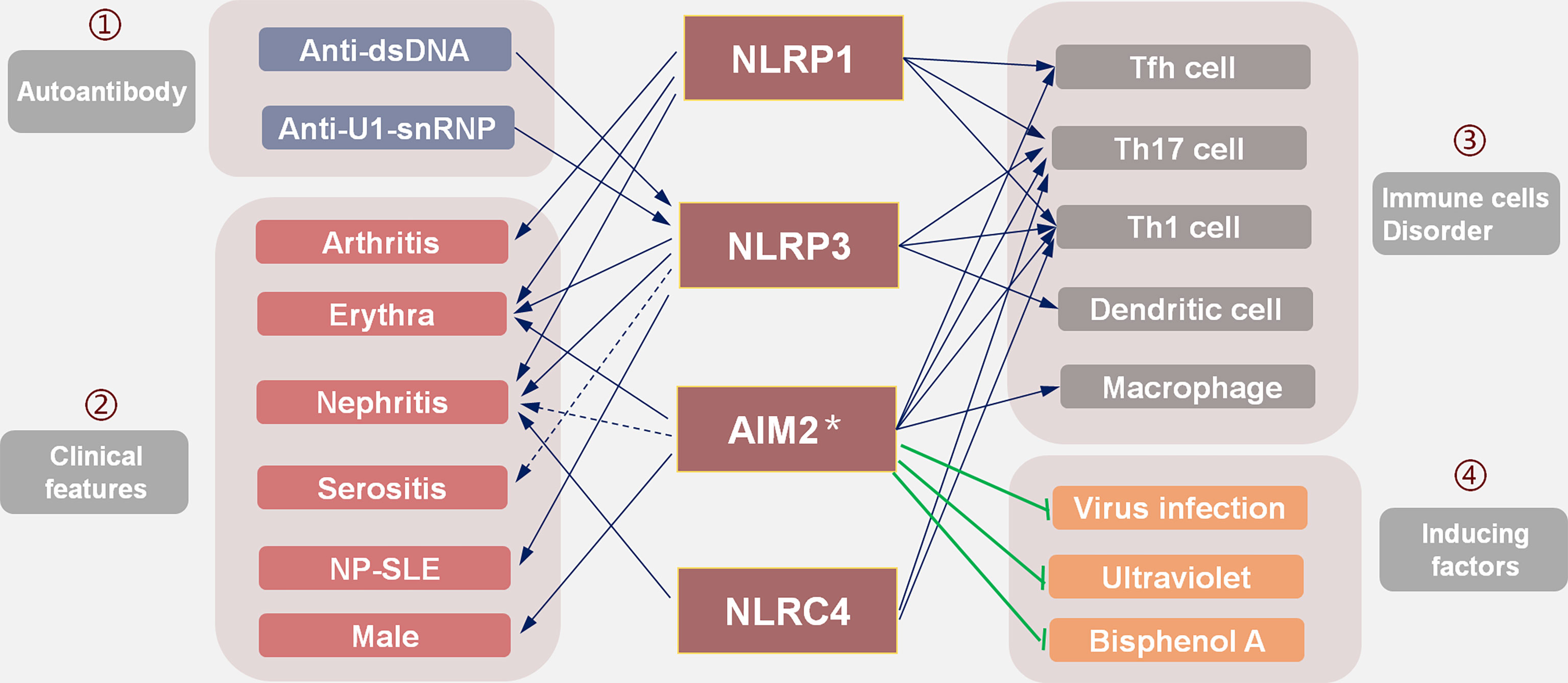
Figure 3 The selection strategies of targeted inflammasome inhibitors in SLE: existing evidence suggests that (1) inflammasomes mediate the pathogenicity of autoantibodies; (2) different organ damage is related to the activation of corresponding inflammasomes; (3) inflammasomes affect the differentiation and proliferation of immune cells and participate in SLE; (4) induced factors such as viral infection, ultraviolet rays, and BPA all play a role through IFN-I, while the AIM2 inflammasome may play a protective role. In the selection of inflammasome inhibitors, many factors should be considered to screen out the inflammasome that plays the main pathogenic role.
Selection of Inflammasome Inhibitors Based on Clinical Characteristics
Different inflammasomes appear to be associated with different organ damage in SLE (Figure 2). The NLRP3 inflammasome is involved in erythema, nephritis, NP-SLE, and other organ damage, so inhibition of the NLRP3 inflammasome may have surprising effects. Since specific inflammasomes are related to organ damage, the appropriate inflammasome inhibitor might be selected according to the clinical picture based on organ involvement in the specific patient. For example, an NLRP1 inflammasome inhibitor may be effective in patients with LN complicated by arthritis or erythema; an NLRP3 inflammasome inhibitor may be more effective in LN patients with NP-SLE.
Laboratory tests are helpful in the selection of inflammasome inhibitors. During the onset of SLE, anti-dsDNA antibodies activate the NLRP3 (18, 19) and the NLRC4 inflammasomes (38), while anti-U1 snRNP antibodies activate the NLRP3 inflammasome (39, 40). Therefore, NLRP3 inflammasome inhibitors should be selected when anti-dsDNA and anti-U1 snRNP antibodies are detected in patients.
Although the AIM2 inflammasome is involved in the pathogenesis of SLE, it seems to be harmful only to male patients (104, 105). The AIM2 inflammasome also has a protective effect on the pathogenicity caused by IFN-I (100). Therefore, AIM2 inflammasome inhibitors are likely more suitable for male or non-IFN-I based SLE patients.
Selection of Inflammasome Inhibitors Based on the Immune Cell Disorder
The strategy of selecting inflammasome inhibitors based on a patient’s clinical presentation is relatively convenient and straightforward in the average clinical setting. However, due to the complexity of SLE, different mechanisms may underlie the same clinical manifestation; SLE involves various immune cell disorders that can be clinically similar. Disordered immune cell function in SLE patients is regulated by the inflammasome. Therefore, the selection of inflammasome inhibitors should be based on the specific immune disorder.
NLRP3, NLRP1, NLRC4, and AIM2 inflammasome inhibitors should be considered in SLE patients who show disordered Th1/Tfh or Th17/Treg profiles; the NLRP3 inhibitors may be helpful for patients with dendritic cell disorders (22). Because the expression of the AIM2 inflammasome is closely related to macrophage activation (106), this inflammasome can promote Th17 cell differentiation (28). Therefore, AIM2 inflammasome inhibitors may be useful in patients with these cell disorders.
Selection of Inflammasome Inhibitors Based on Inducing Factors
We lack effective methods to comprehensively profile immune abnormalities in clinical settings. However, with an emerging understanding of SLE pathogenesis, the immune abnormalities caused by different inducing factors are becoming clearer. Therefore, understanding the main mechanism involved in the pathogenesis of SLE based on disease-inducing events can also help in the selection of inflammasome inhibitors. For example, virus infection is a common cause of recurrence or aggravation and involves increased IFN levels. It is recommended not to use AIM2 inflammasome inhibitors if there is a clear viral infection, as the AIM2 inflammasome has a protective effect on IFN-I (100). Ultraviolet radiation is a common factor that triggers SLE. Studies have shown that ultraviolet radiation caused the onset of SLE through abnormal activation of IFN-I signaling pathways (125, 126). Likewise, it is recommended not to treat such cases with AIM2 inflammasome inhibitors. In mice and human bone marrow cells, it has been shown that bisphenol A (BPA)-induced signals stimulate IFN-I signaling and further promote the pathogenesis of SLE (127). Therefore, SLE patients exposed to BPA should not use AIM2 inflammasome inhibitors.
Future Prospects
The inflammasome is involved in the immune mechanism of SLE, but it does not cause SLE. Considering that the inflammasome is positively correlated with disease activity, especially with inflammation related to IL-1 and IL-18, the inflammasome plays an important role in the adaptive immune imbalance. Therefore, inflammasome antagonists could play an important role in controlling disease activity and preventing recurrence.
Due to the paucity of relevant research and the fact that most studies involve animal models, we are yet to fully understand the role and clinical characteristics of inflammasomes in SLE. At present, studies have mainly focused on the NLRP3 inflammasome but this does not dismiss the potential effectiveness of other inflammasome antagonists in the treatment of SLE. The evidence behind inflammasome inhibitor selection is currently rudimentary but intended to serve as a conceptual foundation to stimulate further research in this field. Considering the complexity of SLE pathogenesis, further clinical trials are needed. We also need to continue pre-clinical mechanism-based research in parallel, which will ultimately allow us to identify biomarkers of precision treatments in the clinical–basic–clinical research cycle. In addition, because inflammasomes are activated in many pathways and interact with a variety of pathogenic signals in SLE, basic research is more conducive to obtaining accurate and reliable data for the selection of appropriate animal models (128, 129) and adopting different blocking strategies (128) according to the activation mechanism of the inflammasome.
The selection strategy of targeted inflammasome inhibitors provided in this paper is not perfect, but it provides a clear process for current clinical applications and research. Given the heterogeneity of SLE, most of the targeted therapy drugs do not perform well in clinical trials (1). A selection strategy of inflammasome inhibitors based on specific mechanisms will divide patients into clinical and pathophysiological subtypes, ensuring several uniform pathogenesis groupings that should improve the success rate of clinical trials and increase the efficacy of patient treatments.
Author Contributions
YL and XT wrote the manuscript. JT contributed to providing the general idea and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81771774) and the Anhui Provincial Key Research and Development Plan (201904a07020103).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to express their gratitude to Edit Springs (https://www.editsprings.cn) for the expert linguistic services provided.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.894847/full#supplementary-material
References
1. Davis LS, Reimold AM. Research and Therapeutics-Traditional and Emerging Therapies in Systemic Lupus Erythematosus. Rheumatol (Oxford England) (2017) 56(suppl_1):i100–i13. doi: 10.1093/rheumatology/kew417
2. Varga G, Foell D. Anti-Inflammatory Monocytes-Interplay of Innate and Adaptive Immunity. Mol Cell Pediatr (2018) 5(1):5. doi: 10.1186/s40348-018-0083-4
3. Sun L, Wang X, Saredy J, Yuan Z, Yang X, Wang H. Innate-Adaptive Immunity Interplay and Redox Regulation in Immune Response. Redox Biol (2020) 37:101759. doi: 10.1016/j.redox.2020.101759
4. Evavold CL, Kagan JC. How Inflammasomes Inform Adaptive Immunity. J Mol Biol (2018) 430(2):217–37. doi: 10.1016/j.jmb.2017.09.019
5. Camell CD, Günther P, Lee A, Goldberg EL, Spadaro O, Youm YH, et al. Aging Induces an Nlrp3 Inflammasome-Dependent Expansion of Adipose B Cells That Impairs Metabolic Homeostasis. Cell Metab (2019) 30(6):1024–39.e6. doi: 10.1016/j.cmet.2019.10.006
6. Voronov E, Dayan M, Zinger H, Gayvoronsky L, Lin JP, Iwakura Y, et al. Il-1 Beta-Deficient Mice Are Resistant to Induction of Experimental Sle. Eur Cytokine Netw (2006) 17(2):109–16.
7. Migliorini P, Anzilotti C, Pratesi F, Quattroni P, Bargagna M, Dinarello CA, et al. Serum and Urinary Levels of Il-18 and Its Inhibitor Il-18bp in Systemic Lupus Erythematosus. Eur Cytokine Netw (2010) 21(4):264–71. doi: 10.1684/ecn.2010.0210
8. Mende R, Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Lin E, Chang J, et al. Analysis of Serum Interleukin (Il)-1β and Il-18 in Systemic Lupus Erythematosus. Front Immunol (2018) 9:1250. doi: 10.3389/fimmu.2018.01250
9. Wu CY, Yang HY, Yao TC, Liu SH, Huang JL. Serum Il-18 as Biomarker in Predicting Long-Term Renal Outcome Among Pediatric-Onset Systemic Lupus Erythematosus Patients. Med (Baltimore) (2016) 95(40):e5037. doi: 10.1097/md.0000000000005037
10. Kahlenberg JM, Yalavarthi S, Zhao W, Hodgin JB, Reed TJ, Tsuji NM, et al. An Essential Role of Caspase 1 in the Induction of Murine Lupus and Its Associated Vascular Damage. Arthritis Rheumatol (Hoboken NJ) (2014) 66(1):152–62. doi: 10.1002/art.38225
11. Zhao J, Wang H, Dai C, Wang H, Zhang H, Huang Y, et al. P2x7 Blockade Attenuates Murine Lupus Nephritis by Inhibiting Activation of the Nlrp3/Asc/Caspase 1 Pathway. Arthritis Rheum (2013) 65(12):3176–85. doi: 10.1002/art.38174
12. Li M, Shi X, Qian T, Li J, Tian Z, Ni B, et al. A20 Overexpression Alleviates Pristine-Induced Lupus Nephritis by Inhibiting the Nf-Kb and Nlrp3 Inflammasome Activation in Macrophages of Mice. Int J Clin Exp Med (2015) 8(10):17430–40.
13. Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 Attenuates Murine Lupus Nephritis Via Inhibiting Nlrp3 Inflammasome and Nf-Kb Activation. Int Immunopharmacol (2013) 17(1):116–22. doi: 10.1016/j.intimp.2013.05.027
14. Kahlenberg JM, Thacker SG, Berthier CC, Cohen CD, Kretzler M, Kaplan MJ. Inflammasome Activation of Il-18 Results in Endothelial Progenitor Cell Dysfunction in Systemic Lupus Erythematosus. J Immunol (Baltimore Md 1950) (2011) 187(11):6143–56. doi: 10.4049/jimmunol.1101284
15. Pontillo A, Reis EC, Liphaus BL, Silva CA, Carneiro-Sampaio M. Inflammasome Polymorphisms in Juvenile Systemic Lupus Erythematosus. Autoimmunity (2015) 48(7):434–7. doi: 10.3109/08916934.2015.1064399
16. da Cruz HLA, Cavalcanti CAJ, de Azêvedo Silva J, de Lima CAD, Fragoso TS, Barbosa AD, et al. Differential Expression of the Inflammasome Complex Genes in Systemic Lupus Erythematosus. Immunogenetics (2020) 72(4):217–24. doi: 10.1007/s00251-020-01158-6
17. Alcocer-Gómez E, Castejón-Vega B, Cordero MD. Stress-Induced Nlrp3 Inflammasome in Human Diseases. Adv Protein Chem Struct Biol (2017) 108:127–62. doi: 10.1016/bs.apcsb.2017.02.002
18. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti-Dsdna Antibodies Bind to Tlr4 and Activate Nlrp3 Inflammasome in Lupus Monocytes/Macrophages. J Transl Med (2016) 14(1):156. doi: 10.1186/s12967-016-0911-z
19. Shin MS, Kang Y, Lee N, Wahl ER, Kim SH, Kang KS, et al. Self Double-Stranded (Ds)DNA Induces Il-1β Production From Human Monocytes by Activating Nlrp3 Inflammasome in the Presence of Anti-Dsdna Antibodies. J Immunol (Baltimore Md 1950) (2013) 190(4):1407–15. doi: 10.4049/jimmunol.1201195
20. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil Extracellular Trap-Associated Protein Activation of the Nlrp3 Inflammasome Is Enhanced in Lupus Macrophages. J Immunol (Baltimore Md 1950) (2013) 190(3):1217–26. doi: 10.4049/jimmunol.1202388
21. Clark KL, Reed TJ, Wolf SJ, Lowe L, Hodgin JB, Kahlenberg JM. Epidermal Injury Promotes Nephritis Flare in Lupus-Prone Mice. J Autoimmun (2015) 65:38–48. doi: 10.1016/j.jaut.2015.08.005
22. Westerterp M, Gautier EL, Ganda A, Molusky MM, Wang W, Fotakis P, et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab (2017) 25(6):1294–304.e6. doi: 10.1016/j.cmet.2017.04.005
23. Muhammad Yusoff F, Wong KK, Mohd Redzwan N. Th1, Th2, and Th17 Cytokines in Systemic Lupus Erythematosus. Autoimmunity (2020) 53(1):8–20. doi: 10.1080/08916934.2019.1693545
24. Wang H, Wei G, Cheng S, Wang D, Ma J, Xin S. Circulatory Cd4-Positive T-Lymphocyte Imbalance Mediated by Homocysteine-Induced Aim2 and Nlrp1 Inflammasome Upregulation and Activation Is Associated With Human Abdominal Aortic Aneurysm. J Vasc Res (2020) 57(5):276–90. doi: 10.1159/000508077
25. Zwicker S, Hattinger E, Bureik D, Batycka-Baran A, Schmidt A, Gerber PA, et al. Th17 Micro-Milieu Regulates Nlrp1-Dependent Caspase-5 Activity in Skin Autoinflammation. PLoS One (2017) 12(4):e0175153. doi: 10.1371/journal.pone.0175153
26. Noroozi S, Meimand HAE, Arababadi MK, Nakhaee N, Asadikaram G. The Effects of Ifn-B 1a on the Expression of Inflammasomes and Apoptosis-Associated Speck-Like Proteins in Multiple Sclerosis Patients. Mol Neurobiol (2017) 54(4):3031–7. doi: 10.1007/s12035-016-9864-8
27. Ip WK, Medzhitov R. Macrophages Monitor Tissue Osmolarity and Induce Inflammatory Response Through Nlrp3 and Nlrc4 Inflammasome Activation. Nat Commun (2015) 6:6931. doi: 10.1038/ncomms7931
28. Ji J, Xu J, Zhao S, Liu F, Qi J, Song Y, et al. Myeloid-Derived Suppressor Cells Contribute to Systemic Lupus Erythaematosus by Regulating Differentiation of Th17 Cells and Tregs. Clin Sci (Lond) (2016) 130(16):1453–67. doi: 10.1042/cs20160311
29. Yu Y, Fu S, Zhang X, Wang L, Zhao L, Wan W, et al. Leptin Facilitates the Differentiation of Th17 Cells From Mrl/Mp-Fas Lpr Lupus Mice by Activating Nlrp3 Inflammasome. Innate Immun (2020) 26(4):294–300. doi: 10.1177/1753425919886643
30. Leal VNC, Reis EC, Fernandes FP, Soares J, Oliveira IGC, Souza de Lima D, et al. Common Pathogen-Associated Molecular Patterns Induce the Hyper-Activation of Nlrp3 Inflammasome in Circulating B Lymphocytes of Hiv-Infected Individuals. AIDS (London England) (2021) 35(6):899–910. doi: 10.1097/qad.0000000000002833
31. Ali MF, Dasari H, Van Keulen VP, Carmona EM. Canonical Stimulation of the Nlrp3 Inflammasome by Fungal Antigens Links Innate and Adaptive B-Lymphocyte Responses by Modulating Il-1β and Igm Production. Front Immunol (2017) 8:1504. doi: 10.3389/fimmu.2017.01504
32. Yang M, Long D, Hu L, Zhao Z, Li Q, Guo Y, et al. Aim2 Deficiency in B Cells Ameliorates Systemic Lupus Erythematosus by Regulating Blimp-1-Bcl-6 Axis-Mediated B-Cell Differentiation. Signal Transduct Target Ther (2021) 6(1):341. doi: 10.1038/s41392-021-00725-x
33. Fousert E, Toes R, Desai J. Neutrophil Extracellular Traps (Nets) Take the Central Stage in Driving Autoimmune Responses. Cells (2020) 9(4):915. doi: 10.3390/cells9040915
34. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J Immunol (Baltimore Md 1950) (2011) 187(1):538–52. doi: 10.4049/jimmunol.1100450
35. Frangou E, Chrysanthopoulou A, Mitsios A, Kambas K, Arelaki S, Angelidou I, et al. Redd1/Autophagy Pathway Promotes Thromboinflammation and Fibrosis in Human Systemic Lupus Erythematosus (Sle) Through Nets Decorated With Tissue Factor (Tf) and Interleukin-17a (Il-17a). Ann Rheum Dis (2019) 78(2):238–48. doi: 10.1136/annrheumdis-2018-213181
36. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6676. doi: 10.1126/sciimmunol.aar6676
37. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D Plays a Vital Role in the Generation of Neutrophil Extracellular Traps. Sci Immunol (2018) 3(26):eaar6689. doi: 10.1126/sciimmunol.aar6689
38. Yang F, Yang Y, Zeng W. Anti-Dsdna Blockade Ameliorates Systemic Lupus Erythematosus in Mrl/Faslpr Mice Through Ameliorating Inflammation Via Pkcδ-Nlrc4 Axis. Biochem Cell Biol (2020) 99(3):313–21. doi: 10.1139/bcb-2020-0265
39. Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, et al. U1-Small Nuclear Ribonucleoprotein Activates the Nlrp3 Inflammasome in Human Monocytes. J Immunol (Baltimore Md 1950) (2012) 188(10):4769–75. doi: 10.4049/jimmunol.1103355
40. Shin MS, Kang Y, Wahl ER, Park HJ, Lazova R, Leng L, et al. Macrophage Migration Inhibitory Factor Regulates U1 Small Nuclear Rnp Immune Complex-Mediated Activation of the Nlrp3 Inflammasome. Arthritis Rheumatol (Hoboken NJ) (2019) 71(1):109–20. doi: 10.1002/art.40672
41. Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB. Type I Interferon in the Pathogenesis of Systemic Lupus Erythematosus. Curr Opin Immunol (2020) 67:87–94. doi: 10.1016/j.coi.2020.10.014
42. Crow MK. Type I Interferon in the Pathogenesis of Lupus. J Immunol (Baltimore Md 1950) (2014) 192(12):5459–68. doi: 10.4049/jimmunol.1002795
43. Niewold TB. Interferon Alpha as a Primary Pathogenic Factor in Human Lupus. J Interferon Cytokine Res (2011) 31(12):887–92. doi: 10.1089/jir.2011.0071
44. Satoh M, Reeves WH. Induction of Lupus-Associated Autoantibodies in Balb/C Mice by Intraperitoneal Injection of Pristane. J Exp Med (1994) 180(6):2341–6. doi: 10.1084/jem.180.6.2341
45. Pothlichet J, Meunier I, Davis BK, Ting JP, Skamene E, von Messling V, et al. Type I Ifn Triggers Rig-I/Tlr3/Nlrp3-Dependent Inflammasome Activation in Influenza a Virus Infected Cells. PLoS Pathog (2013) 9(4):e1003256. doi: 10.1371/journal.ppat.1003256
46. Fang R, Hara H, Sakai S, Hernandez-Cuellar E, Mitsuyama M, Kawamura I, et al. Type I Interferon Signaling Regulates Activation of the Absent in Melanoma 2 Inflammasome During Streptococcus Pneumoniae Infection. Infect Immun (2014) 82(6):2310–7. doi: 10.1128/iai.01572-14
47. Liu J, Berthier CC, Kahlenberg JM. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated Via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol (Hoboken NJ) (2017) 69(9):1840–9. doi: 10.1002/art.40166
48. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity (2011) 34(2):213–23. doi: 10.1016/j.immuni.2011.02.006
49. Yu X, Du Y, Cai C, Cai B, Zhu M, Xing C, et al. Inflammasome Activation Negatively Regulates Myd88-Irf7 Type I Ifn Signaling and Anti-Malaria Immunity. Nat Commun (2018) 9(1):4964. doi: 10.1038/s41467-018-07384-7
50. Yang Q, Yu C, Yang Z, Wei Q, Mu K, Zhang Y, et al. Deregulated Nlrp3 and Nlrp1 Inflammasomes and Their Correlations With Disease Activity in Systemic Lupus Erythematosus. J Rheumatol (2014) 41(3):444–52. doi: 10.3899/jrheum.130310
51. Liu J, Huang X, Hao S, Wang Y, Liu M, Xu J, et al. Peli1 Negatively Regulates Noncanonical Nf-Kb Signaling to Restrain Systemic Lupus Erythematosus. Nat Commun (2018) 9(1):1136. doi: 10.1038/s41467-018-03530-3
52. Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, Anzelon-Mills A, et al. Alternative and Classical Nf-Kappa B Signaling Retain Autoreactive B Cells in the Splenic Marginal Zone and Result in Lupus-Like Disease. Immunity (2006) 25(3):403–15. doi: 10.1016/j.immuni.2006.07.010
53. Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol EB, et al. Nf-Kb Inducing Kinase Is a Therapeutic Target for Systemic Lupus Erythematosus. Nat Commun (2018) 9(1):179. doi: 10.1038/s41467-017-02672-0
54. Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The Ubiquitin Modifying Enzyme A20 Restricts B Cell Survival and Prevents Autoimmunity. Immunity (2010) 33(2):181–91. doi: 10.1016/j.immuni.2010.07.017
55. Zhao LD, Liang D, Wu XN, Li Y, Niu JW, Zhou C, et al. Contribution and Underlying Mechanisms of Cxcr4 Overexpression in Patients With Systemic Lupus Erythematosus. Cell Mol Immunol (2017) 14(10):842–9. doi: 10.1038/cmi.2016.47
56. Ma X, Nakayamada S, Kubo S, Sakata K, Yamagata K, Miyazaki Y, et al. Expansion of T Follicular Helper-T Helper 1 Like Cells Through Epigenetic Regulation by Signal Transducer and Activator of Transcription Factors. Ann Rheum Dis (2018) 77(9):1354–61. doi: 10.1136/annrheumdis-2017-212652
57. Zhang CJ, Wang C, Jiang M, Gu C, Xiao J, Chen X, et al. Act1 Is a Negative Regulator in T and B Cells Via Direct Inhibition of Stat3. Nat Commun (2018) 9(1):2745. doi: 10.1038/s41467-018-04974-3
58. Huang N, Perl A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol (2018) 39(7):562–76. doi: 10.1016/j.it.2018.04.006
59. Katsuyama T, Li H, Comte D, Tsokos GC, Moulton VR. Splicing Factor Srsf1 Controls T Cell Hyperactivity and Systemic Autoimmunity. J Clin Invest (2019) 129(12):5411–23. doi: 10.1172/jci127949
60. Rozo C, Chinenov Y, Maharaj RK, Gupta S, Leuenberger L, Kirou KA, et al. Targeting the Rhoa-Rock Pathway to Reverse T-Cell Dysfunction in Sle. Ann Rheum Dis (2017) 76(4):740–7. doi: 10.1136/annrheumdis-2016-209850
61. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil Extracellular Traps (Nets) in Autoimmune Diseases: A Comprehensive Review. Autoimmun Rev (2017) 16(11):1160–73. doi: 10.1016/j.autrev.2017.09.012
62. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin Suppresses Nlrp3 Inflammasome Activation Via Tlr4/Myd88/Nf-Kb Signaling in Pma-Stimulated Thp-1 Monocytes. Biomed Pharmacother = Biomedec Pharmacother (2016) 82:167–72. doi: 10.1016/j.biopha.2016.04.043
63. Zhao W, Ma L, Cai C, Gong X. Caffeine Inhibits Nlrp3 Inflammasome Activation by Suppressing Mapk/Nf-Kb and A2ar Signaling in Lps-Induced Thp-1 Macrophages. Int J Biol Sci (2019) 15(8):1571–81. doi: 10.7150/ijbs.34211
64. Dong L, Hu R, Yang D, Zhao J, Kan H, Tan J, et al. Fine Particulate Matter [Pm(2.5)] Upregulates Expression of Inflammasome Nlrp1 Via Ros/Nf-Kb Signaling in Hacat Cells. Int J Med Sci (2020) 17(14):2200–6. doi: 10.7150/ijms.46962
65. Pan J, Han L, Guo J, Wang X, Liu D, Tian J, et al. Aim2 Accelerates the Atherosclerotic Plaque Progressions in Apoe-/- Mice. Biochem Biophys Res Commun (2018) 498(3):487–94. doi: 10.1016/j.bbrc.2018.03.005
66. Su WJ, Zhang Y, Chen Y, Gong H, Lian YJ, Peng W, et al. Nlrp3 Gene Knockout Blocks Nf-Kb and Mapk Signaling Pathway in Cums-Induced Depression Mouse Model. Behav Brain Res (2017) 322(Pt A):1–8. doi: 10.1016/j.bbr.2017.01.018
67. Li Y, Liu C, Wan XS, Li SW. Nlrp1 Deficiency Attenuates Diabetic Retinopathy (Dr) in Mice Through Suppressing Inflammation Response. Biochem Biophys Res Commun (2018) 501(2):351–7. doi: 10.1016/j.bbrc.2018.03.148
68. Goropevšek A, Holcar M, Avčin T. The Role of Stat Signaling Pathways in the Pathogenesis of Systemic Lupus Erythematosus. Clin Rev Allergy Immunol (2017) 52(2):164–81. doi: 10.1007/s12016-016-8550-y
69. Furuya MY, Asano T, Sumichika Y, Sato S, Kobayashi H, Watanabe H, et al. Tofacitinib Inhibits Granulocyte-Macrophage Colony-Stimulating Factor-Induced Nlrp3 Inflammasome Activation in Human Neutrophils. Arthritis Res Ther (2018) 20(1):196. doi: 10.1186/s13075-018-1685-x
70. Lee KM, Kang JH, Yun M, Lee SB. Quercetin Inhibits the Poly(Da:Dt)-Induced Secretion of Il-18 Via Down-Regulation of the Expressions of Aim2 and Pro-Caspase-1 by Inhibiting the Jak2/Stat1 Pathway in Ifn-Γ-Primed Human Keratinocytes. Biochem Biophys Res Commun (2018) 503(1):116–22. doi: 10.1016/j.bbrc.2018.05.191
71. Marín-Aguilar F, Lechuga-Vieco AV, Alcocer-Gómez E, Castejón-Vega B, Lucas J, Garrido C, et al. Nlrp3 Inflammasome Suppression Improves Longevity and Prevents Cardiac Aging in Male Mice. Aging Cell (2020) 19(1):e13050. doi: 10.1111/acel.13050
72. Qiu Q, Yang Z, Cao F, Yang C, Hardy P, Yan X, et al. Activation of Nlrp3 Inflammasome by Lymphocytic Microparticles Via Tlr4 Pathway Contributes to Airway Inflammation. Exp Cell Res (2020) 386(2):111737. doi: 10.1016/j.yexcr.2019.111737
73. Qiu Y, Huang X, Huang L, Tang L, Jiang J, Chen L, et al. 5-Ht(1a) Receptor Antagonist Improves Behavior Performance of Delirium Rats Through Inhibiting Pi3k/Akt/Mtor Activation-Induced Nlrp3 Activity. IUBMB Life (2016) 68(4):311–9. doi: 10.1002/iub.1491
74. Chen J, Wang Z, Yu S. Aim2 Regulates Viability and Apoptosis in Human Colorectal Cancer Cells Via the Pi3k/Akt Pathway. Onco Targets Ther (2017) 10:811–7. doi: 10.2147/ott.S125039
75. Xie S, Yang T, Wang Z, Li M, Ding L, Hu X, et al. Astragaloside Iv Attenuates Sepsis-Induced Intestinal Barrier Dysfunction Via Suppressing Rhoa/Nlrp3 Inflammasome Signaling. Int Immunopharmacol (2020) 78:106066. doi: 10.1016/j.intimp.2019.106066
76. Curran CS, Gupta S, Sanz I, Sharon E. Pd-1 Immunobiology in Systemic Lupus Erythematosus. J Autoimmun (2019) 97:1–9. doi: 10.1016/j.jaut.2018.10.025
77. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of Cyclic Gmp-Amp Synthase in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol (Hoboken NJ) (2017) 69(4):800–7. doi: 10.1002/art.40002
78. Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-Derived Membrane Vesicles Drive the Cgas-Sting Pathway and Enhance Type I Ifn Production in Systemic Lupus Erythematosus. Ann Rheum Dis (2018) 77(10):1507–15. doi: 10.1136/annrheumdis-2018-212988
79. Tian Y, Bao Z, Ji Y, Mei X, Yang H. Epigallocatechin-3-Gallate Protects H(2)O(2)-Induced Nucleus Pulposus Cell Apoptosis and Inflammation by Inhibiting Cgas/Sting/Nlrp3 Activation. Drug Des Devel Ther (2020) 14:2113–22. doi: 10.2147/dddt.S251623
80. Wang W, Hu D, Wu C, Feng Y, Li A, Liu W, et al. Sting Promotes Nlrp3 Localization in Er and Facilitates Nlrp3 Deubiquitination to Activate the Inflammasome Upon Hsv-1 Infection. PLoS Pathog (2020) 16(3):e1008335. doi: 10.1371/journal.ppat.1008335
81. Yan S, Shen H, Lian Q, Jin W, Zhang R, Lin X, et al. Deficiency of the Aim2-Asc Signal Uncovers the Sting-Driven Overreactive Response of Type I Ifn and Reciprocal Depression of Protective Ifn-Γ Immunity in Mycobacterial Infection. J Immunol (Baltimore Md 1950) (2018) 200(3):1016–26. doi: 10.4049/jimmunol.1701177
82. Li X, Liu F, Zhang X, Shi G, Ren J, Ji J, et al. Notch-Hes-1 Axis Controls Tlr7-Mediated Autophagic Death of Macrophage Via Induction of P62 in Mice With Lupus. Cell Death Dis (2016) 7(8):e2341. doi: 10.1038/cddis.2016.244
83. Teachey DT, Seif AE, Brown VI, Bruno M, Bunte RM, Chang YJ, et al. Targeting Notch Signaling in Autoimmune and Lymphoproliferative Disease. Blood (2008) 111(2):705–14. doi: 10.1182/blood-2007-05-087353
84. Lee S, Kim SK, Park H, Lee YJ, Park SH, Lee KJ, et al. Contribution of Autophagy-Notch1-Mediated Nlrp3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts. Int J Mol Sci (2020) 21(21):8050. doi: 10.3390/ijms21218050
85. Li X, Zhang X, Pan Y, Shi G, Ren J, Fan H, et al. Mtor Regulates Nlrp3 Inflammasome Activation Via Reactive Oxygen Species in Murine Lupus. Acta Biochim Biophys Sin (Shanghai) (2018) 50(9):888–96. doi: 10.1093/abbs/gmy088
86. Barsalou J, Blincoe A, Fernandez I, Dal-Soglio D, Marchitto L, Selleri S, et al. Rapamycin as an Adjunctive Therapy for Nlrc4 Associated Macrophage Activation Syndrome. Front Immunol (2018) 9:2162. doi: 10.3389/fimmu.2018.02162
87. Shi X, Wang L, Ren L, Li J, Li S, Cui Q, et al. Dihydroartemisinin, an Antimalarial Drug, Induces Absent in Melanoma 2 Inflammasome Activation and Autophagy in Human Hepatocellular Carcinoma Hepg2215 Cells. Phytother Res (2019) 33(5):1413–25. doi: 10.1002/ptr.6332
88. Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals Via the Adaptor Myd88 in B Cells and Dcs Make Distinct and Synergistic Contributions to Immune Activation and Tissue Damage in Lupus. Immunity (2013) 38(3):528–40. doi: 10.1016/j.immuni.2012.11.017
89. Kong F, Ye B, Cao J, Cai X, Lin L, Huang S, et al. Curcumin Represses Nlrp3 Inflammasome Activation Via Tlr4/Myd88/Nf-Kb and P2x7r Signaling in Pma-Induced Macrophages. Front Pharmacol (2016) 7:369. doi: 10.3389/fphar.2016.00369
90. Huang W, Jiao J, Liu J, Huang M, Hu Y, Ran W, et al. Mfg-E8 Accelerates Wound Healing in Diabetes by Regulating "Nlrp3 Inflammasome-Neutrophil Extracellular Traps" Axis. Cell Death Discov (2020) 6:84. doi: 10.1038/s41420-020-00318-7
91. Molad Y, Amit-Vasina M, Bloch O, Yona E, Rapoport MJ. Increased Erk and Jnk Activities Correlate With Disease Activity in Patients With Systemic Lupus Erythematosus. Ann Rheum Dis (2010) 69(1):175–80. doi: 10.1136/ard.2008.102780
92. Chei S, Oh HJ, Song JH, Seo YJ, Lee K, Kim KJ, et al. Spirulina Maxima Extract Prevents Activation of the Nlrp3 Inflammasome by Inhibiting Erk Signaling. Sci Rep (2020) 10(1):2075. doi: 10.1038/s41598-020-58896-6
93. Liu LB, Shen HF, Cha W, Jin ZJ, Xia HJ, Liu JJ, et al. Sxbx Pill Suppresses Homocysteine-Induced Vascular Smooth Muscle Cells Dedifferentiation by Inhibiting Nlrp3 Inflammasomes Activation Via Erk/P38 Mapk Pathways. Am J Transl Res (2019) 11(2):806–18.
94. Chu X, Wang C, Wu Z, Fan L, Tao C, Lin J, et al. Jnk/C-Jun-Driven Nlrp3 Inflammasome Activation in Microglia Contributed to Retinal Ganglion Cells Degeneration Induced by Indirect Traumatic Optic Neuropathy. Exp Eye Res (2020) 202:108335. doi: 10.1016/j.exer.2020.108335
95. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The Lysosome Rupture-Activated Tak1-Jnk Pathway Regulates Nlrp3 Inflammasome Activation. J Biol Chem (2014) 289(47):32926–36. doi: 10.1074/jbc.M114.579961
96. Guo H, Sun L, Ling S, Xu JW. Levistilide a Ameliorates Nlrp3 Expression Involving the Syk-P38/Jnk Pathway and Peripheral Obliterans in Rats. Mediators Inflamm (2018) 2018:7304096. doi: 10.1155/2018/7304096
97. Chen S, Zuo Y, Huang L, Sherchan P, Zhang J, Yu Z, et al. The Mc(4) Receptor Agonist Ro27-3225 Inhibits Nlrp1-Dependent Neuronal Pyroptosis Via the Ask1/Jnk/P38 Mapk Pathway in a Mouse Model of Intracerebral Haemorrhage. Br J Pharmacol (2019) 176(9):1341–56. doi: 10.1111/bph.14639
98. Lin C, Wu F, Zheng T, Wang X, Chen Y, Wu X. Kaempferol Attenuates Retinal Ganglion Cell Death by Suppressing Nlrp1/Nlrp3 Inflammasomes and Caspase-8 Via Jnk and Nf-Kb Pathways in Acute Glaucoma. Eye (Lond) (2019) 33(5):777–84. doi: 10.1038/s41433-018-0318-6
99. Fu R, Xia Y, Li M, Mao R, Guo C, Zhou M, et al. Pim-1 as a Therapeutic Target in Lupus Nephritis. Arthritis Rheumatol (Hoboken NJ) (2019) 71(8):1308–18. doi: 10.1002/art.40863
100. Choubey D, Panchanathan R. Interferon (Ifn)-Inducible Absent in Melanoma 2 Proteins in the Negative Regulation of the Type I Ifn Response: Implications for Lupus Nephritis. Cytokine (2020) 132:154682. doi: 10.1016/j.cyto.2019.03.008
101. Choubey D, Panchanathan R. Absent in Melanoma 2 Proteins in Sle. Clin Immunol (2017) 176:42–8. doi: 10.1016/j.clim.2016.12.011
102. Panchanathan R, Shen H, Bupp MG, Gould KA, Choubey D. Female and Male Sex Hormones Differentially Regulate Expression of Ifi202, an Interferon-Inducible Lupus Susceptibility Gene Within the Nba2 Interval. J Immunol (Baltimore Md 1950) (2009) 183(11):7031–8. doi: 10.4049/jimmunol.0802665
103. Yin Q, Sester DP, Tian Y, Hsiao YS, Lu A, Cridland JA, et al. Molecular Mechanism for P202-Mediated Specific Inhibition of Aim2 Inflammasome Activation. Cell Rep (2013) 4(2):327–39. doi: 10.1016/j.celrep.2013.06.024
104. Panchanathan R, Duan X, Arumugam M, Shen H, Liu H, Choubey D. Cell Type and Gender-Dependent Differential Regulation of the P202 and Aim2 Proteins: Implications for the Regulation of Innate Immune Responses in Sle. Mol Immunol (2011) 49(1-2):273–80. doi: 10.1016/j.molimm.2011.08.022
105. Yang CA, Huang ST, Chiang BL. Sex-Dependent Differential Activation of Nlrp3 and Aim2 Inflammasomes in Sle Macrophages. Rheumatol (Oxford England) (2015) 54(2):324–31. doi: 10.1093/rheumatology/keu318
106. Zhang W, Cai Y, Xu W, Yin Z, Gao X, Xiong S. Aim2 Facilitates the Apoptotic DNA-Induced Systemic Lupus Erythematosus Via Arbitrating Macrophage Functional Maturation. J Clin Immunol (2013) 33(5):925–37. doi: 10.1007/s10875-013-9881-6
107. Yang F, Zhai Z, Luo X, Luo G, Zhuang L, Zhang Y, et al. Bioinformatics Identification of Key Candidate Genes and Pathways Associated With Systemic Lupus Erythematosus. Clin Rheumatol (2020) 39(2):425–34. doi: 10.1007/s10067-019-04751-7
108. Ding L, Dong G, Zhang D, Ni Y, Hou Y. The Regional Function of Cgas/Sting Signal in Multiple Organs: One of Culprit Behind Systemic Lupus Erythematosus? Med Hypotheses (2015) 85(6):846–9. doi: 10.1016/j.mehy.2015.09.026
109. Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive Oligodeoxynucleotides Delay the Onset of Glomerulonephritis and Prolong Survival in Lupus-Prone Nzb X Nzw Mice. Arthritis Rheum (2005) 52(2):651–8. doi: 10.1002/art.20810
110. Pontillo A, Girardelli M, Kamada AJ, Pancotto JA, Donadi EA, Crovella S, et al. Polimorphisms in Inflammasome Genes Are Involved in the Predisposition to Systemic Lupus Erythematosus. Autoimmunity (2012) 45(4):271–8. doi: 10.3109/08916934.2011.637532
111. Bonomini F, Dos Santos M, Veronese FV, Rezzani R. Nlrp3 Inflammasome Modulation by Melatonin Supplementation in Chronic Pristane-Induced Lupus Nephritis. Int J Mol Sci (2019) 20(14):3466. doi: 10.3390/ijms20143466
112. Li X, Wang M, Hong H, Luo C, Liu Z, Yang R. Sophocarpine Attenuates Murine Lupus Nephritis Via Inhibiting Nlrp3 Inflammasome and Nf-Kb Activation. Immunol Res (2018) 66(4):521–7. doi: 10.1007/s12026-018-9012-9
113. Zhao J, Wang J, Zhou M, Li M, Li M, Tan H. Curcumin Attenuates Murine Lupus Via Inhibiting Nlrp3 Inflammasome. Int Immunopharmacol (2019) 69:213–6. doi: 10.1016/j.intimp.2019.01.046
114. Su B, Ye H, You X, Ni H, Chen X, Li L. Icariin Alleviates Murine Lupus Nephritis Via Inhibiting Nf-Kb Activation Pathway and Nlrp3 Inflammasome. Life Sci (2018) 208:26–32. doi: 10.1016/j.lfs.2018.07.009
115. Huang T, Yin H, Ning W, Wang X, Chen C, Lin W, et al. Expression of Inflammasomes Nlrp1, Nlrp3 and Aim2 in Different Pathologic Classification of Lupus Nephritis. Clin Exp Rheumatol (2020) 38(4):680–90.
116. Guo C, Fu R, Zhou M, Wang S, Huang Y, Hu H, et al. Pathogenesis of Lupus Nephritis: Rip3 Dependent Necroptosis and Nlrp3 Inflammasome Activation. J Autoimmun (2019) 103:102286. doi: 10.1016/j.jaut.2019.05.014
117. Ehtesham N, Zare Rafie M, Esmaeilzadeh E, Dehani M, Davar S, Mosallaei M, et al. Three Functional Variants in the Nlrp3 Gene Are Associated With Susceptibility and Clinical Characteristics of Systemic Lupus Erythematosus. Lupus (2021) 30(8):1273–82. doi: 10.1177/09612033211014273
118. Tang L, Zhou F. Inflammasomes in Common Immune-Related Skin Diseases. Front Immunol (2020) 11:882. doi: 10.3389/fimmu.2020.00882
119. Hu S, Yu F, Ye C, Huang X, Lei X, Dai Y, et al. The Presence of P2rx7 Single Nuclear Polymorphism Is Associated With a Gain of Function in P2x7 Receptor and Inflammasome Activation in Sle Complicated With Pericarditis. Clin Exp Rheumatol (2020) 38(3):442–9.
120. Furini F, Giuliani AL, Parlati ME, Govoni M, Di Virgilio F, Bortoluzzi A. P2x7 Receptor Expression in Patients With Serositis Related to Systemic Lupus Erythematosus. Front Pharmacol (2019) 10:435. doi: 10.3389/fphar.2019.00435
121. Spel L, Martinon F. Inflammasomes Contributing to Inflammation in Arthritis. Immunol Rev (2020) 294(1):48–62. doi: 10.1111/imr.12839
122. Jeltsch-David H, Muller S. Neuropsychiatric Systemic Lupus Erythematosus and Cognitive Dysfunction: The Mrl-Lpr Mouse Strain as a Model. Autoimmun Rev (2014) 13(9):963–73. doi: 10.1016/j.autrev.2014.08.015
123. Gulinello M, Putterman C. The Mrl/Lpr Mouse Strain as a Model for Neuropsychiatric Systemic Lupus Erythematosus. J BioMed Biotechnol (2011) 2011:207504. doi: 10.1155/2011/207504
124. He J, Sun M, Tian S. Procyanidin B2 Prevents Lupus Nephritis Development in Mice by Inhibiting Nlrp3 Inflammasome Activation. Innate Immun (2018) 24(5):307–15. doi: 10.1177/1753425918780985
125. Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The Early Local and Systemic Type I Interferon Responses to Ultraviolet B Light Exposure Are Cgas Dependent. Sci Rep (2020) 10(1):7908. doi: 10.1038/s41598-020-64865-w
126. Wolf SJ, Estadt SN, Theros J, Moore T, Ellis J, Liu J, et al. Ultraviolet Light Induces Increased T Cell Activation in Lupus-Prone Mice Via Type I Ifn-Dependent Inhibition of T Regulatory Cells. J Autoimmun (2019) 103:102291. doi: 10.1016/j.jaut.2019.06.002
127. Panchanathan R, Liu H, Leung YK, Ho SM, Choubey D. Bisphenol a (Bpa) Stimulates the Interferon Signaling and Activates the Inflammasome Activity in Myeloid Cells. Mol Cell Endocrinol (2015) 415:45–55. doi: 10.1016/j.mce.2015.08.003
128. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological Inhibitors of the Nlrp3 Inflammasome. Front Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
Keywords: inflammasome, treatment, systemic lupus erythematosus (SLE), target, strategy
Citation: Liu Y, Tao X and Tao J (2022) Strategies of Targeting Inflammasome in the Treatment of Systemic Lupus Erythematosus. Front. Immunol. 13:894847. doi: 10.3389/fimmu.2022.894847
Received: 12 March 2022; Accepted: 19 April 2022;
Published: 18 May 2022.
Edited by:
Jian Gao, Shanghai Children’s Medical Center, ChinaReviewed by:
Lihua Duan, Jiangxi Provincial People’s Hospital, ChinaSarang Tartey, IGM Biosciences, United States
Huaxiang Wu, Zhejiang University, China
Copyright © 2022 Liu, Tao and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhui Tao, taojinhui@ustc.edu.cn
†These authors have contributed equally to this work