 Erik P. Lillehoj
Erik P. Lillehoj Irina G. Luzina2,3
Irina G. Luzina2,3 Sergei P. Atamas
Sergei P. Atamas- 1Department of Pediatrics, University of Maryland School of Medicine, Baltimore, MD, United States
- 2Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, United States
- 3Research Service, Baltimore Veterans Affairs (VA) Medical Center, Baltimore, MD, United States
Mammalian neuraminidases (NEUs), also known as sialidases, are enzymes that cleave off the terminal neuraminic, or sialic, acid resides from the carbohydrate moieties of glycolipids and glycoproteins. A rapidly growing body of literature indicates that in addition to their metabolic functions, NEUs also regulate the activity of their glycoprotein targets. The simple post-translational modification of NEU protein targets—removal of the highly electronegative sialic acid—affects protein folding, alters protein interactions with their ligands, and exposes or covers proteolytic sites. Through such effects, NEUs regulate the downstream processes in which their glycoprotein targets participate. A major target of desialylation by NEUs are mucins (MUCs), and such post-translational modification contributes to regulation of disease processes. In this review, we focus on the regulatory roles of NEU-modified MUCs as coordinators of disease pathogenesis in fibrotic, inflammatory, infectious, and autoimmune diseases. Special attention is placed on the most abundant and best studied NEU1, and its recently discovered important target, mucin-1 (MUC1). The role of the NEU1 - MUC1 axis in disease pathogenesis is discussed, along with regulatory contributions from other MUCs and other pathophysiologically important NEU targets.
Introduction
Protein glycosylation is a biologically ubiquitous, enzymatic post-translational modification involving the attachment of one or more carbohydrate moieties to a protein molecule, typically at the nitrogen atom of the side chain amide group of an Asn residue (N-linked glycosylation) or the oxygen atom of the side chain hydroxyl group of a Ser or Thr residue (O-linked glycosylation). Glycosyltransferases catalyze the transfer of the glycan to the protein acceptor, while glycosidases catalyze the reverse reaction. Sialyltransferases and neuramindases (NEUs), also known as sialidases, are subclasses of glycosyltransferases and glycosidases, respectively, that specifically add or remove sialic acids to/from the terminal position of glycan chains (Figure 1). Sialic acids comprise a family of more than 50 closely related, but structurally diverse, nine-carbon monosaccharides (1–3). N-acetylneuraminic acid (Neu5Ac or NANA) is the predominant sialic acid in most mammalian cells and is often used interchangeably with sialic acid. Most commonly, the C-2 carbon atom of the α-anomer of Neu5Ac forms a covalent bond with the C-3 hydroxyl group of galactose (Gal), or the C-6 hydroxyl group of Gal or N-acetylgalactosamine (GalNAc), to form α(2,3)- and α(2,6)-linked structures. In a small number of proteins, polysialic acid has been identified as a unique, α(2,8)-linked homopolymer of up to 150 sialic acid residues that confers an exceptionally high electronegative charge (4). With their terminal location and negative charge at physiological pH, sialic acids are strategically positioned to influence intermolecular and intercellular interactions through steric hindrance and/or electronic effects. Consequently, sialic acids, and the enzymes that add or remove them from glycan chains, influence protein tertiary conformation, bioactivity, and proteolysis.
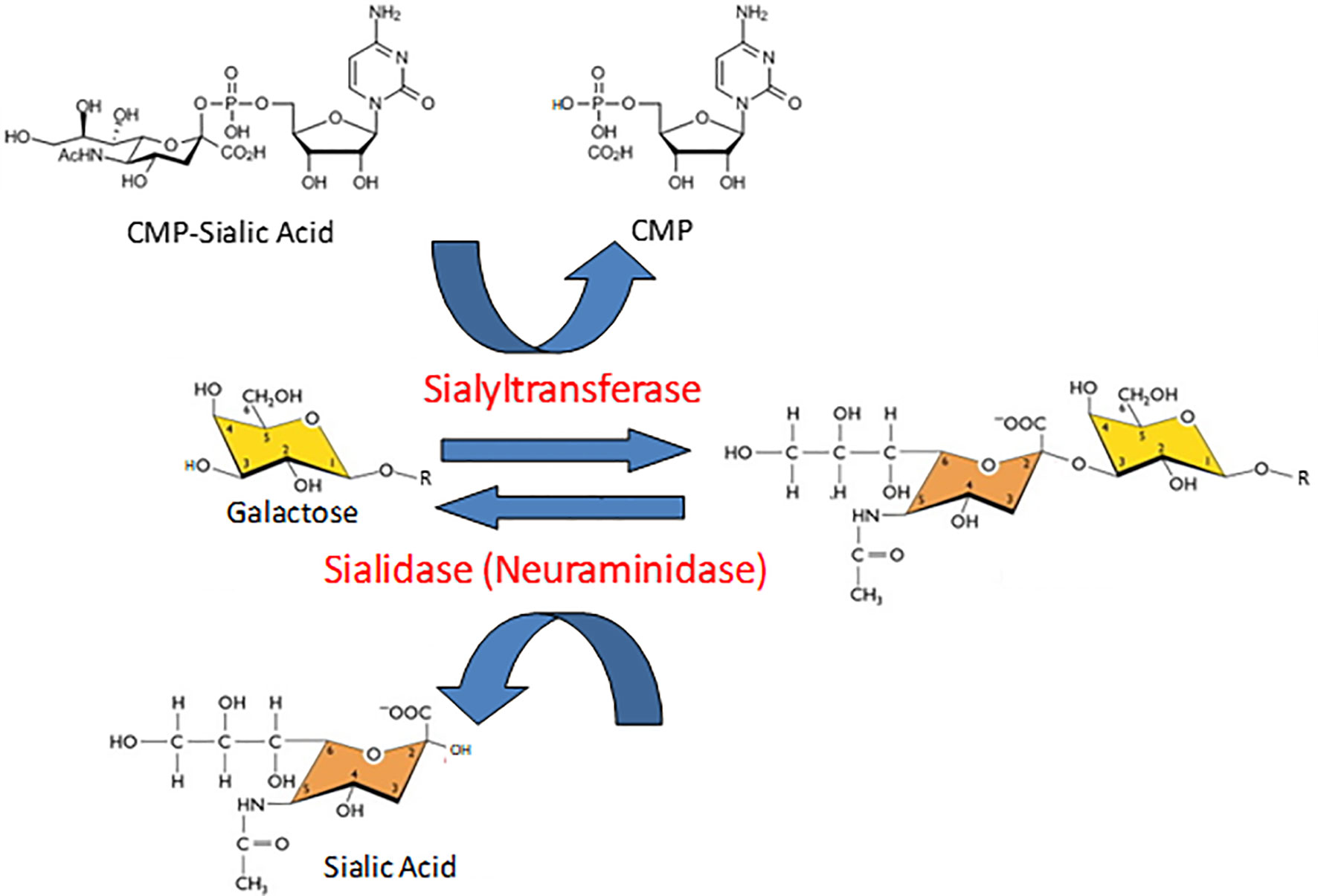
Figure 1 Opposing catalytic activities of sialyltransferases and sialidases. In the reaction catalyzed by sialyltransferases, sialic acid is transferred from its activated nucleotide sugar donor, cytidine 5’-monophosphate (CMP)-sialic acid, to the penultimate galactose (illustrated here) or N-acetylgalactosamine residue of a glycan chain, thereby releasing free CMP. The resulting sialic acid-galactose disaccharide is illustrated in an α(2-3) linkage. In the sialidase-catalyzed reaction, the sialic acid-galactose/N-acetylgalactosamine covalent bond is hydrolyzed to released release free sialic acid.
NEUs – General Features
As early as 1960, mammalian sialidase activity was detected in fractioned human serum (5). Later, crude sialidase activity was reported in a variety of eukaryotic cell and tissue extracts (6–11). Prior to isolation and characterization of mammalian NEU genes, the biochemical characterization and enrichment/purification of these various sialidases demonstrated the presence of enzymes with different molecular weights, subcellular localization, substrate specificities, and pH optimums in vitro (12). Klotho, a protein originally associated with senescence and aging, also has been reported to exhibit sialidase activity, although it lacks conserved structural domains characteristic of canonical NEU proteins (13).
NEUs are found in organisms of diverse evolutionary lineages, from viruses to mammals, and functional consequences of their enzymatic activity across and within taxa are remarkably broad and complex. There are four mammalian NEUs, NEU1 – NEU4, and they are all exosialidases, meaning that they remove the terminal sialic residues from the glycan moieties of glycoproteins and glycolipids. Mammalian NEUs are differentially expressed across tissues and differentially distributed at the subcellular level. Their relative expression levels are usually different, with NEU1 being the predominant isoform, often closely followed by the relatively abundant NEU3, whereas NEU2 and NEU4 are commonly expressed minimally if at all; this generalization is not always supported as it may be hindered by specifics of a particular disease process, tissue involved, or differentiation state of the cells engaged. Based on their catalytic function, NEUs are primarily metabolic enzymes; mutations of the NEU1 gene lead to a metabolic disorder, sialidosis (14). The research of the past two decades has revealed that NEUs also regulate important biological functions and are involved in disease pathogenesis (12, 15–17). This is not surprising, considering that many homeostatic mediators involved in metabolic regulation, development, reproduction, and immune tolerance, as well as pathophysiological regulators involved in immune and inflammatory diseases, fibrosis, atherosclerosis, cancer, and other diseases are sialylated glycoproteins. NEU targets and binding partners include multiple glycoproteins known to play important roles in the mechanisms of immunity, inflammation and fibrosis (Table 1).

Table 1 Selected confirmed and putative glycoprotein substrates of NEUs and NEU interactors with regulatory contributions to immune, inflammatory, and fibrotic processes.
At the mechanistic level, the downstream effects of desialylation are substantially defined by the properties of sialic acid, which is a highly electronegative sugar. Desialylation affects the charge of the target molecules and even the often much sialylated cell surface. At the molecular level, desialylation can directly unmask or mask glycoproteins’ bindings sites for their molecular partners. Desialylation can also affect intramolecular interactions and, subsequently, protein folding, again revealing or sequestering binding sites for low molecular weight ligands or high molecular weight interactors, thus affecting the target glycoproteins’ functions as enzymes, receptors, or signal transducers. More specifically, several mechanistic pathways can be envisioned and have been experimentally validated (12, 15–17) through which neuraminidases control their glycoprotein targets and their downstream functioning. Desialylation also affects the target glycoproteins’ folding, thus additionally regulating protein-protein interactions. Desialylation of the entire cell surface has far-going consequences for cell-cell interactions, thus affecting pathophysiological processes on the large scale. Such changes in folding may induce transitions between functionally active and inactive states of enzymes, attenuation or augmentation of ligand binding by receptors, and masking or unmasking of proteolytic sites. On a more direct level, siglecs bind to sialic acid of glycoproteins, whereas galectins bind to Gal residues which become exposed following desialylation, as Gal is typically the subterminal carbohydrate to which the terminal sialic acid is attached in the glycan portions of glycoproteins and glycolipids. Thus, NEU-mediated desialylation diminishes siglec binding and allows for galectin binding to glycoproteins, affecting such interactions that are known to regulate immunity and inflammation.
Originally, the four mammalian NEUs were characterized by their characteristic subcellular localization, including lysosomal NEU1, cytosolic NEU2, plasma membrane NEU3, and lysosomal, mitochondrial, and endoplasmic reticular NEU4 (12). In the lysosome, NEU1 is associated with its chaperone/transport protein, protective protein/cathepsin A (PPCA), and β-galactosidase (60). It became quickly recognized that in addition to such predominant localization, each NEU may be distributed more broadly. For example, in addition to the predominantly membrane-targeted NEU3, the other NEUs can be found on the cell surface, including NEU1 (20, 41, 44–47, 53, 55, 56, 60–75), NEU2 (76–78), and NEU4 (79, 80). NEU1 on the plasma membrane has been reported either in the presence (62, 64–66, 72, 75) or absence (68, 71) of PPCA. Membrane localization allows NEUs to act as structural and functional modulators of extracellular soluble and membrane-bound molecules (17). Further information on the subcellular, cellular, and tissue distribution, substrate specificity, catalytic properties, and amino acid homologies of the four mammalian NEUs can be found in prior review articles (12, 15, 81–87).
MUCs – General Features
Mucus is a complex mixture of ions, salts, peptides, proteins, glycoconjugates, and water covering the surface of mucosal tissues. The primary protein component of mucus are mucins, high molecular weight, extensively glycosylated proteins containing variable numbers of tandem repeats (VNTRs) in which Ser, Thr, and Pro amino acids are highly enriched. Mucin glycosylation primarily occurs through O-glycosidic linkages between the first GalNAc residue of the oligosaccharide side chains and Ser and Thr amino acids of the polypeptide backbone. More than 20 mucin glycoproteins have been identified and are broadly subdivided into secreted and cell membrane-tethered molecules (88, 89). Secreted mucins are further subdivided into gel-forming and non-gel-forming mucins. Examples of membrane-bound mucins include MUC1, MUC3A, MUC3B, MUC4, MUC13, and MUC16, while secreted mucins include MUC2, MUC5AC, MUC5B, MUC6, MUC7, MUC19, and MUC20.
MUC1 is the prototype membrane-tethered mucin. The MUC1 glycoprotein consists of a large extracellular ectodomain (MUC1-ED), a single-pass transmembrane domain, and an intracellular cytoplasmic domain (MUC1-CD) (90–92). The MUC1-ED is primarily composed of varying numbers (25–125) of highly O-glycosylated, 20-amino acid VNTRs, with the number of repeats being a polymorphic genetic trait. The MUC1-CD contains multiple Ser, Thr, and Tyr residues as potential phosphorylation sites. MUC1 is initially synthesized as a single polypeptide chain, but autoproteolytic cleavage at a Gly-Ser peptide bond in the endoplasmic reticulum generates two subunits that are together transported to the cell surface. The larger subunit (> 250 kDa) is derived from most of the MUC1-ED and the smaller subunit (20-30 kDa) contains a juxtamembrane region of the MUC1-ED, the membrane-spanning domain, and the MUC1-CD (93). MUC1 autoproteolysis occurs within its SEA (sea urchin sperm protein, enterokinase, agrin) domain, a 120-amino acid region highly conserved in glycosylated, mucin-like proteins (94, 95). While the two autoproteolytic cleavage fragments constitute separate polypeptide chains, they remain tightly associated on the cell surface, although they are not linked through disulfide bonds, and are only separated under extreme dissociating conditions, for example in the presence of sodium dodecyl sulfate (96). However, the MUC1-ED can be untethered from the cell surface under physiologic conditions by additional proteolytic cleavage by proteases such as neutrophil elastase (97, 98), TNF-α converting enzyme (TACE) (99), matrix metalloprotease-14 (MMP-14) (100, 101), and γ-secretase (102). Finally, an extracellular form of the MUC1-ED, MUC1-SEC, can be produced through alternative splicing of the MUC1 mRNA to introduce a translation stop codon prior to its transmembrane region (103).
The 72-amino acid MUC1-CD contains 9 Ser, 6 Thr, and 7 Tyr residues as potential phosphorylation sites. Many of these residues are located within consensus sequence binding motifs for signaling kinases and adapter proteins. Among the most well-characterized of these are phosphatidylinositol 3-kinase (PI3K), Shc, phospholipase Cγ (PLCγ), protein kinase Cδ (PKCδ), glycogen synthase kinase-3β (GSK3β), c-Src, epidermal growth factor receptor (EGFR), β-catenin, and Grb-2 (104–107). The pattern of MUC1-CD Tyr phosphorylation is similar to that of some cytokine and growth factor receptors, but unlike cytokine/growth factor receptors, the MUC1-CD is not capable of autophosphorylation.
Carbohydrate analysis of MUC1 glycans reveals a high content of sialic acid (108, 109). Because MUC1 is the major membrane-bound mucin expressed in the airways and NEU1 is the major sialidase expressed in the respiratory tract (40), studies were performed to determine the molecular relationship between NEU1 and MUC1 in airway epithelial cells. By immunohistochemical staining, airway expression of NEU1 matches with that seen for MUC1 in these same tissues, i.e. at the superficial surface of airway epithelia, including the brush border of the trachea and bronchus, but not in subepithelial tissues (40, 110, 111). Overexpression of NEU1 in cultured human airway epithelial cells increased its association with MUC1 and decreased the sialic acid content of the MUC1-ED as assessed by lectin blotting with the sialic acid-reactive lectin, Maakia amurensis lectin (40). In contrast, NEU1 silencing in these cells had the opposite effects. Thus, the MUC1-ED is a substrate for NEU1. However, it is unknown whether NEU1 desialylates MUCs other than MUC1, and whether NEUs other than NEU1 desialylate MUC1.
NEU1 desialylates not only MUC1, but also EGFR (40). Because MUC1 forms a molecular complex with EGFR (112–114), and NEU1 plays a role in programmed cell death (115, 116), studies were performed to examine the relationship between NEU1, MUC1, and EGFR in autophagy (117). Immunohistochemically, human triple-negative breast cancer cells expressed high levels of EGFR, but low levels of MUC1 and NEU1. The levels of two autophagy pathway proteins, PI3K and beclin-1, were positively correlated with both NEU1 and MUC1-ED levels, while only PI3K correlated with the MUC1-CD. These results led to the authors to speculate that a NEU1 – MUC1 – EGFR axis is dysregulated in breast cancer cells exhibiting reduced autophagy.
KL-6
In 1988, a mouse monoclonal antibody, Krebs von den Lungen-6 (KL-6) (from the German “cancer from the lungs”) was described that was raised against human pulmonary adenocarcinoma cells (118). In addition to the immunizing cells, the KL-6 antibody reacted with normal human type 2 alveolar pneumocytes and bronchiolar epithelial cells, and with an unidentified high molecular weight mucin-like serum glycoprotein whose levels were increased in patients with lung adenocarcinoma. The epitope on the KL-6 antigen recognized by the KL-6 antibody was initially defined as a sialylated glycan because treatment of the purified antigen with sialidase diminished its reactivity with the cognate antibody (118). Carbohydrate analysis identified Gal, GalNAc, and Neu5Ac (sialic acid) in the KL-6 epitope (119). Subsequently, Ohyabu et al. (120) reported that the minimal antigenic structure of the KL-6 epitope consisted of the glycan, Neu5Acα2,3Galβ1,3GalNAcα, attached to the Thr residue in the peptide, Pro-Asp-Thr-Arg-Pro-Ala-Pro (Figure 2). This carbohydrate structure also is referred to as the 2,3-sialyl T antigen of the core 1-type O-glycan. More recent analysis suggests that the KL-6 antibody also recognizes a 6′-sulfo-Gal/GalNAc glycan epitope (121). Interestingly, the KL-6 epitope only is expressed in humans and apes and not other mammals (122).
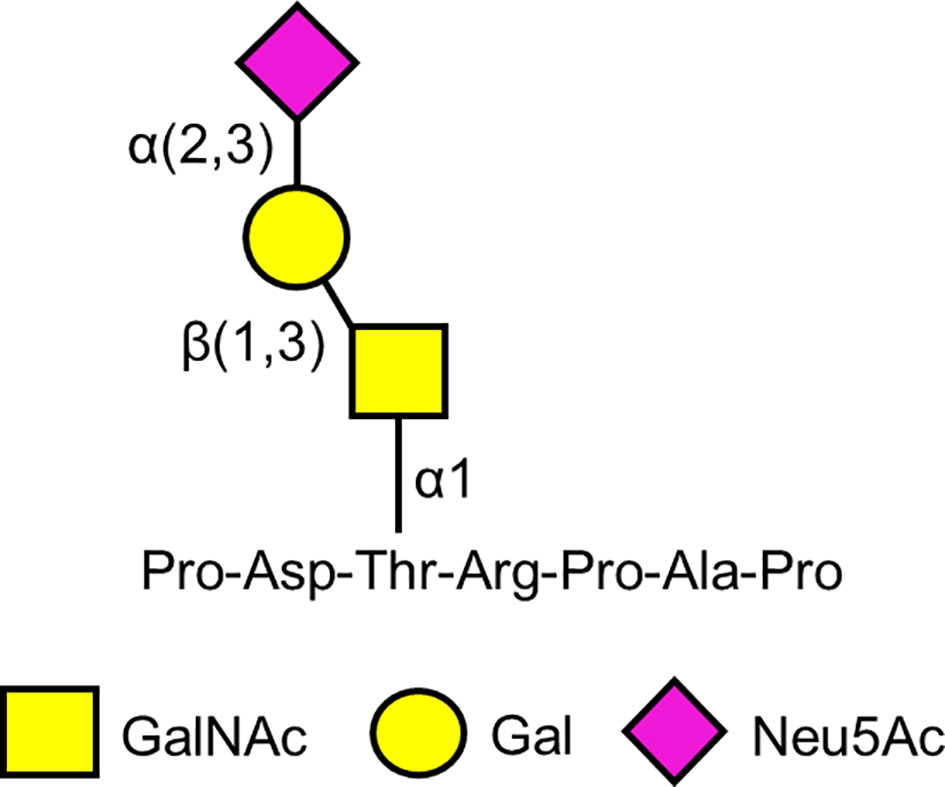
Figure 2 Proposed minimal antigenic structure of the KL-6 epitope based on Ohyabu et al. (120).
On the basis of flow cytometric and immunohistochemical studies, KL-6 was initially categorized as a Cluster 9/MUC1 antigen in the lung tumor and differentiation antigen classification system (123). Biochemical analysis of purified KL-6 established its identity with MUC1 (119). Amino acid analysis indicated a relatively high content of Pro (19.1 mol %), Thr (13.7%), and Ser (12.3%), while carbohydrate analysis revealed enrichment of Neu5Ac (30.2% per weight), Gal (18.9%), and GalNAc (11.9%). Most convincingly, in Western blot analysis the purified KL-6 glycoprotein reacted with the established MUC1 antibody, DF3. Because the KL-6 antibody reacts with the surface of type II pneumocytes and bronchiolar epithelial cells, and KL-6 is present in bronchoalveolar lavage fluid (BALF) and serum, the mechanism through which KL-6 is transported between the cell – BALF – blood compartments has been speculated (124, 125). While it is unclear how KL-6 is released from the cell surface into the alveolar fluid that is collected as BALF, the identification of two established MUC1-ED sheddases, MMP-14 and γ-secretase, in airway epithelial cells (51) suggests a proteolytic mechanism of KL-6 shedding.
NEUs and MUCs in Pulmonary Fibrosis
Exaggerated, often irreversible, and sometimes relentless scarring, known as fibrosis, can affect any organ or tissue. Excessive activation of fibroblasts, manifesting as their transdifferentiation into collagen-depositing, apoptosis-resistant, myofibroblasts underlies fibrosis. The ensuing replacement of the functional parenchyma with the scar tissue can become functionally and cosmetically debilitating in the skin or outright deadly when fibrosis develops in the lung, heart, kidney, or liver. There is no cure for fibrosis, and existing therapies do not consistently prevent fibrosis progression. The mechanistic involvement of NEUs in fibrosis has been suggested relatively recently, mostly in the lungs, where NEU activation promotes fibrosis (48, 52, 126–130); but also heart, with reports suggesting a profibrotic role for NEU1 (131) and an antifibrotic role for NEU3 (132). Mucins have also been suggested as mechanistic contributors to pulmonary fibrosis and, furthermore, NEUs and MUCs interplay mechanistically, as reviewed in detail below. Therefore, the following discussion focuses mostly on pulmonary fibrosis. The mechanisms of fibrosis are highly diverse, complex, redundant, and organ-specific, yet there are numerous unifying features across organ- and tissue-specific fibroses. In relevance to pulmonary fibrosis, these mechanisms include, but are not limited to, activation of transforming growth factor-β (TGF-β) (133, 134), changes in the levels of other cytokines regulating both inflammation and fibrosis (135–142), oxidative stress (143, 144), coagulation disturbances (145, 146), changes in biomechanical forces (147, 148), cellular senescence (149–155), defective autophagy (156, 157), and dysregulated epithelial cell – fibroblast crosstalk (158–161).
Pulmonary fibrosis is usually considered in the context of interstitial lung disease (ILD), an umbrella entity that comprises a large and diverse group of human disorders characterized by variously proportioned pulmonary inflammation and scarring. Idiopathic pulmonary fibrosis (IPF) represents the most severe, debilitating, and poorly understood ILD (162, 163). Connective tissue disease related ILD, especially in patients with scleroderma and rheumatoid arthritis, also poses a serious biomedical problem (164–166). Although the roles for NEU contributions to ILD have been suggested recently, early indications were observed in the past. Lambré et al. (167) first reported sialidase activity in the BALF from patients with IPF, while BALF from healthy subjects and serum from both IPF patients and controls had no such activity. Later, NEU1 mRNA and protein levels were shown to be increased in lung epithelial and endothelial cells, and fibroblasts, of IPF patients compared with healthy controls (128). In the same study, NEU1 overexpression in cultured lung fibroblasts increased the levels of collagen types I and III and MMP-14 degradation products, while NEU1 overexpression in mice increased lung collagen and TGF-β levels and promoted lung leukostasis of CD8+, and to a lesser extent CD4+, T cells. A later report from the same group demonstrated that selective pharmacological inhibition of NEU1 using C9-butyl-amide-2-deoxy-2,3-dehydro-N-acetylneuraminic acid (C9-BA-DANA), attenuated collagen accumulation in the lungs and pulmonary lymphocytosis in the acute and chronic bleomycin models of lung fibrosis (48). Taken together, these results indicated that NEU1 plays a central role in the pathogenesis of pulmonary fibrosis and NEU1-selective inhibition may offer a potential therapeutic intervention for pulmonary fibrotic diseases. Other studies have implicated a role for NEU3 in IPF pathogenesis (52, 126, 127, 129, 130).
NEU1 – MUC1 Axis in ILD
Kohno and colleagues (168) reported that serum levels of the KL-6 antigen positively correlated with disease activity in patients with interstitial pneumonitis, including IPF, hypersensitivity pneumonitis, and sarcoidosis. Elevated levels of KL-6 also were observed in the BALF of patients with ILD and there was a positive correlation between KL-6 serum and BALF levels (125, 169). Serum KL-6 levels were demonstrated to be proportionally higher in interstitial pneumonitis patients compared with other biomarkers of disease activity, such as type III procollagen N-terminal peptide (PIIIP) and type IV collagen 7S (7S collagen), while the latter two were more sensitive markers of alveolar pneumonia (170). In subsequent investigations, elevated serum levels of KL-6 were shown to be present in a greater proportion of patients with ILD compared with other reported disease biomarkers, including sialyl-Lewis a (sLea), sialyl-Lewis x (sLex), surfactant protein-A, surfactant protein-D, monocyte chemoattractant protein-1 (MCP-1), and C-C motif chemokine ligand 18 (CCL18) (171–174). In addition to its value as a diagnostic biomarker of ILD, KL-6 serum levels have been reported to be of prognostic value for predicting disease severity, acute exacerbations, and patient survival (125, 175, 176). Further, the value of KL-6 as a biomarker for ILD extends to diverse ethnic populations throughout the world, despite the fact that polymorphisms in the MUC1 gene affect serum levels of KL-6 (177–179). Based on these collective studies, KL-6 has been approved by the Japanese Health Insurance Program as a clinical diagnostic biomarker for ILD (124). Several relevant review articles can be consulted for further information on the diagnostic and prognostic value of KL-6 in IPF/ILD (125, 180–183).
KL-6 has been implicated in the pathogenesis of IPF. Purified KL-6 increased the chemotaxis of human lung and skin fibroblasts in vitro, which was augmented by fibronectin (119). KL-6 also promoted lung fibroblast proliferation and migration, and inhibited apoptosis, which were synergized by TGF-β (184). In a mouse model of bleomycin-induced pulmonary fibrosis, administration of anti-KL-6 antibody following bleomycin administration reduced the number of BALF inflammatory leukocytes, diminished the content of hydroxyproline in lung tissues, reduced the expression of collagen type I, TGF-β, and KL-6, increased hepatocyte growth factor (HGF) expression, and inhibited airway epithelial cell apoptosis (185). Bleomycin-induced lung fibrosis was decreased in MUC1-deficient mice, and in vitro silencing of MUC1 expression diminished fibroblast proliferation (186). In vitro cultures of lung fibroblasts and epithelial cells treated with the antifibrotic drug, pirfenidone, had reduced TGF-β-induced MUC1-CD phosphorylation at Thr-42 and Tyr-46, and decreased nuclear translocation of the MUC1-CD/SMAD3/β-catenin complex (187). MUC1-ED levels were increased in the BALF of bleomycin-challenged mice, and the NEU1-selective inhibitor, C9-BA-DANA, dose-dependently inhibited bleomycin-induced increases in MUC1-ED levels (48). Further, MUC1-ED in BALF of bleomycin-challenged mice was desialylated, and C9-BA-DANA reduced bleomycin-provoked MUC1-ED desialylation. Transgenic mice expressing human MUC1 had increased serum KL-6 levels following bleomycin challenge (122). Serum KL-6 levels were positively correlated with BALF albumin levels indicating that increases in serum KL-6 after bleomycin administration were associated with disruption of the alveolar-capillary barrier. In an experimental model of silica-induced pulmonary fibrosis using mice expressing the human MUC1 transgene, BALF and serum levels of MUC1 were increased compared with saline controls (188). Unexpectedly, however, MUC1 knockout mice had increased lung collagen deposition and pulmonary inflammation compared with wild type littermates. Collectively, these studies have led us to propose a NEU1 – MUC1 axis as a central component of the pathogenesis of pulmonary fibrotic diseases.
Several unanswered questions remaining concerning the roles of KL-6/MUC1 and NEU1 in the pathogenesis of IPF/ILD. 1) Do all secreted/extracellular forms of MUC1 that arise from different proteolytic cleavages (elastase, TACE, MMP-14, γ-secretase) and/or alternative mRNA splicing (MUC1/SEC) contribute to the appearance of KL-6? 2) Do genetic polymorphisms that dictate the number of MUC1-ED tandem repeats influence the development of IPF? 3) Are KL-6/MUC1 molecules in the serum and BALF of IPF patients desialylated to a similar extent? 4) Do pharmacologic inhibitors that target protease-mediated MUC1-ED shedding inhibit IPF development? 5) And ultimately, what is the molecular mechanism through which NEU1-induced, KL-6/MUC1 desialylation contributes to the profibrotic, proinflammatory phenotype that characterizes IPF?
Increased KL-6 levels in BALF of ILD patients likely reflects the presence of high glycoprotein levels on the surface of regenerating type II pneumocytes (168, 189). Upregulation of MUC1-ED sheddases in these cells during disease manifestation remains an alternative possibility, and elevated levels of MMP-14 have been described in pulmonary fibrosis (190, 191). High KL-6 levels in serum also may be causally related to increased regenerating pneumocytes and/or greater alveolar capillary permeability to proteins following disruption of the alveolar-capillary barrier (124).
Mucins as Substrates for NEU2, NEU3, and NEU4
NEU2 mRNA and sialidase activity were detected in the acellular mucin-enriched capsule of the equine pre-implantation conceptus (192). Recombinant human NEU2, expressed in Escherichia coli and purified to homogeneity, exhibited sialidase activity against bovine submaxillary gland mucin (BSM) (193). Although the particular mucin expressed by the bovine submaxillary gland has yet to be fully characterized, cDNA sequence analysis shows it to be highly similar to MUC19 (194–196). A human NEU4 cDNA, expressed in monkey COS-7 cells, desialylated BSM to a greater extent compared with other NEU substrates, including 4-MU-NANA, sialyllactose, and mixed bovine gangliosides (197). By contrast, while recombinant NEU3 desialylated BSM to a greater extent than 4-MU-NANA, BSM was an inferior NEU3 substrate compared with sialyllactose and bovine gangliosides. Recombinant cDNAs of both the short and long isoforms of human NEU4 (NEU4-S and NEU4-L), expressed in COS-7 cells, also exhibited in vitro sialidase activity against BSM (198). NEU4-S expressed in human embryonic kidney 293T (HEK293T) cells, but not NEU1, NEU2, or NEU3, desialylated the sLea mucin-expressing antigen (79). NEU4-S also exhibited enzymatic activity against the sLex antigen, while NEU3 was moderately active and NEU1 and NEU2 were unreactive against sLex. Compared with untreated controls, treatment of human THP-1 macrophages with the phytochemical, thymoquinone, stimulated an increase in endogenous NEU4 activity that exhibited sialidase activity against BSM (80, 199). Therefore, in addition to the established role of NEU1 in desialylating MUC1, as discussed previously, other NEUs are capable of desialylating other MUCs. Several such mucins—potential desialylation targets of mammalian NEUs—have been suggested or demonstrated to play a role in ILD. A specific single nucleotide polymorphic (SNP) MUC2 gene variant was significantly associated with IPF (200), and the expression levels of MUC2 protein were decreased in patients with IPF (201). Similarly, a specific SNP variant of the MUC5AC gene was associated with IPF (202). However, the expression of MUC5AC was reported to be either increased (203) or decreased (201, 204) in patients with pulmonary fibrosis. In a mouse model, overexpression of MUC5AC protected against inflammation and fibrosis through several mechanisms (205). These apparent discrepant results might be explained, in part, by differences in disease etiologies, patient demographics, and/or lung sampling techniques between the various studies (201, 203, 204). The expression levels of MUC4 and MUC16 were elevated in patients with IPF, with evidence for each MUC4 and MUC16 promoting the fibrotic process in collaboration with TGF-β (206–209). Of particular prominence is a strong association of a gain-of-function MUC5B gene promoter variant (rs35705950) with pulmonary fibrosis (210–215). To date, the MUC5B rs35705950 SNP remains the strongest and most reproducible risk factor for development of IPF. It should be expected that specific details of the interplay between NEU isozymes and these MUCs might transpire in the future.
NEUs and MUCs in Infectious Diseases
NEU1 and MUC1 in Pseudomonas aeruginosa Infection
P. aeruginosa is a Gram-negative, motile, opportunistic pathogen responsible for a wide range of human infections (216). In the respiratory tract, P. aeruginosa contributes to the morbidity and mortality of cystic fibrosis, chronic obstructive pulmonary disease, and bronchiectasis. P. aeruginosa bacteria isolated from the airway secretions of cystic fibrosis patients are tightly bound to mucins (217–219). In in vitro cell adhesion assays, the MUC1-ED bound to P. aeruginosa through bacterial flagellin, the major structural protein of the flagellar filament (220, 221). Flagellin binding to the MUC1-ED stimulates two distinct molecular events, Tyr phosphorylation of the MUC1-CD leading to activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signal transduction pathway and NEU1-catalyzed MUC1-ED shedding from the cell surface as a soluble decoy receptor (40, 49, 51, 222). MUC1-CD diminishes the host inflammatory response to P. aeruginosa lung infection through a mechanism involving EGFR-catalyzed Tyr phosphorylation of the MUC1-CD and inhibition of TLR signaling (104, 105, 107, 112, 223–229). As discussed above, EGFR and TLRs are established NEU1 substrates, but it is unclear to what extent NEU1-mediated desialylation of EGFR and/or TLRs impacts P. aeruginosa-initiated, MUC1-CD-inhibited airway inflammation.
Overexpression of NEU1 in cultured human airway epithelial cells increased P. aeruginosa adhesion to MUC1-expressing airway epithelia, and promoted MUC1-ED desialylation and flagellin-stimulated, MUC1-dependent ERK1/2 phosphorylation, while NEU1 silencing had the opposite effects (40). To extend these results to a physiologically relevant context, additional studies were performed to assess whether P. aeruginosa flagellin, the MUC1 ligand, might regulate NEU1-mediated MUC1-ED desialylation and/or P. aeruginosa adhesion to airway epithelial cells (51). NEU1 overexpression in cultured human airway epithelial cells increased MUC1-dependent adhesion of flagellin-expressing P. aeruginosa to the cells, but not the adhesion of a flagellin-deficient P. aeruginosa isogenic mutant (fliC¯). Treatment of these cells with purified P. aeruginosa flagellin dose-dependently increased bacterial adhesion, which was abolished by NEU1 silencing. In coimmunoprecipitation assays, P. aeruginosa flagellin increased MUC1 association with both NEU1 and the NEU1 chaperone, PPCA, in a dose-dependent manner. Airway epithelial cells treated with P. aeruginosa flagellin also dose-dependently increased MUC1-ED desialylation, which was abrogated by NEU1 knockdown. Because sialic acid residues can mask protease recognition sites (230), additional studies were conducted to assess the role of NEU1 and P. aeruginosa flagellin in MUC1-ED shedding from the cell surface (51). NEU1 overexpression and flagellin stimulation dose-dependently increased shedding of desialylated MUC1-ED into the supernatants of cultured airway epithelial cells. Moreover, the desialylated MUC1-ED that was shed in response to flagellin stimulation decreased P. aeruginosa adhesion to airway epithelial cell-associated MUC1-ED. Thus, the MUC1 ligand, P. aeruginosa flagellin, increased MUC1 association with NEU1, leading to desialylation and shedding of the MUC1-ED from the airway epithelial cell surface, and the shed MUC1-ED competitively blocked P. aeruginosa adhesion to MUC1-ED residing on the cell surface.
Using an intact, physiologically relevant, mouse model of P. aeruginosa pneumonia, further studies were performed to assess whether P. aeruginosa-derived flagellin might stimulate NEU1-dependent MUC1-ED desialylation and shedding (49). Mice administered intranasally with viable bacteria or purified flagellin exhibited both dose- and time-dependent increases in shed MUC1-ED in their BALF. No increases in BALF levels of MUC1-ED were evident in mice administered with the flagellin-deficient P. aeruginosa fliC¯ mutant strain. P. aeruginosa lung infection of mice also increased NEU1-MUC1 and PPCA-MUC1 association and MUC1-ED desialylation, and inhibition of NEU1 activity in vivo with the NEU1-selective inhibitor, C9-BA-DANA (231), abrogated both of these effects. Shed, desialylated MUC1-ED purified from the BALF of P. aeruginosa-infected mice diminished both flagellin and bacterial binding to cultured airway epithelial cells, and a similar effect was observed using a nonglycosylated recombinant MUC1-ED protein expressed in E. coli. Co-administration of the recombinant MUC1-ED protein with P. aeruginosa to mice also decreased bacterial lung burden, diminished BALF levels of keratinocyte-derived chemokine (KC) and tumor necrosis factor-α (TNF-α), inhibited pulmonary leukostasis, and increased 5-day mouse survival from 0% to 75%.
To extend these results to an in vivo human setting, studies were conducted to determine whether shed MUC1-ED levels might be elevated in BALF of patients with P. aeruginosa lung infection, and whether the recombinant MUC1-ED protein might influence in vitro parameters of the host proinflammatory response to bacterial infection (232). BALF from patients with P. aeruginosa pneumonia contained higher levels of MUC1-ED, compared with either BALF from noninfected patients or patients infected with microorganisms other than P. aeruginosa, and shed MUC1-ED in the BALF of P. aeruginosa-infected patients was desialylated, presumably from the action of NEU1. P. aeruginosa-derived flagellin also was detected in the BALF of P. aeruginosa-infected patients, and a positive correlation existed between BALF levels of MUC1-ED and flagellin. Finally, E. coli-expressed recombinant MUC1-ED dose-dependently decreased P. aeruginosa motility and biofilm formation, diminished P. aeruginosa-stimulated interleukin-8 (IL-8) production by airway epithelial cells, and promoted neutrophil-mediated bacterial phagocytosis. Taken together, these results indicate that not only do BALF levels of shed MUC1-ED provide a diagnostic biomarker of P. aeruginosa lung infection, but that NEU1-mediated desialylation of the MUC1-ED generates a flagellin-targeting releasable decoy that provides a protective component of the proinflammatory response to P. aeruginosa at the human airway epithelial surface (Figure 3).
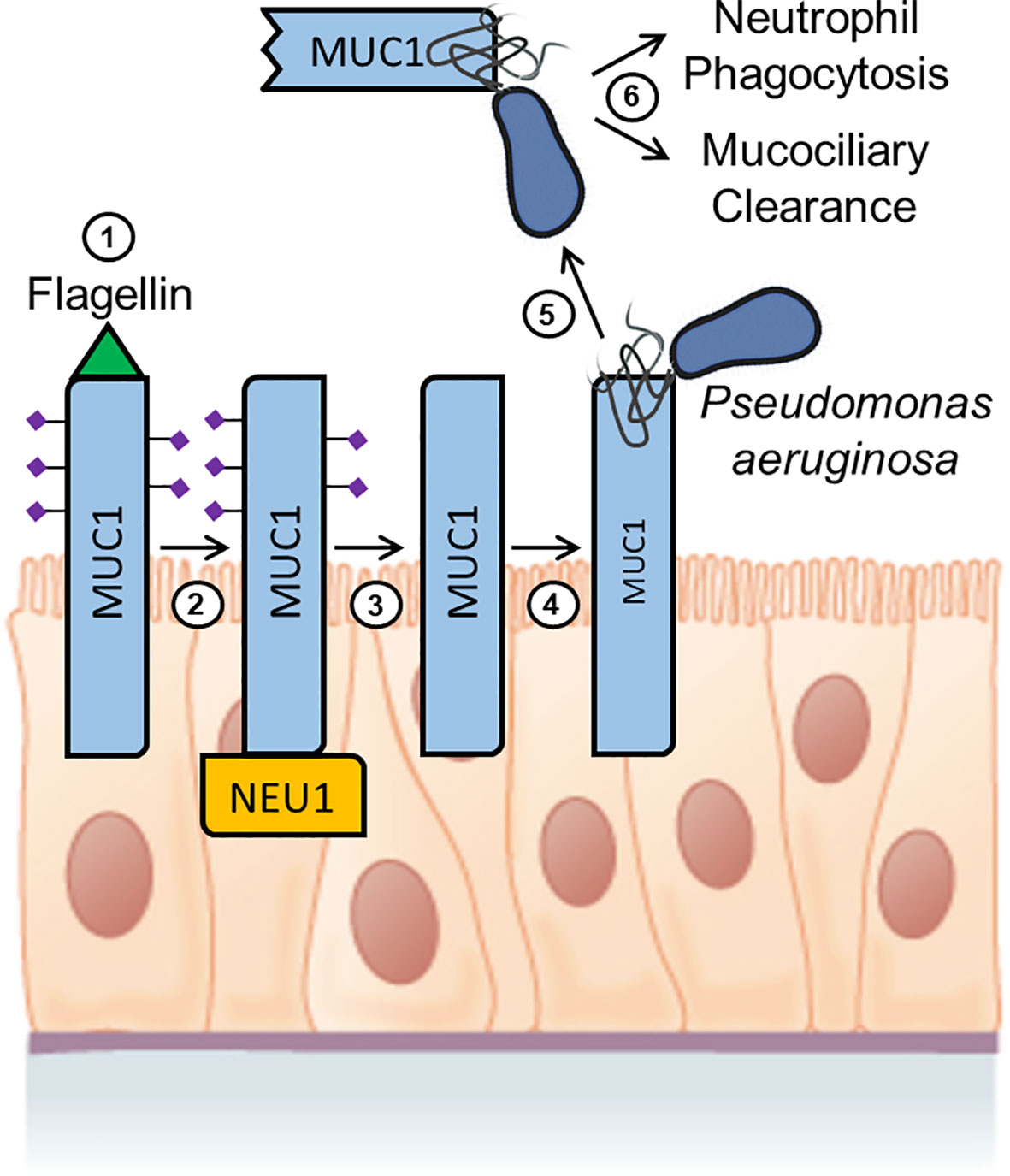
Figure 3 Hypothetical model for Pseudomonas aeruginosa flagellin-induced, NEU1-mediated MUC1-ED desialylation and shedding. Step 1. P. aeruginosa flagellin engages cell-associated MUC1-ED in the airway epithelium. Step 2. NEU1 is recruited to the MUC1-CD. Step 3. NEU1 desialylates the MUC1-ED. Step 4. Desialylated, cell-associated MUC1-ED binds to P. aeruginosa through its flagella. Step 5. The P. aeruginosa-MUC1-ED complex is proteolytically released from the cell surface. Step 6. The P. aeruginosa-MUC1-ED complex is removed from the lungs by the combined actions of neutrophil phagocytosis and mucociliary clearance. Image of the pseudostratified columnar epithelium from Anatomy and Physiology, Chapter 4: The Tissue Level of Organization, Section 4.2: Epithelial Tissue by OpenStax College, Rice University (https://openstax.org/details/books/anatomy-and-physiology) and used under Creative Commons Attribution 4.0 International (CC BY) license/Modified from the original.
In subsequent studies, coimmunoprecipitation and glutathione-S-transferase (GST) pull down assays were performed to identify the biochemical mechanism through which NEU1 associates with MUC1 (233). Unexpectedly, NEU1 associated with the MUC1-CD, but not with the sialic acid-containing MUC1-ED. Inhibition of NEU1 enzymatic activity using C9-BA-DANA did not affect NEU1-MUC1-CD association. The NEU1 binding site was localized to the membrane-proximal 36 amino acids of the MUC1-CD, while the MUC1-CD binding site was mapped to NEU1 amino acids 1-139. NEU1-MUC1-CD association was direct and did not require the NEU1 chaperone protein, PPCA. NEU1-mediated MUC1-ED desialylation in response to P. aeruginosa flagellin was equivalent in cells either expressing or not expressing the MUC1-CD, indicating that NEU1-MUC1-CD interaction was not required for MUC1-ED desialylation. Finally, overexpression of NEU1 in airway epithelial cells reduced MUC1-CD association with PI3K and diminished downstream Akt phosphorylation. These results indicate that NEU1 associates with the juxtamembrane region of the MUC1-CD to inhibit PI3K-Akt signaling.
Several unanswered questions remain concerning the NEU1 – MUC1 axis. 1) What is the molecular mechanism through which engagement of P. aeruginosa flagellin by the MUC1-ED leads to recruitment of NEU1? 2) Do other flagellated respiratory pathogens other than P. aeruginosa activate the flagellin – MUC1 – NEU1 axis? 3) Can the E. coli-expressed recombinant MUC1-ED offer a clinically useful therapeutic intervention to treat P. aeruginosa lung infections either alone or in combination with anti-pseudomonal antibiotics? 4) Can measurement of MUC1-ED and/or flagellin levels in BALF be developed into a dependable and rapid diagnostic assay to identify patients with P. aeruginosa lung infections without the need for bacterial culture or genotyping techniques?
NEUs and MUCs in Other Bacterial Infections
In mouse models of E. coli-induced sepsis or S. enterica serovar Typhimurium-induced colitis, or in mice administered intraperitoneally with E. coli lipopolysaccharide (LPS), circulating levels of both NEU1 and NEU3 were increased compared with saline controls (234). No increases in NEU1 or NEU3 were observed in TLR4 knockout mice. Streptococcus pneumoniae-induced murine sepsis also increased serum NEU1 and NEU3 levels, but these increases were similar in both wild type and TLR4-deficient mice, suggesting an effect of the bacterial-encoded neuraminidase, NanA. Mechanistic studies revealed that increased NEU1/NEU3 levels promoted desialylation and clearance of alkaline phosphatase by the Ashwell-Morell receptor, thereby decreasing LPS dephosphorylation and increasing TLR4-dependent host inflammatory responses. In other studies, repeated gastric administration of S. enterica Typhimurium or LPS increased intestinal sialidase activity and NEU3 mRNA and protein levels in the gut in a TLR4-dependent manner with a corresponding increase in alkaline phosphatase desialylation and decrease phosphatase activity (235). Oral administration of zanamivir (Relenza), an influenza virus NEU inhibitor, blocked the bacterial-induced increased intestinal NEU activity, maintained alkaline phosphatase sialylation, and prevented disease. No increases in intestinal sialidase activity, NEU expression, or alkaline phosphatase desialylation were seen following gastric administration of S. enterica Typhimurium to NEU3 knockout mice (236). NEU3-deficient, S. enterica Typhimurium-infected mice also demonstrated decreases in LPS phosphorylation, intestinal levels of proinflammatory cytokines, and gut infiltration of CD3+ T cells, Gr1+ neutrophils, and F4/80+ monocyte/macrophages, and protected against the development of bacterial colitis. The NEU inhibitor oseltamivir (Tamiflu), originally developed against influenza virus NEU, but also active against NEU1 (41, 237–239), reduced murine lung neutrophil infiltration, IL-17 and TNF-α levels in BALF and serum, and mortality in both experimental E. coli sepsis and the cecal ligation puncture model of sepsis (240).
MUC1 serves as a cell surface receptor for Helicobacter pylori in gastric epithelia (100, 241–246). Here, the MUC1-ED is released as a soluble decoy receptor to limit H. pylori infection (100, 247, 248), but the role of NEU1 in H. pylori-stimulated MUC1-ED shedding is unclear. MUC1 expression downregulates H. pylori-driven gastric inflammation (244, 249–256). MUC1 expression also influences a variety of other human bacterial infections, including those by E. coli, S. enterica Typhimurium, S. pneumoniae, Staphylococcus aureus, Bacillus subtilis, Campylobacter jejuni, and Haemophilus influenzae (257–267). Based on these studies and the documented role of the NEU1 – MUC1 axis in regulating airway inflammation in response to P. aeruginosa (40, 49, 51, 232), it is likely that this same pathway impacts the interaction of a broad array of pathogenic microorganisms with the human host. In summary, these collective studies suggest that NEUs and MUCs play important roles in regulating bacterial infections.
NEUs and MUCs in Viral Infections
DNA methylation analysis of primary nasal epithelial cells from children with or without asthma implicated NEU1 in the host immune response to human rhinovirus-16 infection (268). Compared with non-asthmatic controls, NEU1 mRNA levels were decreased in asthma subjects with rhinovirus-16 infection, and a positive correlation between NEU1 gene methylation and NEU1 transcript levels was observed. The hepatitis B virus core protein (HBc) increased NEU1 gene promoter activity through NF-κB binding sites, leading to activation of ERK1/2 and NF-κB signaling pathways, and increased HBc-mediated migration and proliferation of hepatoma cells (269). Knockdown of NEU2 expression in monkey Vero or human HEp-2 cells increased hepatitis A virus replication by up to 200-fold, however the mechanism was not elucidated (270). Overexpression of mouse NEU3 in COS-7 cells diminished Newcastle Disease Virus (NDV) infection and propagation through cell-cell fusion (271). Because reduced cell surface levels of GD1a also were decreased upon NEU3 overexpression, this ganglioside was suggested to constitute an NDV receptor on host cells. The expression of NEU1, but not NEU3 or NEU4, was increased in upper and lower respiratory tract cells, and in lung infiltrating neutrophils, of SARS-CoV-2-infected patients (240). Interestingly, two recent reports have documented increased MUC1 protein levels in the airways of SARS-Cov-2-infected patients compared with healthy controls (272, 273). MUC1 also has been shown to play a critical role in several other virus infections, including those by respiratory syncytial virus, adenovirus, influenza virus, and human immunodeficiency virus (274–280). Future studies are needed to elucidate the role of NEUs, if any, in regulating the MUC1-dependent responses to virus infections. In conclusion, these investigations reveal that a variety of human and animal virus infections are impacted by the expression and activity of NEU1, NEU2, NEU3, and MUC1.
NEU1 in Parasite Infections
In vitro infection of mouse J774.A1 macrophage cells with the obligate intracellular protozoan parasite, Leishmania donovani, reduced cell surface expression of NEU1 compared with uninfected controls (55). Parasite infection enhanced TLR4 sialylation and reduced TLR4-NEU1 and TLR4-MyD88 association. NEU1 overexpression in L. donovani-infected murine macrophages increased TLR4 desialylation, TLR4-NEU1 and TLR4-MyD88 association, activation of the JNK, ERK1/2, p38, and NF-κB pathways, and expression of IL-12, interferon-γ (IFN-γ), and inducible nitric oxide synthase (NOS) proinflammatory mediators, while decreasing expression of IL-4, IL-10, and TGF-β and reducing parasite burden, all compared with mock-transfected cells. In other studies, NEU1 overexpression in L. donovani-infected mouse macrophages reduced TLR4 ubiquitination and TLR4-siglec-E and siglec-E-SHP1 association, but increased IL-1β, IL-6, and TNF-α levels compared with nontransfected cells (281).
NEUs in Innate and Adaptive Immunity
NEUs and TLRs
Toll-like receptors (TLRs) are cell surface sialoglycoproteins that recognize broadly-distributed pathogen-encoded molecules to initiate MyD88- and TRIF-dependent intracellular signaling pathways that activate host innate immune responses (282). It is now established that sialidases, principally NEU1, regulate TLR-dependent innate immunity (Table 2).
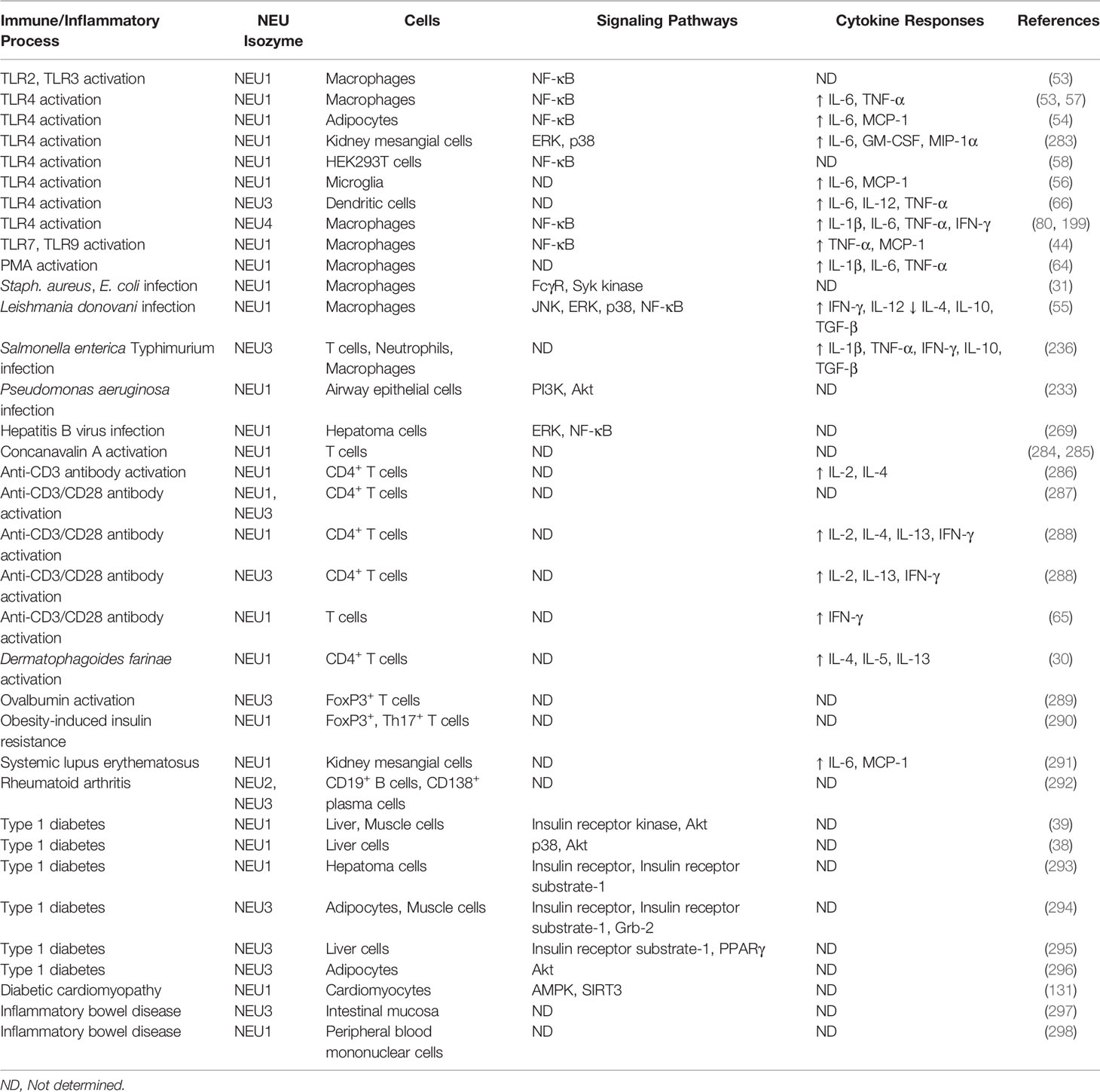
Table 2 NEU-associated cells, signaling pathways, and cytokine responses for selected immune/inflammatory processes.
TLR ligands, including LPS, poly(I:C), Mycobacterium butyricum, and zymosan, induced sialidase activity in mouse BMC-2 and primary bone marrow macrophages that was associated with NF-κB activation and the production of nitric oxide, IL-6, and TNF-α (53). In these same studies, NEU1, but not NEU2, NEU3, or NEU4, colocalized with TLR2, TLR3, and TLR4 on the cell surface of unstimulated mouse macrophages, which increased following stimulation with zymosan, poly(I:C), or LPS, respectively. NEU1 knockout mice had reduced granulocyte-colony stimulating factor (G-CSF), IL-1Rα, IL-6, KC, and macrophage inflammatory protein-2 (MIP-2) production in response to LPS administration compared with NEU1-expressing mice. In other studies, NEU1- and NEU4-deficient macrophages had reduced LPS-induced TLR4-MyD88 complex formation and NF-κB activation compared with wild type littermates (57, 80, 199). Following LPS stimulation of mouse dendritic cells (DCs), NEU1 translocated from the lysosome to the cell surface where it associated with and desialylated TLR4, and NEU1 silencing diminished LPS-stimulated TNF-α production (70). In these same studies, NEU1 deficiency in murine hematopoietic cells or in vivo treatment with the sialidase pharmacologic inhibitor, 2-deoxy-2,3-dehydro-N-glycolylneuraminic acid (Neu5Gc2en), protected mice against lethal LPS challenge. NEU1 silencing in mouse 3T3-L1 adipocytes increased TLR4 sialylation and decreased LPS-stimulated NF-κB nuclear translocation and IL-6 and monocyte chemoattractant protein-1 (MCP-1) production (54). NEU1 pharmacologic inhibition with oseltamivir decreased LPS-stimulated IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and MIP-1α production in mouse primary renal mesangial cells through a TLR4 → p38/ERK pathway (283). Treatment of mouse RAW264.7 macrophages with LPS decreased NEU1 levels on the cell surface, and NEU1 silencing promoted LPS-induced tolerance, while NEU1 overexpression had the opposite effect (73). Finally, treatment of RAW264.7 cells with TLR7 or TLR9 agonists promoted TLR7-NEU1 and TLR9-NEU1 association, NF-κB activation, and TNF-α and MCP-1 production, all of which were reversed by NEU1 silencing or pharmacologic inhibition with oseltamivir (44).
In human studies, NEU4 was identified as the strongest locus associated with TLR4 stimulation in a genetic analysis comparing LPS-treated primary monocytes with untreated cells (299). Overexpression of NEU1, but not NEU3, in HEK293T cells dose-dependently increased LPS-stimulated, TLR4-mediated NF-κB activation and treatment with the broad spectrum sialidase inhibitor, 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (Neu5Ac2en or 2-deoxy-NANA) reduced NF-κB activation (58). Mechanistically, it was proposed that NEU1-mediated desialylation of TLR4 unmasked TLR dimerization sites to promote receptor activation and initiation of the host proinflammatory response (53, 57). Additional studies revealed that NEU1, MMP-9, and G protein-coupled receptors (GPCRs) formed a multiprotein complex on the cell surface wherein GPCR agonists activated GPCR signaling to stimulate MMP-9-dependent NEU1 activation through removal of the elastin-binding protein, thereby allowing for TLR desialylation and initiation of host inflammation (44, 45, 47). Further investigations revealed NEU1 – MMP-9 cross-talk in combination with the GPCR agonist, neuromedin B, as necessary for EGFR activation and downstream intracellular signaling (41). LPS activation of mouse microglial BV-2 cells and rat primary microglia increased cell surface sialidase activity and desialylation of the cell surface (300, 301), which was abolished by NEU1 knockdown (56). NEU1 knockdown in microglia also reduced LPS-stimulated IL-6 and MCP-1 expression, while NEU1 overexpression had the opposite effects (56). Using a cell surface biotinylation assay, NEU1 and TLR4 were localized in close proximity (within 100 nm) on the cell surface of LPS-activated BV-2 cells, and LPS treatment of the cells increased TLR4 desialylation. Collectively, studies in both human and mouse experimental systems indicate that NEU1, and to a lesser extent NEU4, regulate TLR-driven intracellular signaling and host innate immunity.
NEUs and PMNs
Polymorphonuclear leukocytes (PMNs) are blood-borne leukocytes that play a critical role in the host innate immune response to infectious pathogens through microbial phagocytosis, release of soluble anti-microbial compounds, and generation of neutrophil extracellular traps (NETs) (302). Sialidase activity was identified in the primary and secondary granules of human PMNs as well as on the plasma membrane, and PMN activation induced translocation of the enzymatically-active sialidase from the granules to the cell surface (303). In addition, PMN homotypic aggregation and adhesion to nylon or plastic surfaces was inhibited by 2-deoxy-NANA. In other studies, a rabbit antibody prepared against the Clostridium perfringens NEU cross-reacted with a sialidase expressed on the surface of IL-8-stimulated human PMNs, but not on unstimulated cells, and with sialidase proteins in PMN lysates and granule preparations (304). This same antibody inhibited human PMN sialidase activity in vitro and diminished both pulmonary leukostasis in mice administered with cobra venom factor and intrapulmonary transendothelial migration of PMNs into the bronchoalveolar compartment of IL8-administered mice. In subsequent experiments, the sialidase antibody was shown to recognize recombinant human NEU3, but not NEU1, and inhibited NEU3 sialidase activity against the synthetic fluorogenic substrate, 4-MU-NANA (305). In other studies, NEU1, NEU2, and NEU3 mRNAs were detected in unstimulated murine PMNs and IL-1β stimulation of human PMNs increased NEU1 and NEU2, but not NEU3, expression (288). LPS-stimulated human PMNs had decreased amounts of sialic acid on their surface that was inhibited by oseltamivir and zanamivir (240). Both NEU1 inhibitors also reduced PMN-mediated phagocytosis and killing of E. coli, LPS-driven PMN activation, phorbol 12-myristate 13-acetate (PMA)-stimulated reactive oxygen species production, and LPS-mediated NET formation.
Utilizing the NEU3 cross-reactive antibody, Sakarya et al. (306) reported reduced, antibody-dependent PMN adhesion to pulmonary vascular endothelial cell monolayers in vitro, which was associated with PMN activation. Preincubation of endothelial cell monolayers with activated PMNs rendered the cells hyperadhesive for freshly added PMNs via PMN sialidase-dependent autodesialylation. Desialylation of both the CD11b and CD18 protein subunits of β2-integrin, through mobilization of an endogenous PMN sialidase, unmasked activation epitopes on both proteins and increased binding to intracellular adhesion molecule-1 (ICAM-1) (21). NEU1 was expressed on the surface of PMNs and colocalized with CD18, the latter being increased following PMN activation. Desialylation of immobilized ICAM-1 increased leukocyte arrest in vivo and treatment with 2-deoxy-NANA diminished LPS-stimulated leukocyte adhesion to ICAM-1. Taken together, these results indicate that NEU1, NEU2, and NEU3 regulate the activities of PMNs.
NEUs and Monocytes, Macrophages, and Dendritic Cells
Macrophages isolated from NEU1 knockout mice exhibited reduced phagocytosis of Gram positive (S. aureus) and Gram negative (E. coli) bacteria and phosphorylation of FcγR and Syk kinase, and treatment of cultured NEU1-deficient macrophages with purified mouse NEU1 restored their phagocytic activity (31). NEU1-knockout macrophages displayed increased lysosomal exocytosis as a consequence of reduced NEU1-mediated desialylation of lysosomal-associated membrane protein-1 (LAMP-1) (34). High levels of NEU1 mRNA were detected in unstimulated human monocytes, while NEU2 and NEU3 were expressed at much lower levels (288). In vitro stimulation of human monocytes with GM-CSF to promote differentiation into macrophages temporally increased sialidase activity for the 4-MU-NANA substrate up to 14-fold after 7 days in culture (307). Correspondingly, NEU1 and NEU3, but not NEU4, mRNAs and proteins were increased following monocyte-to-macrophage differentiation. Following PMA-stimulated differentiation of human primary monocytes and the THP-1 monocyte cell line to macrophages, NEU1, but not NEU3, protein levels increased and NEU1 translocated from the lysosomes to the plasma membrane (64). NEU1 silencing reduced E. coli phagocytosis by PMA-induced macrophages and diminished their production of IL-1β, IL-6, and TNF-α. Differentiation of human monocytes into DCs following in vitro culture in the presence of GM-CSF and IL-4 increased sialidase activity and elevated the mRNA and protein levels of NEU1 and NEU3, but not NEU4 (66). Further, monocyte-to-DC differentiation in the presence of zanamivir or 2-deoxy-NANA to inhibit NEU1 and NEU3 activities decreased LPS-stimulated IL-6, IL-12, and TNF-α production. Overexpression of NEU1 in THP-1 macrophages increased IL-1β and TNF-α production while NEU1 silencing had the opposite effect (308). In these same studies, NEU1 silencing in human primary M1 macrophages suppressed IL-1β and TNF-α production. PMA-induced differentiation of THP-1 cells to M0 macrophages increased intracellular sialidase activity and diminished sialoglycoconjugate levels on the cell surface (308). However, further polarization of M0 macrophages into M1 or M2 macrophages did not affect the amount of sialic acid on the cell surface.
NEU1 expression was increased in human circulating monocytes isolated from patients with myocardial infarction compared with healthy controls, and high NEU1 levels were observed in macrophages isolated from atherosclerotic plaques (308). In an animal model of atherosclerosis, NEU1-deficient mice exhibited reduced monocyte and lymphocyte infiltration into atherosclerotic lesions compared with NEU1-sufficient mice (309). Reductions in serum and liver nonhigh-density lipoprotein (nonHDL) cholesterol levels, atherosclerotic lesion size, and lesion macrophage numbers were observed in apolipoprotein E (ApoE) knockout mice expressing hypomorphic levels of NEU1 (NEUhypo) compared with ApoE-/- littermates (310). NEU1hypo ApoE knockout mice also had reduced circulating PMNs, but increased numbers of M2 macrophages, compared with ApoE knockout mice expressing normal NEU1 levels. Finally, treatment of ApoE-/- mice with the general sialidase inhibitor, 2-deoxy-NANA, reduced liver nonHDL cholesterol levels and atherosclerotic lesion size, compared with saline controls. In another study, recombinant human NEU1 and NEU3 desialylated apolipoprotein B 100 in human low-density lipoprotein (LDL) in vitro, and ApoE-NEU1 and ApoE-NEU3 double knockout mice, or ApoE knockout mice treated with pharmacologic inhibitors of NEU1 or NEU3, exhibited reduced atherosclerosis compared with ApoE single knockout mice or ApoE null mice treated with vehicle control (18). In a mouse model of ischemia/reperfusion (I/R) cardiac injury, elevated sialidase activity, and NEU1, PPCA, and β-galactosidase mRNA and protein levels, were seen in the heart compared with sham controls (311). These authors also reported that NEUhypo mice subjected to I/R injury had diminished proinflammatory and increased anti-inflammatory macrophage numbers compared with controls. Further, cardiomyocyte-specific NEU1 overexpression in vivo was associated with cardiac hypertrophy, but no changes in cardiac inflammation, following I/R injury compared with wild type littermates. Binding of pro-atherogenic elastin-derived peptides (EDP) to the elastin receptor complex (ERC), a heterotrimeric association of NEU1, PPCA, and the elastin binding protein, a splice variant of β-galactosidase (312), led to NEU1-mediated desialylation of the CD36 scavenger receptor and augmented uptake of oxidized LDL by human macrophages in vitro (22). In a subsequent report, EDP binding to the ERC also promoted NEU1-β2-integrin association and NEU1-driven β2-integrin desialylation in human monocytes, as well as NEU1-ICAM-1 association and NEU1-mediated ICAM-1 desialylation in human umbilical vein endothelial cells (23). As a result, EDP binding to the ERC promoted monocyte adhesion to and migration across endothelial cell monolayers through NEU1. In conclusion, these studies strongly suggest that NEU1 regulates monocyte, macrophage, and DC differentiation and function (Table 2).
NEUs and Lymphocytes
Landolfi and coworkers (284) reported that mouse T lymphocytes possess an endogenous sialidase activity that increased following in vitro stimulation with the T cell mitogen, concanavalin A, and sialidase activity was controlled by the NEU1 locus in the murine major histocompatibility complex. Mouse strains carrying the H-2v haplotype (SM/J, B10.SM) exhibited reduced sialidase activity compared with other haplotypes. Previously, the SM/J mouse strain had been reported to possess deficient sialidase activity (313). Subsequent analysis revealed that a point mutation in the NEU1 gene, resulting in a Leu-to-Ile substitution, was responsible for the deficient sialidase activity in the SM/J stain (314). Increased sialidase activity following T cell activation was associated with desialylation of cell surface glycoproteins (285). Mouse T cell activation through a mixed lymphocyte reaction (MLR) with allogeneic B cells also resulted in increased sialidase activity and desialylation of T cell surface glycoproteins (315). Wipfler et al. (316) reported that total sialidase activity was greatest in unstimulated human peripheral blood lymphocytes (PBLs), intermediate in thymic T cells, and lowest in tonsillar T and B cells. At the mRNA level, NEU1 was expressed at approximately 2.5-fold greater levels in PBLs compared with thymocytes and tonsillar T and B cells, while NEU3 was expressed at up to 125-fold greater levels in thymic T cells compared with PBLs and tonsillar T and B cells. In a different study, NEU1 and NEU3 transcripts were identified in murine thymic T cells (20). Crude membrane fractions of mouse thymocytes had sialidase activity that was inhibited by 2-deoxy-NANA and the NEU1-specific inhibitor, C9-BA-DANA. CD5 was identified as a substrate for thymocyte cell surface NEU1 by peanut agglutinin (PNA) lectin blotting and anti-CD5 antibody immunoprecipitation studies. A subpopulation of murine thymic lymphocytes was identified that co-expressed surface IgG, β2-integrin, and NEU1 (Neu-medullocytes) (317, 318). Real-time PCR studies revealed that NEU2 was the major sialidase expressed in Neu-medullocytes with NEU1 detected at low levels (319). Of five NEU2 mRNAs arising as a result of alternative mRNA splicing (variants A, B, C, D, N), only variant B was identified in murine thymocytes (320). Up to 40% of NEU2 variant B sialidase activity was found in the membrane fraction of transfected COS-7 cells. An analysis of mouse thymic lysates prepared with different detergents suggested that NEU2 was a peripheral membrane protein, rather than a transmembrane or GPI‐anchored protein, with enzymatic activity at pH 7.0 but not pH 4.5 (78). It should be noted that other studies have established NEU2 as a cytosolic enzyme with a pH optimum of 6.0-6.5 and an amino acid sequence lacking protein domains typical of transmembrane or GPI-anchored proteins (12, 84, 85).
The levels of IL-2 and a T helper 2 (Th2) cytokine, IL-4, were increased following in vitro anti-CD3 antibody stimulation of murine spleen cells, and reduced levels of IL-4, but not IL-2 or IFN-γ, were seen upon anti-CD3 antibody activation of SM/J splenocytes (286). Addition of exogenous IL-4 during CD4+ T cell activation enhanced NEU1 sialidase activity and cell surface levels of asialoGM1 ganglioside, while activation of T cells in the presence of 2-deoxy-NANA diminished IL-4 production. In other studies, treatment of IL-4-primed CD4+ T cells with the GM3 ganglioside diminished IL-4, IL-5, and IL-13 production, but had no effect on IL-2 or IFN-γ, through a mechanism involving mobilization of intracellular calcium (321). Further, anti-CD3 antibody stimulation of NEU1-deficient B10.SM strain CD4+ T cells reduced intracellular calcium levels compared with unstimulated cells. A COOH-terminal tetrapeptide, Tyr-Gly-Thr-Leu, was suggested to constitute an internalization signal in the NEU1 protein that targets it to the lysosome in human lymphocytes, fibroblasts, and COS-7 cells (62). Upon Tyr phosphorylation of the tetrapeptide in activated lymphocytes, NEU1 sialidase activity was unchanged, but the sialidase was redistributed from the lysosome to the cell surface. NEU1 and NEU3 enzyme activities and mRNA levels were increased in human CD4+ T cells activated with anti-CD3 and anti-CD28 antibodies (288). NEU3 was the major sialidase expressed in the activated T cells. Overexpression of NEU1 in human Jurkat T cells increased IL-2, IL-4, IL-13, and IFN-γ expression following CD3/CD28 costimulation, while these same cytokines, with the exception of IL-4, were produced in NEU3-overexpressing cells. CD3/CD28 costimulation of human primary T cells increased IFN-γ production, which was inhibited by zanamivir and 2-deoxy-NANA (65). CD3/CD28 costimulation also increased NEU1 mRNA levels and sialidase activity with minimal increase in NEU3 activity, heightened NEU1 and PPCA cell surface expression, and increased the levels of desialylated cell surface glycoconjugates, all compared with unstimulated cells. While these studies suggested that NEU1, and possibly NEU3, might be linked to Th2 immunity, they also highlight the apparent contradiction to the studies reviewed above implicating NEU1 in the Th1 response (65, 66, 308).
Altered expression of surface sialoglycoconjugates is a characteristic feature of apoptosis (287). Treatment of Jurkat T cells with the proapoptotic drug, etoposide, increased sialidase activity and desialylated cell surface glycoconjugates during the early phase of apoptosis, which was reversed by 2-deoxy-NANA (322). In subsequent studies, etoposide-induced apoptosis was associated with increased cell surface GM3, reduced GD3, and increased NEU3, but not NEU1, mRNA levels (323). Changes in GM3, GD3, and NEU3 levels were blocked by caspase inhibition. Phytohemagglutinin/IL-2-induced human T cell blasts undergoing UV- or staurosporine-induced apoptosis exhibited decreased surface staining for terminal α2,3- and α2,6-linked sialic acids (324). Desialylation of surface glycoconjugates also was observed on microparticles derived from apoptotic lymphoblast membranes. Plasma membrane-derived apoptotic microparticles had increased sialidase activity on their surface (325). Treatment of viable Jurkat cell lysates with caspase-3 increased sialidase activity to the same degree as that of apoptotic cell lysates, consistent with a predicted caspase-3 proteolytic cleavage site at Asp-135 in the NEU1 protein. Further, the caspase inhibitor, zVAD, decreased sialidase activity in etoposide-induced apoptotic Jurkat cells. Not only was cell surface NEU1 responsible for desialylating Jurkat T cell surface glycoconjugates in a cis-acting manner, but also in trans on the surface of co-incubated human erythrocytes (67).
In a mite allergen model of allergic asthma, in vitro exposure of Dermatophagoides farinae (Derf)-sensitized CD4+ T cells to the antigen increased desialylation of cell surface glycoconjugates, augmented binding of hyaluronic acid (HA) to its cognate receptor, CD44, and heightened NEU1, but not NEU2 or NEU3, transcript levels (30). In NEU1-deficient cells from SM/J mice, CD44 receptor activity for HA was reduced, as were reductions in BALF eosinophil and Th2 cell numbers, Th2 cytokine levels, and airway hyperresponsiveness following Derf challenge, compared with cells expressing normal NEU1 levels. Subsequent studies using in vitro-differentiated ovalbumin-specific CD4+ T cells revealed that NEU1 and NEU3 mRNA levels were greater in Th2, compared with Th1, cells (326). Collectively, these studies indicated that NEU1 is required for HA-CD44 interaction and development of acute asthmatic inflammation, and suggested that NEU1-mediated desialylation of CD44 on CD4+ T cells alters the ability of CD44 to interact with HA (28). In subsequent studies, analysis of mouse CD4+ T cell subsets revealed that while NEU1 mRNA was expressed at approximately equal amounts in Th1, Th2, Th17, and induced regulatory T (iTreg) cells, NEU3 transcript levels were greater in iTreg cells compared with the other cell subsets (289). CD4+ activated Treg (FoxP3+CD62L−) spleen cells expressed higher levels of NEU3 transcripts compared with resting Treg (FoxP3+CD62L+) and FoxP3− cells, and overexpression of NEU3 in naïve CD4+ T cells upregulated FoxP3 expression. In a mouse model of obesity-induced insulin resistance, the percentages of CD4+FoxP3+ Treg cells and NEU1 levels were decreased, while the percentages of CD4+Th17+ cells and the levels of the NEU1-targeting microRNA, miR-23b-3p, were increased, all compared with nonobese controls (290). Administration of the anti-obesity phytochemical, acacetin, reversed these effects, while administration miR-23b-3p exacerbated the outcomes. Co-administration of acacetin and miR-23b-3p restored the obesity-related downregulated levels of NEU1, while NEU1 overexpression offset the effects of miR-23b-3p on the Treg/Th17 cell ratio. The authors concluded that acacetin reverses insulin resistance in obese model mice by regulating the Treg/Th17 cell balance through a miR-23b-3p – NEU1 axis. Finally, in human studies, differential gene expression analysis identified 7 genes related to the lysosomal pathway, including NEU1, that were downregulated in the airways of asthma patients compared with healthy controls, suggesting that lymphocyte lysosomal dysfunction might be involved in the pathogenesis of allergic asthma (327).
Vitamin D is a potent immunomodulator (328).Vitamin D-binding protein (DBP), also referred to as the group-specific component (Gc) protein in humans, is the main transporter of vitamin D in the circulation. Part of the immune-modulating effects of vitamin D are mediated through the conversion of Gc/DBP to macrophage activating factor (MAF) (329). In humans, the Gc protein contains a mucin-type O-linked trisaccharide composed of Thr-linked GalNAc with dibranched sialic acid and Gal termini, while MAF contains only the Thr-linked monosaccharide GalNAc (330). A two-step sequential action of β-galactosidase on the surface of B cells followed by surface-expressed NEU1 on T cells was identified as being responsible for the conversion of Gc to MAF (331–333). In mice, DBP is not sialylated and only the action of β-galactosidase on the Gal-GalNAc disaccharide was required to deglycosylate DBP to MAF. Taken together, these results indicate that NEU1, and to a lesser extent NEU3, play in important role in regulation of adaptive immunity through their effects on T and B lymphocytes (Table 2).
NEUs and MUCs in Autoimmune Diseases
Autoimmune diseases comprise a diverse collection of pathological conditions originating from a dysregulated immune response to a self tissue or organ. As discussed above, NEUs play a critical role in the activity of Treg cells (289, 290) suggesting the possible involvement of NEUs in the pathogenesis of autoimmunity. Further, MUC1 has been suggested as a checkpoint inhibitor on T cells (334), implicating MUC1, and possibly other membrane-bound mucins, in the development of autoimmune diseases. This section reviews the role of NEUs and MUCs in the context of four selected autoimmune diseases, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), type 1 diabetes (T1D), and inflammatory bowel disease (IBD).
Systemic Lupus Erythematosus
SLE is an autoimmune disease typically characterized by the presence of anti-nuclear antibodies (ANA) and anti-extractable nuclear antigen (anti-ENA) antibodies with immune complex deposition in multiple organs, particularly the kidneys (335). A growing body of evidence suggests a role for NEU1 in the pathogenesis of lupus nephritis (283, 336–338). More specifically, NEU1 expression and catalytic activity were increased in the kidneys of MRL/lpr lupus mice (339), and kidney mesangial cells from NEU1-haplodeficent mice had reduced cytokine expression and secretion in response to LPS or lupus serum (LS) compared with NEU1-expressing mice (340). In other studies, overexpression of NEU1 in cultured mouse MES13 kidney mesangial cells dose-dependently increased spontaneous IL-6 and MCP-1 production and treatment of primary mesangial cells from MRL/lpr lupus mice with heat-aggregated IgG (HA-IgG), a mimic of immune complex deposition, increased NEU1 expression (291). Moreover, pharmacologic inhibition of NEU1 activity with oseltamivir reduced HA-IgG- or LS-stimulated IL-6 and MCP-1 production. In human studies, proteomic analysis of renal tissues comparing patients with proliferative lupus nephritis vs. healthy controls revealed NEU1 as the greatest up-regulated protein of 48 identified proteins (341). NEU1 immunohistochemical staining and urinary excretion were significantly elevated in lupus nephritis patients compared with controls. Finally, flow cytometric analysis of human B cells from SLE patients revealed a positive correlation between the cell surface ratio of sialyltransferase ST3Gal-1 and NEU3 with disease severity (342).
SLE patients with serositis had greater MUC16 levels and disease duration compared with SLE patients without serositis (343). Pseudo–pseudo Meigs’ syndrome is a rare manifestation in SLE patients presenting with ascites, pleural effusion, and elevated serum MUC16 levels unrelated to malignancy (344–346). MUC20 transcript levels were increased with the progression of lupus in the kidneys of MRL/lpr lupus-prone mice (347). Interestingly, mouse NEU1 (348) and mouse and human MUC20 (347) are highly expressed in kidneys compared with other organs, but it is unknown whether MUC20 might be a NEU1 substrate.
Rheumatoid Arthritis
RA is an autoimmune disease of unknown etiology characterized by synovitis and bone erosion (349). A positive correlation between B cell surface ST3Gal-1/NEU3 ratio and RA disease severity was seen in patients with mild or high disease activity, but not patients in non-remission (342). In a follow-up study, B cell surface NEU3 levels were found to positively correlate with RA disease activity (350). NEU1 catalytic activity was increased in the blood and liver of rats with Freund’s adjuvant-induced RA (351). In a mouse model of bovine collagen type II-induced RA, pharmacologic inhibition of NEU2 and NEU3 activity with zanamivir dose-dependently diminished CD19+ B cells and CD138+/TACI+ plasma cells numbers, anti-collagen type II autoantibody levels, disease activity, and total sialidase activity in arthritic joints (292). Corresponding, increased sialylation of circulating IgG and α2,3-linked surface sialylation of CD138+/TACI+ plasma cells was noted.
MUC1 protein levels were elevated on T cells isolated from the joint fluid of a patient with RA (352). The combination of lung ultrasound and serum MUC1 levels, in addition to current markers, was proposed for screening and following patients with ILD associated with RA (353). A greater number of RA patients expressed MUC3 protein in synovial lining cells and synovial macrophages compared with normal controls (354). The MUC5B gene promoter variant rs5705950 was linked to an increased susceptibility to pulmonary fibrosis in RA patients (213, 215, 355). Increased salivary MUC7 sulfation was detected in RA patients compared with healthy controls (356). Synovial fluid cells from RA patients contain a distinct population of MUC18-positive T cells (357), and these cells were identified as a biomarker for synovial membrane angiogenesis in RA patients (358).
Type 1 Diabetes
T1D is an autoreactive disease arising from immune destruction of insulin-producing pancreatic β cells (359). Both NEU1 and NEU3 have been implicated in the pathogenesis of T1D. NEU1 transcript levels and catalytic activity were diminished in two strains of diabetes-prone mice compared with nondiabetic littermates (360). Treatment of cultured rat L6 myoblasts with purified mouse NEU1 promoted desialylation of both the insulin receptor (IR) and insulin-like growth factor-1 receptor (IGF-1R) compared with untreated controls (36). Mice deficient in NEU1 activity and fed a high fat diet (HFD) developed glucose intolerance and insulin resistance compared with mice expressing normal levels of NEU1, and HEK293 cells overexpressing both NEU1 and the insulin receptor kinase (IRK) exhibited increased IRK-NEU1 association and NEU1-mediated IRK desialylation (39). Liver and muscle cells from NEU1-deficient, HFD-fed mice had decreased IR phosphorylation and downstream Akt activation, and treatment of HEK293 cells overexpressing IR and NEU1 promoted IR-NEU1 association and NEU1-mediated IR desialylation. Administration of EDPs to normal mice expressing the NEU1-containing ERC increased IR-NEU1 association and NEU1-mediated IR desialylation in muscle tissues, both of which were reversed by 2-deoxy-NANA (37). Murine hepatocytes treated with EDPs in the context of nonalcoholic steatohepatitis linked to insulin resistance had decreased hepatic growth factor receptor (HGFR) phosphorylation as a consequence of NEU1-mediated HGFR desialylation (43). Overexpression of NEU1 in HEK293 cells increased IR desialylation and dimerization, and NEU1 overexpression in human HepG2 liver cells reversed palmitate-induced insulin resistance (38). In these same studies, pharmacologic upregulation of NEU1 expression and activity with ambroxol in mice led to desialylation of the IR, increased IR Tyr phosphorylation and Akt signaling, and improved glucose tolerance and insulin resistance in mice exposed to a HFD. NEU1 expression was increased in the spleen, liver, and kidneys of streptozotocin (STZ)-treated diabetic rats (361). Similarly, NEU1 protein levels were increased in cardiomyocytes in the STZ mouse model of diabetic cardiomyopathy, and NEU1 silencing protected against STZ-induced cardiomyopathy and myocardial fibrosis, inflammation, and apoptosis through an AMPK-SIRT3 pathway (131). Inhibition of NEU1 activity with anti-NEU1 antibody or oseltamivir reduced insulin-stimulated IGF-1R and insulin receptor substrate-1 (IRS-1) phosphorylation in human fibroblasts, and promoted IR-NEU1 association in rat HTC hepatoma cells (69). Further, pharmacologic inhibition of MMP-9 in HTC cells dose-dependently reduced insulin-stimulated NEU1 sialidase activity, and treatment of NEU1-deficient human fibroblasts with olanzapine, an antipsychotic drug associated with insulin resistance, increased NEU3 sialidase activity, both compared with vehicle controls. Finally, a variety of different G protein-coupled receptor agonists dose-dependently increased NEU1 sialidase activity, IR activation, and IR and IRS-1 phosphorylation in human IR-overexpressing HTC cells (293).
NEU3-overexpressing mouse 3T3-L1 adipocytes and L6 myocytes, and muscle tissue from human NEU3 transgenic mice, had decreased insulin-stimulated IR signaling, and administration of insulin to NEU3 transgenic mice induced NEU3 Tyr phosphorylation and NEU3-Grb-2 association (294). In vivo overexpression of NEU3 in the livers of insulin-sensitive or insulin-resistant mice increased hepatic levels of glycogen, triglycerides, peroxisome proliferator-activated receptor γ (PPARγ), ganglioside GM1, and IRS-1 phosphorylation, compared with control mice, suggesting that NEU3 regulates hepatic insulin sensitivity and glucose tolerance (295). Obese strain rats fed a HFD had reduced NEU3 expression in adipose tissue through an intracellular palmitate-driven, Akt-dependent pathway (296). Glucose-stimulated insulin release by rat INS-1D pancreatic β cells was increased by 2-deoxy-NANA, and blood insulin levels were greater in NEU3-deficient mice compared with wild type controls (362). The relationship between insulin resistance and intestinal hypoxia was explained, in part, by the activation of hypoxia-inducible factor-2α (HIF-2α) in hypoxic, HFD-fed mice leading to NEU3 activation (363).
MUC1 downregulation is necessary for human and mouse fetal-uterine implantation, and diabetic mice expressed increased MUC1 mRNA and protein levels at fetal-uterine implantation sites compared with nondiabetic controls (364). STZ-induced diabetic mice administered with sodium hyaluronate eye drops for treatment of diabetic ocular surface disease exhibited increased MUC5AC expression in the conjunctival epithelium compared with saline controls (365). STZ-induced diabetic rats had decreased MUC10 protein levels in salivary submandibular glands compared with nondiabetic controls (366). Transcriptome analysis of nucleolar protein p120 (NOP2)-deficient vs. NOP2-expressing human colon cancer cells identified the MUC19 gene as significantly upregulated, and T1D as a gene pathway most significantly affected, by NOP2 silencing (367). Future studies are needed to determine whether NEUs regulate MUC-associated diabetes.
Inflammatory Bowel Disease
IBD comprises a collection of idiopathic, intestinal autoimmune diseases, the most common being Crohn’s disease (CD) and ulcerative colitis (UC) (368). CD typically affects the small and large intestines, while UC primarily involves the colon and rectum. Elevated expression of intestinal NEU3 (297) was correlated with diminished levels of intestinal alkaline phosphatase (IAP) in patients with colitis (369), and human genetic deficiency of IAP is associated with IBD (370), suggesting that NEU3 may play a role in the pathogenesis of IBD. Compared with controls, a 2.0-fold increase in GM3 ganglioside levels and an 8.3-fold increase in NEU3 protein levels were reported in the intestine of IBD patients (297). Because NEU3 is responsible for the conversion of disialylated GD3 to monosialylated GM3, the authors speculated that increasing dietary GD3 intake may be beneficial for treatment of IBD patients. A SNP, rs4947331, encoding a minor T allele located in the 3’ untranslated region of the NEU1 gene was significantly associated with the presence of CD in North Indian and Dutch populations (298). It was suggested that the NEU1 pharmacologic inhibitor, oseltamivir, might be applicable to treat CD patients. Serum sialic acid levels were increased, presumably as a consequence of increased NEU catalytic activity, and positively correlated with established markers of disease activity, in CD patients (371, 372). Meta-analysis of six genome-wide association studies in CD patients identified the MUC1 gene as a candidate for disease susceptibility (373). Elevated expression of multiple mucin gene products, including MUC1, MUC4, and MUC13, were reported in IBD patients compared with controls (374–376). By contrast, another study reported decreased expression of these MUCs in IBD patients (377). MUC1 knockout mice had more severe forms of Th1- and Th2-induced colitis and increased Th17 cell-mediated responses in the colon, compared with MUC1-expressing mice (378). Studies in both human and animal model systems indicated that MUC1 regulates the progression of IBD to colon cancer (379–384). Alterations in MUC2 intestinal expression and glycosylation were reported in IBD patients (385, 386). MUC2 knockout mice had greater intestinal inflammation and reduced body weight gain compared with MUC2-expressing mice, despite equal food intake, and exhibited intestinal microbiome, short chain fatty acid, and inflammatory cytokine profiles similar to that of IBD patients (387, 388). The Winnie mouse strain possessing a MUC2 gene point mutation encoding a Cys-to-Tyr missense mutation develops spontaneous colitis with many of the same pathologic features seen in UC patients (389). Other MUCs associated with CD and UC disease include MUC3, MUC5AC, MUC5B, MUC6, MUC16, MUC19, and MUC20 (390–394). Additional studies are needed to establish a causal link between the expression and catalytic activity of NEUs with MUCs in IBD. In summary, the development and progression of SLE, RA, T1D, and IBD are influenced by both NEUs (Table 2) and MUCs.
Discussion
In this review, we have attempted to summarize historical and recent research on the roles of mammalian NEUs and MUCs, both individually and together, in the context of fibrotic and immune-mediated human diseases. First, general features of the 20-member MUC family and 4-member NEU family were summarized, including an updated tabulation of selected known and putative NEU1 glycoprotein substrates relevant to fibrotic and inflammatory processes. Particular attention was paid to the KL-6 glycoprotein, a soluble form of the membrane-bound MUC1-ED. The concept of a NEU1 – MUC1 axis in IPF/ILD was presented. Evidence supporting the NEU1 – MUC1 axis included publications demonstrating that NEU1 regulates pulmonary collagen deposition, lymphocytosis, and fibrosis in human and mouse models of IPF (128), KL-6 serves as a diagnostic and prognostic biomarker for, and mediator of, IPF/ILD (125, 180–183), and most importantly, NEU1 pharmacologic inhibition reverses bleomycin-induced increases in MUC1-ED desialylation and shedding from the cell surface (48). Finally, the influence of NEU1 on human infections by bacterial, viral, and parasitic pathogens, and the impact of NEUs and MUCs on four selected human autoimmune diseases was discussed.
A limitation of our analysis was that the expanse and complexity of this field prevented us from an in-depth exploration of the broader NEU/MUC biology. We utilized a reductionist approach, which on the one hand, allowed for a focused analysis, but on the other hand, limited the scope by excluding related topics. Only mammalian NEUs and MUCs were considered. Bacterial or viral exosialidases, endosialidases, or trans-sialidases were not discussed despite their direct relevance to disease processes. We did not consider catalytic mechanisms of NEU activity but instead emphasized the functional consequences relevant to health and disease. Further reducing the complexity and sharpening the focus of this review, only the immune-, inflammation-, and fibrosis-related functions of mammalian NEUs and MUCs were discussed; the effects on other processes, e.g., atherosclerosis, diabetes, or cancer, were mentioned as far as they related to the immune, inflammatory, or connective tissue aspects of those processes. Moreover, the complexity of the mechanisms controlling NEU1 elevation or decline in the diseases were not considered. We only reviewed the functional consequences of glycoprotein desialylation, with substantially less attention devoted to desialylation of another major group of NEU substrates, glycolipids, and its functional sequelae. We did not reflect on the balancing action of sialyltransferases, which covalently attach sialic acid to glycans in contrast to NEUs, which remove it. The biological effects of free sialic acid were not considered either. An important topic that was not discussed in depth relates to recent developments in discovery and characterization of mammalian NEU isozyme-specific inhibitors; these have been considered here only to the extent that helps in reviewing NEU function. We refer the reader interested in those aspects of NEUs excluded here to numerous review articles that illuminate these topics (12, 15, 17, 81, 395). Even with such reductions, the information summarized in this review is rather voluminous and complicated, yet we hope that it is sufficiently focused thematically to be useful.
In summary, human NEUs and MUCs, particularly NEU1 and MUC1, contribute alone, and in symphony, to the development and progression of a variety of fibrotic, inflammatory, and fibro-inflammatory human pathologies. Future studies focusing on the details of the NEU1 – MUC1 relationship are expected provide new insights into the mechanisms of selected disease processes characterized by dysregulated fibrosis and inflammation that may someday lead to novel bedside therapeutic interventions for the benefit of humankind.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
All authors conceived and designed the review outline, contributed to the writing and critical review of the manuscript, and approved the final version of the manuscript.
Funding
This work was supported by grants from the National Institutes of Allergy and Infectious Diseases (AI-144497) and the U.S. Department of Defense (W81XWH1910056) to EL and the U.S. Department of Defense (PR202031) to IL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Varki A. Sialic Acids in Human Health and Disease. Trends Mol Med (2008) 14(8):351–60. doi: 10.1016/j.molmed.2008.06.002
2. Schauer R. Sialic Acids as Regulators of Molecular and Cellular Interactions. Curr Opin Struct Biol (2009) 19(5):507–14. doi: 10.1016/j.sbi.2009.06.003
3. Varki A, Schnaar RL, Schauer R, Cummings RD, Esko JD, Stanley P, et al. Sialic Acids and Other Nonulosonic Acids. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, editors. Essentials of Glycobiology. NY: Cold Spring Harbor Laboratory Press (2015). p. 179–95.
4. Finne J. Occurrence of Unique Polysialosyl Carbohydrate Units in Glycoproteins of Developing Brain. J Biol Chem (1982) 257(20):11966–70. doi: 10.1016/S0021-9258(18)33661-5
5. Warren L, Spearing CW. Mammalian Sialidase (Neuraminidase). Biochem Biophys Res Commun (1960) 3:489–92. doi: 10.1016/0006-291x(60)90161-3
6. Corfield AP, Veh RW, Wember M, Michalski JC, Schauer R. The Release of N-Acetyl- and N-Glycolloyl-Neuraminic Acid From Soluble Complex Carbohydrates and Erythrocytes by Bacterial, Viral and Mammalian Sialidases. Biochem J (1981) 197(2):293–9. doi: 10.1042/bj1970293
7. Kishore CS, Tulsiani DR, Bhavanandan VP, Carubelli R. Membrane-Bound Neuraminidases of Rat Liver. Neuraminidase Activity in Golgi Apparatus. J Biol Chem (1975) 250(7):2655–9.
8. Schengrund CL, Rosenberg A. Intracellular Location and Properties of Bovine Brain Sialidase. J Biol Chem (1970) 245(22):6196–200. doi: 10.1016/S0021-9258(18)62678-X
9. Mahadevan S, Nduguba JC, Tappel AL. Sialidase of Rat Liver and Kidney. J Biol Chem (1967) 242(19):4409–13. doi: 10.1016/S0021-9258(18)99554-2
10. Sandhoff K, Jatkewitz H. A Particle-Bound Sialyl Lactosidoceramide Splitting Mammalian Sialidase. Biochim Biophys Acta (1967) 141(2):442–4. doi: 10.1016/0304-4165(67)90124-9
11. Carubelli R, Trucco RE, Caputto R. Neuraminidase Activity in Mammalian Organs. Biochim Biophys Acta (1962) 60:196–7. doi: 10.1016/0006-3002(62)90392-x
12. Miyagi T, Yamaguchi K. Mammalian Sialidases: Physiological and Pathological Roles in Cellular Functions. Glycobiology (2012) 22(7):880–96. doi: 10.1093/glycob/cws057
13. Dalton GD, Xie J, An SW, Huang CL. New Insights Into the Mechanism of Action of Soluble Klotho. Front Endocrinol (Lausanne) (2017) 8:323. doi: 10.3389/fendo.2017.00323
14. Khan A, Sergi C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics (Basel) (2018) 8(2):29. doi: 10.3390/diagnostics8020029
15. Glanz VY, Myasoedova VA, Grechko AV, Orekhov AN. Sialidase Activity in Human Pathologies. Eur J Pharmacol (2019) 842:345–50. doi: 10.1016/j.ejphar.2018.11.014
16. Bennasroune A, Romier-Crouzet B, Blaise S, Laffargue M, Efremov RG, Martiny L, et al. Elastic Fibers and Elastin Receptor Complex: Neuraminidase-1 Takes the Center Stage. Matrix Biol (2019) 84:57–67. doi: 10.1016/j.matbio.2019.06.007
17. Pshezhetsky AV, Ashmarina LI. Desialylation of Surface Receptors as a New Dimension in Cell Signaling. Biochem (Mosc) (2013) 78(7):736–45. doi: 10.1134/S0006297913070067
18. Demina EP, Smutova V, Pan X, Fougerat A, Guo T, Zou C, et al. Neuraminidases 1 and 3 Trigger Atherosclerosis by Desialylating Low-Density Lipoproteins and Increasing Their Uptake by Macrophages. J Am Heart Assoc (2021) 10(4):e018756. doi: 10.1161/JAHA.120.018756
19. Satyavarapu EM, Nath S, Mandal C. Desialylation of Atg5 by Sialidase (Neu2) Enhances Autophagosome Formation to Induce Anchorage-Dependent Cell Death in Ovarian Cancer Cells. Cell Death Discov (2021) 7(1):26. doi: 10.1038/s41420-020-00391-y
20. Kijimoto-Ochiai S, Matsumoto-Mizuno T, Kamimura D, Murakami M, Kobayashi M, Matsuoka I, et al. Existence of Neu1 Sialidase on Mouse Thymocytes Whose Natural Substrate Is Cd5. Glycobiology (2018) 28(5):306–17. doi: 10.1093/glycob/cwy009
21. Feng C, Zhang L, Almulki L, Faez S, Whitford M, Hafezi-Moghadam A, et al. Endogenous Pmn Sialidase Activity Exposes Activation Epitope on Cd11b/Cd18 Which Enhances Its Binding Interaction With Icam-1. J Leukoc Biol (2011) 90(2):313–21. doi: 10.1189/jlb.1210708
22. Kawecki C, Bocquet O, Schmelzer CEH, Heinz A, Ihling C, Wahart A, et al. Identification of Cd36 as a New Interaction Partner of Membrane Neu1: Potential Implication in the Pro-Atherogenic Effects of the Elastin Receptor Complex. Cell Mol Life Sci (2019) 76(4):791–807. doi: 10.1007/s00018-018-2978-6
23. Bocquet O, Tembely D, Rioult D, Terryn C, Romier B, Bennasroune A, et al. Characterization of Novel Interactions With Membrane Neu1 Highlights New Regulatory Functions for the Elastin Receptor Complex in Monocyte Interaction With Endothelial Cells. Cell Biosci (2021) 11(1):206. doi: 10.1186/s13578-021-00718-x
24. Howlader MA, Li C, Zou C, Chakraberty R, Ebesoh N, Cairo CW. Neuraminidase-3 Is a Negative Regulator of Lfa-1 Adhesion. Front Chem (2019) 7:791. doi: 10.3389/fchem.2019.00791
25. Lee C, Liu A, Miranda-Ribera A, Hyun SW, Lillehoj EP, Cross AS, et al. Neu1 Sialidase Regulates the Sialylation State of Cd31 and Disrupts Cd31-Driven Capillary-Like Tube Formation in Human Lung Microvascular Endothelia. J Biol Chem (2014) 289(13):9121–35. doi: 10.1074/jbc.M114.555888
26. Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, et al. Desialylation Is a Mechanism of Fc-Independent Platelet Clearance and a Therapeutic Target in Immune Thrombocytopenia. Nat Commun (2015) 6:7737. doi: 10.1038/ncomms8737
27. Jansen AJ, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, et al. Desialylation Accelerates Platelet Clearance After Refrigeration and Initiates Gpibalpha Metalloproteinase-Mediated Cleavage in Mice. Blood (2012) 119(5):1263–73. doi: 10.1182/blood-2011-05-355628
28. Katoh S. Critical Involvement of Cd44 in T Helper Type 2 Cell-Mediated Eosinophilic Airway Inflammation in a Mouse Model of Acute Asthma. Front Immunol (2021) 12:811600. doi: 10.3389/fimmu.2021.811600
29. Faller CE, Guvench O. Terminal Sialic Acids on Cd44 N-Glycans Can Block Hyaluronan Binding by Forming Competing Intramolecular Contacts With Arginine Sidechains. Proteins (2014) 82(11):3079–89. doi: 10.1002/prot.24668
30. Katoh S, Maeda S, Fukuoka H, Wada T, Moriya S, Mori A, et al. A Crucial Role of Sialidase Neu1 in Hyaluronan Receptor Function of Cd44 in T Helper Type 2-Mediated Airway Inflammation of Murine Acute Asthmatic Model. Clin Exp Immunol (2010) 161(2):233–41. doi: 10.1111/j.1365-2249.2010.04165.x
31. Seyrantepe V, Iannello A, Liang F, Kanshin E, Jayanth P, Samarani S, et al. Regulation of Phagocytosis in Macrophages by Neuraminidase 1. J Biol Chem (2010) 285(1):206–15. doi: 10.1074/jbc.M109.055475
32. Uemura T, Shiozaki K, Yamaguchi K, Miyazaki S, Satomi S, Kato K, et al. Contribution of Sialidase Neu1 to Suppression of Metastasis of Human Colon Cancer Cells Through Desialylation of Integrin Beta4. Oncogene (2009) 28(9):1218–29. doi: 10.1038/onc.2008.471
33. Zanoteli E, van de Vlekkert D, Bonten EJ, Hu H, Mann L, Gomero EM, et al. Muscle Degeneration in Neuraminidase 1-Deficient Mice Results From Infiltration of the Muscle Fibers by Expanded Connective Tissue. Biochim Biophys Acta (2010) 1802(7-8):659–72. doi: 10.1016/j.bbadis.2010.04.002
34. Yogalingam G, Bonten EJ, van de Vlekkert D, Hu H, Moshiach S, Connell SA, et al. Neuraminidase 1 Is a Negative Regulator of Lysosomal Exocytosis. Dev Cell (2008) 15(1):74–86. doi: 10.1016/j.devcel.2008.05.005
35. Hinek A, Bodnaruk TD, Bunda S, Wang Y, Liu K. Neuraminidase-1, a Subunit of the Cell Surface Elastin Receptor, Desialylates and Functionally Inactivates Adjacent Receptors Interacting With the Mitogenic Growth Factors Pdgf-Bb and Igf-2. Am J Pathol (2008) 173(4):1042–56. doi: 10.2353/ajpath.2008.071081
36. Arabkhari M, Bunda S, Wang Y, Wang A, Pshezhetsky AV, Hinek A. Desialylation of Insulin Receptors and Igf-1 Receptors by Neuraminidase-1 Controls the Net Proliferative Response of L6 Myoblasts to Insulin. Glycobiology (2010) 20(5):603–16. doi: 10.1093/glycob/cwq010
37. Blaise S, Romier B, Kawecki C, Ghirardi M, Rabenoelina F, Baud S, et al. Elastin-Derived Peptides Are New Regulators of Insulin Resistance Development in Mice. Diabetes (2013) 62(11):3807–16. doi: 10.2337/db13-0508
38. Fougerat A, Pan X, Smutova V, Heveker N, Cairo CW, Issad T, et al. Neuraminidase 1 Activates Insulin Receptor and Reverses Insulin Resistance in Obese Mice. Mol Metab (2018) 12:76–88. doi: 10.1016/j.molmet.2018.03.017
39. Dridi L, Seyrantepe V, Fougerat A, Pan X, Bonneil E, Thibault P, et al. Positive Regulation of Insulin Signaling by Neuraminidase 1. Diabetes (2013) 62(7):2338–46. doi: 10.2337/db12-1825
40. Lillehoj EP, Hyun SW, Feng C, Zhang L, Liu A, Guang W, et al. Neu1 Sialidase Expressed in Human Airway Epithelia Regulates Epidermal Growth Factor Receptor (Egfr) and Muc1 Protein Signaling. J Biol Chem (2012) 287(11):8214–31. doi: 10.1074/jbc.M111.292888
41. Gilmour AM, Abdulkhalek S, Cheng TS, Alghamdi F, Jayanth P, O’Shea LK, et al. A Novel Epidermal Growth Factor Receptor-Signaling Platform and Its Targeted Translation in Pancreatic Cancer. Cell Signal (2013) 25(12):2587–603. doi: 10.1016/j.cellsig.2013.08.008
42. Mozzi A, Forcella M, Riva A, Difrancesco C, Molinari F, Martin V, et al. Neu3 Activity Enhances Egfr Activation Without Affecting Egfr Expression and Acts on Its Sialylation Levels. Glycobiology (2015) 25(8):855–68. doi: 10.1093/glycob/cwv026
43. Romier B, Ivaldi C, Sartelet H, Heinz A, Schmelzer CEH, Garnotel R, et al. Production of Elastin-Derived Peptides Contributes to the Development of Nonalcoholic Steatohepatitis. Diabetes (2018) 67(8):1604–15. doi: 10.2337/db17-0490
44. Abdulkhalek S, Szewczuk MR. Neu1 Sialidase and Matrix Metalloproteinase-9 Cross-Talk Regulates Nucleic Acid-Induced Endosomal Toll-Like Receptor-7 and -9 Activation, Cellular Signaling and Pro-Inflammatory Responses. Cell Signal (2013) 25(11):2093–105. doi: 10.1016/j.cellsig.2013.06.010
45. Abdulkhalek S, Amith SR, Franchuk SL, Jayanth P, Guo M, Finlay T, et al. Neu1 Sialidase and Matrix Metalloproteinase-9 Cross-Talk Is Essential for Toll-Like Receptor Activation and Cellular Signaling. J Biol Chem (2011) 286(42):36532–49. doi: 10.1074/jbc.M111.237578
46. Jayanth P, Amith SR, Gee K, Szewczuk MR. Neu1 Sialidase and Matrix Metalloproteinase-9 Cross-Talk Is Essential for Neurotrophin Activation of Trk Receptors and Cellular Signaling. Cell Signal (2010) 22(8):1193–205. doi: 10.1016/j.cellsig.2010.03.011
47. Abdulkhalek S, Guo M, Amith SR, Jayanth P, Szewczuk MR. G-Protein Coupled Receptor Agonists Mediate Neu1 Sialidase and Matrix Metalloproteinase-9 Cross-Talk to Induce Transactivation of Toll-Like Receptors and Cellular Signaling. Cell Signal (2012) 24(11):2035–42. doi: 10.1016/j.cellsig.2012.06.016
48. Luzina IG, Lillehoj EP, Lockatell V, Hyun SW, Lugkey KN, Imamura A, et al. Therapeutic Effect of Neuraminidase-1-Selective Inhibition in Mouse Models of Bleomycin-Induced Pulmonary Inflammation and Fibrosis. J Pharmacol Exp Ther (2021) 376(1):136–46. doi: 10.1124/jpet.120.000223
49. Lillehoj EP, Guang W, Hyun SW, Liu A, Hegerle N, Simon R, et al. Neuraminidase 1-Mediated Desialylation of the Mucin 1 Ectodomain Releases a Decoy Receptor That Protects Against Pseudomonas Aeruginosa Lung Infection. J Biol Chem (2019) 294(2):662–78. doi: 10.1074/jbc.RA118.006022
50. Hyun SW, Liu A, Liu Z, Cross AS, Verceles AC, Magesh S, et al. The Neu1-Selective Sialidase Inhibitor, C9-Butyl-Amide-Dana, Blocks Sialidase Activity and Neu1-Mediated Bioactivities in Human Lung In Vitro and Murine Lung In Vivo. Glycobiology (2016) 26(8):834–49. doi: 10.1093/glycob/cww060
51. Lillehoj EP, Hyun SW, Liu A, Guang W, Verceles AC, Luzina IG, et al. Neu1 Sialidase Regulates Membrane-Tethered Mucin (Muc1) Ectodomain Adhesiveness for Pseudomonas Aeruginosa and Decoy Receptor Release. J Biol Chem (2015) 290(30):18316–31. doi: 10.1074/jbc.M115.657114
52. Karhadkar TR, Meek TD, Gomer RH. Inhibiting Sialidase-Induced Tgf-Beta1 Activation Attenuates Pulmonary Fibrosis in Mice. J Pharmacol Exp Ther (2021) 376(1):106–17. doi: 10.1124/jpet.120.000258
53. Amith SR, Jayanth P, Franchuk S, Siddiqui S, Seyrantepe V, Gee K, et al. Dependence of Pathogen Molecule-Induced Toll-Like Receptor Activation and Cell Function on Neu1 Sialidase. Glycoconj J (2009) 26(9):1197–212. doi: 10.1007/s10719-009-9239-8
54. Natori Y, Nasui M, Edo K, Sato S, Sakurai T, Kizaki T, et al. Neu1 Sialidase Controls Gene Expression and Secretion of Il-6 and Mcp-1 Through Nf-Kappab Pathway in 3t3-L1 Adipocytes. J Biochem (2017) 162(2):137–43. doi: 10.1093/jb/mvx006
55. Karmakar J, Roy S, Mandal C. Modulation of Tlr4 Sialylation Mediated by a Sialidase Neu1 and Impairment of Its Signaling in Leishmania Donovani Infected Macrophages. Front Immunol (2019) 10:2360. doi: 10.3389/fimmu.2019.02360
56. Allendorf DH, Franssen EH, Brown GC. Lipopolysaccharide Activates Microglia Via Neuraminidase 1 Desialylation of Toll-Like Receptor 4. J Neurochem (2020) 155(4):403–16. doi: 10.1111/jnc.15024
57. Amith SR, Jayanth P, Franchuk S, Finlay T, Seyrantepe V, Beyaert R, et al. Neu1 Desialylation of Sialyl Alpha-2,3-Linked Beta-Galactosyl Residues of Toll-Like Receptor 4 Is Essential for Receptor Activation and Cellular Signaling. Cell Signal (2010) 22(2):314–24. doi: 10.1016/j.cellsig.2009.09.038
58. Feng C, Stamatos NM, Dragan AI, Medvedev A, Whitford M, Zhang L, et al. Sialyl Residues Modulate Lps-Mediated Signaling Through the Toll-Like Receptor 4 Complex. PLoS One (2012) 7(4):e32359. doi: 10.1371/journal.pone.0032359
59. Woronowicz A, Amith SR, De Vusser K, Laroy W, Contreras R, Basta S, et al. Dependence of Neurotrophic Factor Activation of Trk Tyrosine Kinase Receptors on Cellular Sialidase. Glycobiology (2007) 17(1):10–24. doi: 10.1093/glycob/cwl049
60. Bonten EJ, Annunziata I, d’Azzo A. Lysosomal Multienzyme Complex: Pros and Cons of Working Together. Cell Mol Life Sci (2014) 71(11):2017–32. doi: 10.1007/s00018-013-1538-3
61. Pshezhetsky AV, Hinek A. Where Catabolism Meets Signalling: Neuraminidase 1 as a Modulator of Cell Receptors. Glycoconj J (2011) 28(7):441–52. doi: 10.1007/s10719-011-9350-5
62. Lukong KE, Seyrantepe V, Landry K, Trudel S, Ahmad A, Gahl WA, et al. Intracellular Distribution of Lysosomal Sialidase Is Controlled by the Internalization Signal in Its Cytoplasmic Tail. J Biol Chem (2001) 276(49):46172–81. doi: 10.1074/jbc.M104547200
63. Hinek A, Pshezhetsky AV, von Itzstein M, Starcher B. Lysosomal Sialidase (Neuraminidase-1) Is Targeted to the Cell Surface in a Multiprotein Complex That Facilitates Elastic Fiber Assembly. J Biol Chem (2006) 281(6):3698–710. doi: 10.1074/jbc.M508736200
64. Liang F, Seyrantepe V, Landry K, Ahmad R, Ahmad A, Stamatos NM, et al. Monocyte Differentiation Up-Regulates the Expression of the Lysosomal Sialidase, Neu1, and Triggers Its Targeting to the Plasma Membrane Via Major Histocompatibility Complex Class Ii-Positive Compartments. J Biol Chem (2006) 281(37):27526–38. doi: 10.1074/jbc.M605633200
65. Nan X, Carubelli I, Stamatos NM. Sialidase Expression in Activated Human T Lymphocytes Influences Production of Ifn-Gamma. J Leukoc Biol (2007) 81(1):284–96. doi: 10.1189/jlb.1105692
66. Stamatos NM, Carubelli I, van de Vlekkert D, Bonten EJ, Papini N, Feng C, et al. Lps-Induced Cytokine Production in Human Dendritic Cells Is Regulated by Sialidase Activity. J Leukoc Biol (2010) 88(6):1227–39. doi: 10.1189/jlb.1209776
67. Shkandina T, Herrmann M, Bilyy R. Sweet Kiss of Dying Cell: Sialidase Activity on Apoptotic Cell Is Able to Act Toward Its Neighbors. Autoimmunity (2012) 45(8):574–8. doi: 10.3109/08916934.2012.719951
68. D’Avila F, Tringali C, Papini N, Anastasia L, Croci G, Massaccesi L, et al. Identification of Lysosomal Sialidase Neu1 and Plasma Membrane Sialidase Neu3 in Human Erythrocytes. J Cell Biochem (2013) 114(1):204–11. doi: 10.1002/jcb.24355
69. Alghamdi F, Guo M, Abdulkhalek S, Crawford N, Amith SR, Szewczuk MR. A Novel Insulin Receptor-Signaling Platform and Its Link to Insulin Resistance and Type 2 Diabetes. Cell Signal (2014) 26(6):1355–68. doi: 10.1016/j.cellsig.2014.02.015
70. Chen GY, Brown NK, Wu W, Khedri Z, Yu H, Chen X, et al. Broad and Direct Interaction Between Tlr and Siglec Families of Pattern Recognition Receptors and Its Regulation by Neu1. Elife (2014) 3:e04066. doi: 10.7554/eLife.04066
71. Maurice P, Baud S, Bocharova OV, Bocharov EV, Kuznetsov AS, Kawecki C, et al. New Insights Into Molecular Organization of Human Neuraminidase-1: Transmembrane Topology and Dimerization Ability. Sci Rep (2016) 6:38363. doi: 10.1038/srep38363
72. Scandolera A, Odoul L, Salesse S, Guillot A, Blaise S, Kawecki C, et al. The Elastin Receptor Complex: A Unique Matricellular Receptor With High Anti-Tumoral Potential. Front Pharmacol (2016) 7:32. doi: 10.3389/fphar.2016.00032
73. Wu Y, Lan C, Ren D, Chen GY. Induction of Siglec-1 by Endotoxin Tolerance Suppresses the Innate Immune Response by Promoting Tgf-Beta1 Production. J Biol Chem (2016) 291(23):12370–82. doi: 10.1074/jbc.M116.721258
74. Sasaki N, Itakura Y, Toyoda M. Sialylation Regulates Myofibroblast Differentiation of Human Skin Fibroblasts. Stem Cell Res Ther (2017) 8(1):81. doi: 10.1186/s13287-017-0534-1
75. Wahart A, Hocine T, Albrecht C, Henry A, Sarazin T, Martiny L, et al. Role of Elastin Peptides and Elastin Receptor Complex in Metabolic and Cardiovascular Diseases. FEBS J (2019) 286(15):2980–93. doi: 10.1111/febs.14836
76. van der Wal DE, Davis AM, Mach M, Marks DC. The Role of Neuraminidase 1 and 2 in Glycoprotein Ibalpha-Mediated Integrin Alphaiibbeta3 Activation. Haematologica (2020) 105(4):1081–94. doi: 10.3324/haematol.2019.215830
77. Nath S, Mandal C, Chatterjee U, Mandal C. Association of Cytosolic Sialidase Neu2 With Plasma Membrane Enhances Fas-Mediated Apoptosis by Impairing Pi3k-Akt/Mtor-Mediated Pathway in Pancreatic Cancer Cells. Cell Death Dis (2018) 9(2):210. doi: 10.1038/s41419-017-0191-4
78. Kijimoto-Ochiai S, Doi N, Fujii M, Go S, Kabayama K, Moriya S, et al. Possible Association of Neu2 With Plasma Membrane Fraction From Mouse Thymus Exhibited Sialidase Activity With Fetuin at Ph 7.0 But Not at Ph 4.5. Microbiol Immunol (2013) 57(8):569–82. doi: 10.1111/1348-0421.12076
79. Shiozaki K, Yamaguchi K, Takahashi K, Moriya S, Miyagi T. Regulation of Sialyl Lewis Antigen Expression in Colon Cancer Cells by Sialidase Neu4. J Biol Chem (2011) 286(24):21052–61. doi: 10.1074/jbc.M111.231191
80. Finlay TM, Jayanth P, Amith SR, Gilmour A, Guzzo C, Gee K, et al. Thymoquinone From Nutraceutical Black Cumin Oil Activates Neu4 Sialidase in Live Macrophage, Dendritic, and Normal and Type I Sialidosis Human Fibroblast Cells Via Gpcr Galphai Proteins and Matrix Metalloproteinase-9. Glycoconj J (2010) 27(3):329–48. doi: 10.1007/s10719-010-9281-6
81. Achyuthan KE, Achyuthan AM. Comparative Enzymology, Biochemistry and Pathophysiology of Human Exo-Alpha-Sialidases (Neuraminidases). Comp Biochem Physiol B Biochem Mol Biol (2001) 129(1):29–64. doi: 10.1016/s1096-4959(01)00372-4
82. Monti E, Preti A, Venerando B, Borsani G. Recent Development in Mammalian Sialidase Molecular Biology. Neurochem Res (2002) 27(7-8):649–63. doi: 10.1023/A:1020276000901
83. Miyagi T. Aberrant Expression of Sialidase and Cancer Progression. Proc Jpn Acad Ser B Phys Biol Sci (2008) 84(10):407–18. doi: 10.2183/pjab.84.407
84. Monti E, Bonten E, D’Azzo A, Bresciani R, Venerando B, Borsani G, et al. Sialidases in Vertebrates: A Family of Enzymes Tailored for Several Cell Functions. Adv Carbohydr Chem Biochem (2010) 64:403–79. doi: 10.1016/S0065-2318(10)64007-3
85. Monti E, Miyagi T. Structure and Function of Mammalian Sialidases. Top Curr Chem (2015) 366:183–208. doi: 10.1007/128_2012_328
86. Miyagi T, Takahashi K, Yamamoto K, Shiozaki K, Yamaguchi K. Biological and Pathological Roles of Ganglioside Sialidases. Prog Mol Biol Transl Sci (2018) 156:121–50. doi: 10.1016/bs.pmbts.2017.12.005
87. Bourguet E, Figurska S, Fra Czek MM. Human Neuraminidases: Structures and Stereoselective Inhibitors. J Med Chem (2022) 65(4):3002–25. doi: 10.1021/acs.jmedchem.1c01612
88. Ma J, Rubin BK, Voynow JA. Mucins, Mucus, and Goblet Cells. Chest (2018) 154(1):169–76. doi: 10.1016/j.chest.2017.11.008
89. Rose MC, Voynow JA. Respiratory Tract Mucin Genes and Mucin Glycoproteins in Health and Disease. Physiol Rev (2006) 86(1):245–78. doi: 10.1152/physrev.00010.2005
90. Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, et al. Molecular Cloning and Expression of Human Tumor-Associated Polymorphic Epithelial Mucin. J Biol Chem (1990) 265(25):15286–93. doi: 10.1016/S0021-9258(18)77254-2
91. Lan MS, Batra SK, Qi WN, Metzgar RS, Hollingsworth MA. Cloning and Sequencing of a Human Pancreatic Tumor Mucin Cdna. J Biol Chem (1990) 265(25):15294–9. doi: 10.1016/S0021-9258(18)77255-4
92. Ligtenberg MJ, Vos HL, Gennissen AM, Hilkens J. Episialin, a Carcinoma-Associated Mucin, Is Generated by a Polymorphic Gene Encoding Splice Variants With Alternative Amino Termini. J Biol Chem (1990) 265(10):5573–8. doi: 10.1016/S0021-9258(19)39399-8
93. Ligtenberg MJ, Kruijshaar L, Buijs F, van Meijer M, Litvinov SV, Hilkens J. Cell-Associated Episialin Is a Complex Containing Two Proteins Derived From a Common Precursor. J Biol Chem (1992) 267(9):6171–7. doi: 10.1016/S0021-9258(18)42677-4
94. Levitin F, Stern O, Weiss M, Gil-Henn C, Ziv R, Prokocimer Z, et al. The Muc1 Sea Module Is a Self-Cleaving Domain. J Biol Chem (2005) 280(39):33374–86. doi: 10.1074/jbc.M506047200
95. Macao B, Johansson DG, Hansson GC, Hard T. Autoproteolysis Coupled to Protein Folding in the Sea Domain of the Membrane-Bound Muc1 Mucin. Nat Struct Mol Biol (2006) 13(1):71–6. doi: 10.1038/nsmb1035
96. Julian J, Carson DD. Formation of Muc1 Metabolic Complex Is Conserved in Tumor-Derived and Normal Epithelial Cells. Biochem Biophys Res Commun (2002) 293(4):1183–90. doi: 10.1016/S0006-291X(02)00352-2
97. Blalock TD, Spurr-Michaud SJ, Tisdale AS, Gipson IK. Release of Membrane-Associated Mucins From Ocular Surface Epithelia. Invest Ophthalmol Vis Sci (2008) 49(5):1864–71. doi: 10.1167/iovs.07-1081
98. Kim KC, Wasano K, Niles RM, Schuster JE, Stone PJ, Brody JS. Human Neutrophil Elastase Releases Cell Surface Mucins From Primary Cultures of Hamster Tracheal Epithelial Cells. Proc Natl Acad Sci USA (1987) 84(24):9304–8. doi: 10.1073/pnas.84.24.9304
99. Thathiah A, Blobel CP, Carson DD. Tumor Necrosis Factor-Alpha Converting Enzyme/Adam 17 Mediates Muc1 Shedding. J Biol Chem (2003) 278(5):3386–94. doi: 10.1074/jbc.M208326200
100. Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, et al. Muc1 Limits Helicobacter Pylori Infection Both by Steric Hindrance and by Acting as a Releasable Decoy. PLoS Pathog (2009) 5(10):e1000617. doi: 10.1371/journal.ppat.1000617
101. Thathiah A, Carson DD. Mt1-Mmp Mediates Muc1 Shedding Independent of Tace/Adam17. Biochem J (2004) 382(Pt 1):363–73. doi: 10.1042/BJ20040513
102. Julian J, Dharmaraj N, Carson DD. Muc1 Is a Substrate for Gamma-Secretase. J Cell Biochem (2009) 108(4):802–15. doi: 10.1002/jcb.22292
103. Smorodinsky N, Weiss M, Hartmann ML, Baruch A, Harness E, Yaakobovitz M, et al. Detection of a Secreted Muc1/Sec Protein by Muc1 Isoform Specific Monoclonal Antibodies. Biochem Biophys Res Commun (1996) 228(1):115–21. doi: 10.1006/bbrc.1996.1625
104. Kim KC, Lillehoj EP. Muc1 Mucin: A Peacemaker in the Lung. Am J Respir Cell Mol Biol (2008) 39(6):644–7. doi: 10.1165/rcmb.2008-0169TR
105. Lillehoj EP, Kato K, Lu W, Kim KC. Cellular and Molecular Biology of Airway Mucins. Int Rev Cell Mol Biol (2013) 303:139–202. doi: 10.1016/B978-0-12-407697-6.00004-0
106. Lillehoj ER, Kim KC. Airway Mucus: Its Components and Function. Arch Pharm Res (2002) 25(6):770–80. doi: 10.1007/BF02976990
107. Kato K, Lillehoj EP, Lu W, Kim KC. Muc1: The First Respiratory Mucin With an Anti-Inflammatory Function. J Clin Med (2017) 6(12):110. doi: 10.3390/jcm6120110
108. Argueso P, Herreras JM, Calonge M, Citores L, Pastor JC, Girbes T. Analysis of Human Ocular Mucus: Effects of Neuraminidase and Chitinase Enzymes. Cornea (1998) 17(2):200–7. doi: 10.1097/00003226-199803000-00015
109. Patton S, Gendler SJ, Spicer AP. The Epithelial Mucin, Muc1, of Milk, Mammary Gland and Other Tissues. Biochim Biophys Acta (1995) 1241(3):407–23. doi: 10.1016/0304-4157(95)00014-3
110. Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, et al. A Periciliary Brush Promotes the Lung Health by Separating the Mucus Layer From Airway Epithelia. Science (2012) 337(6097):937–41. doi: 10.1126/science.1223012
111. Stonebraker JR, Wagner D, Lefensty RW, Burns K, Gendler SJ, Bergelson JM, et al. Glycocalyx Restricts Adenoviral Vector Access to Apical Receptors Expressed on Respiratory Epithelium In Vitro and In Vivo: Role for Tethered Mucins as Barriers to Lumenal Infection. J Virol (2004) 78(24):13755–68. doi: 10.1128/JVI.78.24.13755-13768.2004
112. Kato K, Lillehoj EP, Kim KC. Pseudomonas Aeruginosa Stimulates Tyrosine Phosphorylation of and Tlr5 Association With the Muc1 Cytoplasmic Tail Through Egfr Activation. Inflammation Res (2016) 65(3):225–33. doi: 10.1007/s00011-015-0908-8
113. Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, et al. The Epidermal Growth Factor Receptor Regulates Interaction of the Human Df3/Muc1 Carcinoma Antigen With C-Src and Beta-Catenin. J Biol Chem (2001) 276(38):35239–42. doi: 10.1074/jbc.C100359200
114. Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic Muc1 Interacts With Epidermal Growth Factor Receptor and Correlates With Mitogen-Activated Protein Kinase Activation in the Mouse Mammary Gland. J Biol Chem (2001) 276(16):13057–64. doi: 10.1074/jbc.M011248200
115. Chen S, Li M, Jiang W, Zheng H, Qi LW, Jiang S. The Role of Neu1 in the Protective Effect of Dipsacoside B on Acetaminophen-Induced Liver Injury. Ann Transl Med (2020) 8(13):823. doi: 10.21037/atm-19-3850
116. Wang W, Liu L, Zhou Y, Ye Q, Yang X, Jiang J, et al. Hydroxychloroquine Enhances the Antitumor Effects of Bc001 in Gastric Cancer. Int J Oncol (2019) 55(2):405–14. doi: 10.3892/ijo.2019.4824
117. Garbar C, Mascaux C, Giustiniani J, Salesse S, Debelle L, Antonicelli F, et al. Autophagy Is Decreased in Triple-Negative Breast Carcinoma Involving Likely the Muc1-Egfr-Neu1 Signalling Pathway. Int J Clin Exp Pathol (2015) 8(5):4344–55.
118. Kohno N, Akiyama M, Kyoizumi S, Hakoda M, Kobuke K, Yamakido M. Detection of Soluble Tumor-Associated Antigens in Sera and Effusions Using Novel Monoclonal Antibodies, Kl-3 and Kl-6, Against Lung Adenocarcinoma. Jpn J Clin Oncol (1988) 18(3):203–16.
119. Hirasawa Y, Kohno N, Yokoyama A, Inoue Y, Abe M, Hiwada K. Kl-6, a Human Muc1 Mucin, Is Chemotactic for Human Fibroblasts. Am J Respir Cell Mol Biol (1997) 17(4):501–7. doi: 10.1165/ajrcmb.17.4.2253
120. Ohyabu N, Hinou H, Matsushita T, Izumi R, Shimizu H, Kawamoto K, et al. An Essential Epitope of Anti-Muc1 Monoclonal Antibody Kl-6 Revealed by Focused Glycopeptide Library. J Am Chem Soc (2009) 131(47):17102–9. doi: 10.1021/ja903361f
121. Seko A, Ohkura T, Ideo H, Yamashita K. Novel O-Linked Glycans Containing 6’-Sulfo-Gal/Galnac of Muc1 Secreted From Human Breast Cancer Ymb-S Cells: Possible Carbohydrate Epitopes of Kl-6(Muc1) Monoclonal Antibody. Glycobiology (2012) 22(2):181–95. doi: 10.1093/glycob/cwr118
122. Sakai M, Kubota T, Ohnishi H, Yokoyama A. A Novel Lung Injury Animal Model Using Kl-6-Measurable Human Muc1-Expressing Mice. Biochem Biophys Res Commun (2013) 432(3):460–5. doi: 10.1016/j.bbrc.2013.01.123
123. Stahel RA, Gilks WR, Lehmann HP, Schenker T. Third International Workshop on Lung Tumor and Differentiation Antigens: Overview of the Results of the Central Data Analysis. Int J Cancer Suppl (1994) 8:6–26. doi: 10.1002/ijc.2910570704
124. Ishikawa N, Hattori N, Yokoyama A, Kohno N. Utility of Kl-6/Muc1 in the Clinical Management of Interstitial Lung Diseases. Respir Investig (2012) 50(1):3–13. doi: 10.1016/j.resinv.2012.02.001
125. Kohno N. Serum Marker Kl-6/Muc1 for the Diagnosis and Management of Interstitial Pneumonitis. J Med Invest (1999) 46(3-4):151–8.
126. Karhadkar TR, Chen W, Gomer RH. Attenuated Pulmonary Fibrosis in Sialidase-3 Knockout (Neu3(-/-)) Mice. Am J Physiol Lung Cell Mol Physiol (2020) 318(1):L165–L79. doi: 10.1152/ajplung.00275.2019
127. Karhadkar TR, Pilling D, Cox N, Gomer RH. Sialidase Inhibitors Attenuate Pulmonary Fibrosis in a Mouse Model. Sci Rep (2017) 7(1):15069. doi: 10.1038/s41598-017-15198-8
128. Luzina IG, Lockatell V, Hyun SW, Kopach P, Kang PH, Noor Z, et al. Elevated Expression of Neu1 Sialidase in Idiopathic Pulmonary Fibrosis Provokes Pulmonary Collagen Deposition, Lymphocytosis, and Fibrosis. Am J Physiol Lung Cell Mol Physiol (2016) 310(10):L940–54. doi: 10.1152/ajplung.00346.2015
129. Pilling D, Sahlberg K, Karhadkar TR, Chen W, Gomer RH. The Sialidase Neu3 Promotes Pulmonary Fibrosis in Mice. bioRxiv (2022) 2022:02.12.480197. doi: 10.1101/2022.02.12.480197
130. Chen W, Karhadkar TR, Ryu C, Herzog EL, Gomer RH. Reduced Sialylation and Bioactivity of the Antifibrotic Protein Serum Amyloid P in the Sera of Patients With Idiopathic Pulmonary Fibrosis. Immunohorizons (2020) 4(6):352–62. doi: 10.4049/immunohorizons.2000043
131. Guo Z, Tuo H, Tang N, Liu FY, Ma SQ, An P, et al. Neuraminidase 1 Deficiency Attenuates Cardiac Dysfunction, Oxidative Stress, Fibrosis, Inflammatory Via Ampk-Sirt3 Pathway in Diabetic Cardiomyopathy Mice. Int J Biol Sci (2022) 18(2):826–40. doi: 10.7150/ijbs.65938
132. Ghiroldi A, Piccoli M, Creo P, Cirillo F, Rota P, D’Imperio S, et al. Role of Sialidase Neu3 and Ganglioside Gm3 in Cardiac Fibroblasts Activation. Biochem J (2020) 477(17):3401–15. doi: 10.1042/BCJ20200360
133. Inui N, Sakai S, Kitagawa M. Molecular Pathogenesis of Pulmonary Fibrosis, With Focus on Pathways Related to Tgf-Beta and the Ubiquitin-Proteasome Pathway. Int J Mol Sci (2021) 22(11):6107. doi: 10.3390/ijms22116107
134. Ye Z, Hu Y. Tgfbeta1: Gentlemanly Orchestrator in Idiopathic Pulmonary Fibrosis (Review). Int J Mol Med (2021) 48(1):132. doi: 10.3892/ijmm.2021.4965
135. Huaux F. Interpreting Immunoregulation in Lung Fibrosis: A New Branch of the Immune Model. Front Immunol (2021) 12:690375. doi: 10.3389/fimmu.2021.690375
136. van Geffen C, Deissler A, Quante M, Renz H, Hartl D, Kolahian S. Regulatory Immune Cells in Idiopathic Pulmonary Fibrosis: Friends or Foes? Front Immunol (2021) 12:663203. doi: 10.3389/fimmu.2021.663203
137. Shenderov K, Collins SL, Powell JD, Horton MR. Immune Dysregulation as a Driver of Idiopathic Pulmonary Fibrosis. J Clin Invest (2021) 131(2):e143226. doi: 10.1172/JCI143226
138. Desai O, Winkler J, Minasyan M, Herzog EL. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front Med (Lausanne) (2018) 5:43. doi: 10.3389/fmed.2018.00043
139. Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune Mechanisms in Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2016) 55(3):309–22. doi: 10.1165/rcmb.2016-0121TR
140. Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T Lymphocytes in Pulmonary Fibrosis. J Leukoc Biol (2008) 83(2):237–44. doi: 10.1189/jlb.0707504
141. Luzina IG, Todd NW, Sundararajan S, Atamas SP. The Cytokines of Pulmonary Fibrosis: Much Learned, Much More to Learn. Cytokine (2015) 74(1):88–100. doi: 10.1016/j.cyto.2014.11.008
142. Todd NW, Luzina IG, Atamas SP. Molecular and Cellular Mechanisms of Pulmonary Fibrosis. Fibrogenesis Tissue Repair (2012) 5(1):11. doi: 10.1186/1755-1536-5-11
143. Estornut C, Milara J, Bayarri MA, Belhadj N, Cortijo J. Targeting Oxidative Stress as a Therapeutic Approach for Idiopathic Pulmonary Fibrosis. Front Pharmacol (2021) 12:794997. doi: 10.3389/fphar.2021.794997
144. Otoupalova E, Smith S, Cheng G, Thannickal VJ. Oxidative Stress in Pulmonary Fibrosis. Compr Physiol (2020) 10(2):509–47. doi: 10.1002/cphy.c190017
145. Lin C, Borensztajn K, Spek CA. Targeting Coagulation Factor Receptors - Protease-Activated Receptors in Idiopathic Pulmonary Fibrosis. J Thromb Haemost (2017) 15(4):597–607. doi: 10.1111/jth.13623
146. Crooks MG, Hart SP. Coagulation and Anticoagulation in Idiopathic Pulmonary Fibrosis. Eur Respir Rev (2015) 24(137):392–9. doi: 10.1183/16000617.00008414
147. Nho RS, Ballinger MN, Rojas MM, Ghadiali SN, Horowitz JC. Biomechanical Force and Cellular Stiffness in Lung Fibrosis. Am J Pathol (2022) S0002-944(22):00050–5. doi: 10.1016/j.ajpath.2022.02.001
148. Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z, et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell (2020) 180(1):107–21 e17. doi: 10.1016/j.cell.2019.11.027
149. Kellogg DL, Kellogg DL Jr, Musi N, Nambiar AM. Cellular Senescence in Idiopathic Pulmonary Fibrosis. Curr Mol Biol Rep (2021) 7(3):31–40. doi: 10.1007/s40610-021-00145-4
150. Hernandez-Gonzalez F, Faner R, Rojas M, Agusti A, Serrano M, Sellares J. Cellular Senescence in Lung Fibrosis. Int J Mol Sci (2021) 22(13):7012. doi: 10.3390/ijms22137012
151. Pardo A, Selman M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol (2021) 64(2):163–72. doi: 10.1165/rcmb.2020-0373PS
152. Merkt W, Bueno M, Mora AL, Lagares D. Senotherapeutics: Targeting Senescence in Idiopathic Pulmonary Fibrosis. Semin Cell Dev Biol (2020) 101:104–10. doi: 10.1016/j.semcdb.2019.12.008
153. Gulati S, Thannickal VJ. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am J Med Sci (2019) 357(5):384–9. doi: 10.1016/j.amjms.2019.02.008
154. Waters DW, Blokland KEC, Pathinayake PS, Burgess JK, Mutsaers SE, Prele CM, et al. Fibroblast Senescence in the Pathology of Idiopathic Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2018) 315(2):L162–L72. doi: 10.1152/ajplung.00037.2018
155. Zank DC, Bueno M, Mora AL, Rojas M. Idiopathic Pulmonary Fibrosis: Aging, Mitochondrial Dysfunction, and Cellular Bioenergetics. Front Med (Lausanne) (2018) 5:10. doi: 10.3389/fmed.2018.00010
156. Zhao H, Wang Y, Qiu T, Liu W, Yao P. Autophagy, an Important Therapeutic Target for Pulmonary Fibrosis Diseases. Clin Chim Acta (2020) 502:139–47. doi: 10.1016/j.cca.2019.12.016
157. O’Dwyer DN, Ashley SL, Moore BB. Influences of Innate Immunity, Autophagy, and Fibroblast Activation in the Pathogenesis of Lung Fibrosis. Am J Physiol Lung Cell Mol Physiol (2016) 311(3):L590–601. doi: 10.1152/ajplung.00221.2016
158. Katzen J, Beers MF. Contributions of Alveolar Epithelial Cell Quality Control to Pulmonary Fibrosis. J Clin Invest (2020) 130(10):5088–99. doi: 10.1172/JCI139519
159. Selman M, Pardo A. The Leading Role of Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Cell Signal (2020) 66:109482. doi: 10.1016/j.cellsig.2019.109482
160. Hewlett JC, Kropski JA, Blackwell TS. Idiopathic Pulmonary Fibrosis: Epithelial-Mesenchymal Interactions and Emerging Therapeutic Targets. Matrix Biol (2018) 71-72:112–27. doi: 10.1016/j.matbio.2018.03.021
161. Kulkarni T, de Andrade J, Zhou Y, Luckhardt T, Thannickal VJ. Alveolar Epithelial Disintegrity in Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2016) 311(2):L185–91. doi: 10.1152/ajplung.00115.2016
162. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med (2018) 378(19):1811–23. doi: 10.1056/NEJMra1705751
163. Moss BJ, Ryter SW, Rosas IO. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu Rev Pathol (2022) 17:515–46. doi: 10.1146/annurev-pathol-042320-030240
164. Akiyama M, Kaneko Y. Pathogenesis, Clinical Features, and Treatment Strategy for Rheumatoid Arthritis-Associated Interstitial Lung Disease. Autoimmun Rev (2022) 21(5):103056. doi: 10.1016/j.autrev.2022.103056
165. Shao T, Shi X, Yang S, Zhang W, Li X, Shu J, et al. Interstitial Lung Disease in Connective Tissue Disease: A Common Lesion With Heterogeneous Mechanisms and Treatment Considerations. Front Immunol (2021) 12:684699. doi: 10.3389/fimmu.2021.684699
166. Perelas A, Silver RM, Arrossi AV, Highland KB. Systemic Sclerosis-Associated Interstitial Lung Disease. Lancet Respir Med (2020) 8(3):304–20. doi: 10.1016/S2213-2600(19)30480-1
167. Lambre CR, Pilatte Y, Le Maho S, Greffard A, De Cremoux H, Bignon J. Sialidase Activity and Antibodies to Sialidase-Treated Autologous Erythrocytes in Bronchoalveolar Lavages From Patients With Idiopathic Pulmonary Fibrosis or Sarcoidosis. Clin Exp Immunol (1988) 73(2):230–5.
168. Kohno N, Kyoizumi S, Awaya Y, Fukuhara H, Yamakido M, Akiyama M. New Serum Indicator of Interstitial Pneumonitis Activity. Sialylated Carbohydrate Antigen Kl-6. Chest (1989) 96(1):68–73. doi: 10.1378/chest.96.1.68
169. Kohno N, Awaya Y, Oyama T, Yamakido M, Akiyama M, Inoue Y, et al. Kl-6, a Mucin-Like Glycoprotein, in Bronchoalveolar Lavage Fluid From Patients With Interstitial Lung Disease. Am Rev Respir Dis (1993) 148(3):637–42. doi: 10.1164/ajrccm/148.3.637
170. Kohno N, Yokoyama A, Hirasawa Y, Kondo K, Fujino S, Abe M, et al. Comparative Studies of Circulating Kl-6, Type Iii Procollagen N-Terminal Peptide and Type Iv Collagen 7s in Patients With Interstitial Pneumonitis and Alveolar Pneumonia. Respir Med (1997) 91(9):558–61. doi: 10.1016/s0954-6111(97)90090-1
171. Hamai K, Iwamoto H, Ishikawa N, Horimasu Y, Masuda T, Miyamoto S, et al. Comparative Study of Circulating Mmp-7, Ccl18, Kl-6, Sp-A, and Sp-D as Disease Markers of Idiopathic Pulmonary Fibrosis. Dis Markers (2016) 2016:4759040. doi: 10.1155/2016/4759040
172. Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, et al. Comparative Study of Kl-6, Surfactant Protein-A, Surfactant Protein-D, and Monocyte Chemoattractant Protein-1 as Serum Markers for Interstitial Lung Diseases. Am J Respir Crit Care Med (2002) 165(3):378–81. doi: 10.1164/ajrccm.165.3.2107134
173. Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, et al. Baseline Kl-6 Predicts Increased Risk for Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Respir Med (2014) 108(7):1031–9. doi: 10.1016/j.rmed.2014.04.009
174. Yokoyama A, Kohno N, Kondo K, Ueda S, Hirasawa Y, Watanabe K, et al. Comparative Evaluation of Sialylated Carbohydrate Antigens, Kl-6, Ca19-9 and Slx as Serum Markers for Interstitial Pneumonia. Respirology (1998) 3(3):199–202. doi: 10.1111/j.1440-1843.1998.tb00121.x
175. Aloisio E, Braga F, Puricelli C, Panteghini M. Prognostic Role of Krebs Von Den Lungen-6 (Kl-6) Measurement in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Clin Chem Lab Med (2021) 59(8):1400–8. doi: 10.1515/cclm-2021-0199
176. Zhang H, Chen L, Wu L, Huang J, Li H, Wang X, et al. Diagnostic and Prognostic Predictive Values of Circulating Kl-6 for Interstitial Lung Disease: A Prisma-Compliant Systematic Review and Meta-Analysis. Med (Baltimore) (2020) 99(16):e19493. doi: 10.1097/MD.0000000000019493
177. Bonella F, Long X, Ohshimo S, Horimasu Y, Griese M, Guzman J, et al. Muc1 Gene Polymorphisms Are Associated With Serum Kl-6 Levels and Pulmonary Dysfunction in Pulmonary Alveolar Proteinosis. Orphanet J Rare Dis (2016) 11:48. doi: 10.1186/s13023-016-0430-2
178. Horimasu Y, Hattori N, Ishikawa N, Kawase S, Tanaka S, Yoshioka K, et al. Different Muc1 Gene Polymorphisms in German and Japanese Ethnicities Affect Serum Kl-6 Levels. Respir Med (2012) 106(12):1756–64. doi: 10.1016/j.rmed.2012.09.001
179. Janssen R, Kruit A, Grutters JC, Ruven HJ, Gerritsen WB, van den Bosch JM. The Mucin-1 568 Adenosine to Guanine Polymorphism Influences Serum Krebs Von Den Lungen-6 Levels. Am J Respir Cell Mol Biol (2006) 34(4):496–9. doi: 10.1165/rcmb.2005-0151OC
180. van den Blink B, Wijsenbeek MS, Hoogsteden HC. Serum Biomarkers in Idiopathic Pulmonary Fibrosis. Pulm Pharmacol Ther (2010) 23(6):515–20. doi: 10.1016/j.pupt.2010.08.001
181. d’Alessandro M, Bergantini L, Cameli P, Vietri L, Lanzarone N, Alonzi V, et al. Krebs Von Den Lungen-6 as a Biomarker for Disease Severity Assessment in Interstitial Lung Disease: A Comprehensive Review. Biomark Med (2020) 14(8):665–74. doi: 10.2217/bmm-2019-0545
182. Zhang Y, Kaminski N. Biomarkers in Idiopathic Pulmonary Fibrosis. Curr Opin Pulm Med (2012) 18(5):441–6. doi: 10.1097/MCP.0b013e328356d03c
183. Vij R, Noth I. Peripheral Blood Biomarkers in Idiopathic Pulmonary Fibrosis. Transl Res (2012) 159(4):218–27. doi: 10.1016/j.trsl.2012.01.012
184. Ohshimo S, Yokoyama A, Hattori N, Ishikawa N, Hirasawa Y, Kohno N. Kl-6, a Human Muc1 Mucin, Promotes Proliferation and Survival of Lung Fibroblasts. Biochem Biophys Res Commun (2005) 338(4):1845–52. doi: 10.1016/j.bbrc.2005.10.144
185. Xu L, Yang D, Zhu S, Gu J, Ding F, Bian W, et al. Bleomycin-Induced Pulmonary Fibrosis Is Attenuated by an Antibody Against Kl-6. Exp Lung Res (2013) 39(6):241–8. doi: 10.3109/01902148.2013.798056
186. Milara J, Ballester B, Montero P, Escriva J, Artigues E, Alos M, et al. Muc1 Intracellular Bioactivation Mediates Lung Fibrosis. Thorax (2020) 75(2):132–42. doi: 10.1136/thoraxjnl-2018-212735
187. Ballester B, Milara J, Cortijo J. Pirfenidone Anti-Fibrotic Effects Are Partially Mediated by the Inhibition of Muc1 Bioactivation. Oncotarget (2020) 11(15):1306–20. doi: 10.18632/oncotarget.27526
188. Kato K, Zemskova MA, Hanss AD, Kim MM, Summer R, Kim KC. Muc1 Deficiency Exacerbates Pulmonary Fibrosis in a Mouse Model of Silicosis. Biochem Biophys Res Commun (2017) 493(3):1230–5. doi: 10.1016/j.bbrc.2017.09.047
189. Ohtsuki Y, Fujita J, Hachisuka Y, Uomoto M, Okada Y, Yoshinouchi T, et al. Immunohistochemical and Immunoelectron Microscopic Studies of the Localization of Kl-6 and Epithelial Membrane Antigen (Ema) in Presumably Normal Pulmonary Tissue and in Interstitial Pneumonia. Med Mol Morphol (2007) 40(4):198–202. doi: 10.1007/s00795-007-0382-7
190. Ballester B, Milara J, Cortijo J. The Role of Mucin 1 in Respiratory Diseases. Eur Respir Rev (2021) 30(159):200149. doi: 10.1183/16000617.0149-2020
191. Pardo A, Cabrera S, Maldonado M, Selman M. Role of Matrix Metalloproteinases in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Respir Res (2016) 17:23. doi: 10.1186/s12931-016-0343-6
192. Klein C, Troedsson M. Equine Pre-Implantation Conceptuses Express Neuraminidase 2–A Potential Mechanism for Desialylation of the Equine Capsule. Reprod Domest Anim (2012) 47(3):449–54. doi: 10.1111/j.1439-0531.2011.01901.x
193. Tringali C, Papini N, Fusi P, Croci G, Borsani G, Preti A, et al. Properties of Recombinant Human Cytosolic Sialidase Hsneu2. The Enzyme Hydrolyzes Monomerically Dispersed Gm1 Ganglioside Molecules. J Biol Chem (2004) 279(5):3169–79. doi: 10.1074/jbc.M308381200
194. Bhargava AK, Woitach JT, Davidson EA, Bhavanandan VP. Cloning and Cdna Sequence of a Bovine Submaxillary Gland Mucin-Like Protein Containing Two Distinct Domains. Proc Natl Acad Sci USA (1990) 87(17):6798–802. doi: 10.1073/pnas.87.17.6798
195. Eckhardt AE, Timpte CS, DeLuca AW, Hill RL. The Complete Cdna Sequence and Structural Polymorphism of the Polypeptide Chain of Porcine Submaxillary Mucin. J Biol Chem (1997) 272(52):33204–10. doi: 10.1074/jbc.272.52.33204
196. Jiang W, Woitach JT, Gupta D, Bhavanandan VP. Sequence of a Second Gene Encoding Bovine Submaxillary Mucin: Implication for Mucin Heterogeneity and Cloning. Biochem Biophys Res Commun (1998) 251(2):550–6. doi: 10.1006/bbrc.1998.9515
197. Seyrantepe V, Landry K, Trudel S, Hassan JA, Morales CR, Pshezhetsky AV. Neu4, a Novel Human Lysosomal Lumen Sialidase, Confers Normal Phenotype to Sialidosis and Galactosialidosis Cells. J Biol Chem (2004) 279(35):37021–9. doi: 10.1074/jbc.M404531200
198. Yamaguchi K, Hata K, Koseki K, Shiozaki K, Akita H, Wada T, et al. Evidence for Mitochondrial Localization of a Novel Human Sialidase (Neu4). Biochem J (2005) 390(Pt 1):85–93. doi: 10.1042/BJ20050017
199. Finlay TM, Abdulkhalek S, Gilmour A, Guzzo C, Jayanth P, Amith SR, et al. Thymoquinone-Induced Neu4 Sialidase Activates Nfkappab in Macrophage Cells and Pro-Inflammatory Cytokines In Vivo. Glycoconj J (2010) 27(6):583–600. doi: 10.1007/s10719-010-9302-5
200. Kishore A, Zizkova V, Kocourkova L, Petrkova J, Bouros E, Nunes H, et al. Association Study for 26 Candidate Loci in Idiopathic Pulmonary Fibrosis Patients From Four European Populations. Front Immunol (2016) 7:274. doi: 10.3389/fimmu.2016.00274
201. Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, et al. Ectopic Respiratory Epithelial Cell Differentiation in Bronchiolised Distal Airspaces in Idiopathic Pulmonary Fibrosis. Thorax (2011) 66(8):651–7. doi: 10.1136/thx.2010.151555
202. Lorenzo-Salazar JM, Ma SF, Jou J, Hou PC, Guillen-Guio B, Allen RJ, et al. Novel Idiopathic Pulmonary Fibrosis Susceptibility Variants Revealed by Deep Sequencing. ERJ Open Res (2019) 5(2):00071–2019. doi: 10.1183/23120541.00071-2019
203. Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The Idiopathic Pulmonary Fibrosis Honeycomb Cyst Contains a Mucocilary Pseudostratified Epithelium. PLoS One (2013) 8(3):e58658. doi: 10.1371/journal.pone.0058658
204. Pang J, Luo Y, Wei D, Cao Z, Qi X, Song M, et al. Comparative Transcriptome Analyses Reveal a Transcriptional Landscape of Human Silicosis Lungs and Provide Potential Strategies for Silicosis Treatment. Front Genet (2021) 12:652901. doi: 10.3389/fgene.2021.652901
205. He L, Feng QQ, Zhang Q, Zhang B, Wu SS, Gong JH. Protective Role of Overexpressed Muc5ac Against Fibrosis in Mhv-68-Induced Combined Pulmonary Fibrosis and Emphysema Mouse Model. J Med Virol (2020) 92(12):3726–35. doi: 10.1002/jmv.26094
206. Hoffmann J, Wilhelm J, Marsh LM, Ghanim B, Klepetko W, Kovacs G, et al. Distinct Differences in Gene Expression Patterns in Pulmonary Arteries of Patients With Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis With Pulmonary Hypertension. Am J Respir Crit Care Med (2014) 190(1):98–111. doi: 10.1164/rccm.201401-0037OC
207. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An Epithelial Biomarker Signature for Idiopathic Pulmonary Fibrosis: An Analysis From the Multicentre Profile Cohort Study. Lancet Respir Med (2017) 5(12):946–55. doi: 10.1016/S2213-2600(17)30430-7
208. Milara J, Ballester B, Safont MJ, Artigues E, Escriva J, Morcillo E, et al. Muc4 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Collaborates With Transforming Growth Factor Beta Inducing Fibrotic Responses. Mucosal Immunol (2021) 14(2):377–88. doi: 10.1038/s41385-020-00343-w
209. Ballester B, Milara J, Montero P, Cortijo J. Muc16 Is Overexpressed in Idiopathic Pulmonary Fibrosis and Induces Fibrotic Responses Mediated by Transforming Growth Factor-Beta1 Canonical Pathway. Int J Mol Sci (2021) 22(12):6502. doi: 10.3390/ijms22126502
210. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A Common Muc5b Promoter Polymorphism and Pulmonary Fibrosis. N Engl J Med (2011) 364(16):1503–12. doi: 10.1056/NEJMoa1013660
211. Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, et al. Idiopathic Pulmonary Fibrosis: A Genetic Disease That Involves Mucociliary Dysfunction of the Peripheral Airways. Physiol Rev (2016) 96(4):1567–91. doi: 10.1152/physrev.00004.2016
212. Schwartz DA. Idiopathic Pulmonary Fibrosis Is a Genetic Disease Involving Mucus and the Peripheral Airways. Ann Am Thorac Soc (2018) 15(Suppl 3):S192–S7. doi: 10.1513/AnnalsATS.201802-144AW
213. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. Muc5b Promoter Variant and Rheumatoid Arthritis With Interstitial Lung Disease. N Engl J Med (2018) 379(23):2209–19. doi: 10.1056/NEJMoa1801562
214. Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b Overexpression Causes Mucociliary Dysfunction and Enhances Lung Fibrosis in Mice. Nat Commun (2018) 9(1):5363. doi: 10.1038/s41467-018-07768-9
215. Juge PA, Solomon JJ, van Moorsel CHM, Garofoli R, Lee JS, Louis-Sydney F, et al. Muc5b Promoter Variant Rs35705950 and Rheumatoid Arthritis Associated Interstitial Lung Disease Survival and Progression. Semin Arthritis Rheum (2021) 51(5):996–1004. doi: 10.1016/j.semarthrit.2021.07.002
216. Pier GB, Ramphal R. Pseudomonas Aeruginosa. In: Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Primciples and Practice Infectious Diseases, 2. 7th ed. Philadelphia, PA: Churchill Livingstone Elsevier (2010). p. 2835–60.
217. Ramphal R, Guay C, Pier GB. Pseudomonas Aeruginosa Adhesins for Tracheobronchial Mucin. Infect Immun (1987) 55(3):600–3. doi: 10.1128/iai.55.3.600-603.1987
218. Reddy MS. Human Tracheobronchial Mucin: Purification and Binding to Pseudomonas Aeruginosa. Infect Immun (1992) 60(4):1530–5. doi: 10.1128/iai.60.4.1530-1535.1992
219. Sajjan U, Reisman J, Doig P, Irvin RT, Forstner G, Forstner J. Binding of Nonmucoid Pseudomonas Aeruginosa to Normal Human Intestinal Mucin and Respiratory Mucin From Patients With Cystic Fibrosis. J Clin Invest (1992) 89(2):657–65. doi: 10.1172/JCI115632
220. Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, et al. Muc1 Mucins on the Cell Surface Are Adhesion Sites for Pseudomonas Aeruginosa. Am J Physiol Lung Cell Mol Physiol (2001) 280(1):L181–7. doi: 10.1152/ajplung.2001.280.1.L181
221. Kato K, Lillehoj EP, Kai H, Kim KC. Muc1 Expression by Human Airway Epithelial Cells Mediates Pseudomonas Aeruginosa Adhesion. Front Biosci (Elite Ed) (2010) 2:68–77. doi: 10.2741/e67
222. Lillehoj EP, Kim H, Chun EY, Kim KC. Pseudomonas Aeruginosa Stimulates Phosphorylation of the Airway Epithelial Membrane Glycoprotein Muc1 and Activates Map Kinase. Am J Physiol Lung Cell Mol Physiol (2004) 287(4):L809–15. doi: 10.1152/ajplung.00385.2003
223. Lu W, Hisatsune A, Koga T, Kato K, Kuwahara I, Lillehoj EP, et al. Cutting Edge: Enhanced Pulmonary Clearance of Pseudomonas Aeruginosa by Muc1 Knockout Mice. J Immunol (2006) 176(7):3890–4. doi: 10.4049/jimmunol.176.7.3890
224. Ueno K, Koga T, Kato K, Golenbock DT, Gendler SJ, Kai H, et al. Muc1 Mucin Is a Negative Regulator of Toll-Like Receptor Signaling. Am J Respir Cell Mol Biol (2008) 38(3):263–8. doi: 10.1165/rcmb.2007-0336RC
225. Williams MA, Bauer S, Lu W, Guo J, Walter S, Bushnell TP, et al. Deletion of the Mucin-Like Molecule Muc1 Enhances Dendritic Cell Activation in Response to Toll-Like Receptor Ligands. J Innate Immun (2010) 2(2):123–43. doi: 10.1159/000254790
226. Choi S, Park YS, Koga T, Treloar A, Kim KC. Tnf-Alpha Is a Key Regulator of Muc1, an Anti-Inflammatory Molecule, During Airway Pseudomonas Aeruginosa Infection. Am J Respir Cell Mol Biol (2011) 44(2):255–60. doi: 10.1165/rcmb.2009-0323OC
227. Kato K, Lillehoj EP, Park YS, Umehara T, Hoffman NE, Madesh M, et al. Membrane-Tethered Muc1 Mucin Is Phosphorylated by Epidermal Growth Factor Receptor in Airway Epithelial Cells and Associates With Tlr5 to Inhibit Recruitment of Myd88. J Immunol (2012) 188(4):2014–22. doi: 10.4049/jimmunol.1102405
228. Umehara T, Kato K, Park YS, Lillehoj EP, Kawauchi H, Kim KC. Prevention of Lung Injury by Muc1 Mucin in a Mouse Model of Repetitive Pseudomonas Aeruginosa Infection. Inflammation Res (2012) 61(9):1013–20. doi: 10.1007/s00011-012-0494-y
229. Milara J, Peiro T, Armengot M, Frias S, Morell A, Serrano A, et al. Mucin 1 Downregulation Associates With Corticosteroid Resistance in Chronic Rhinosinusitis With Nasal Polyps. J Allergy Clin Immunol (2015) 135(2):470–6. doi: 10.1016/j.jaci.2014.07.011
230. Bachert C, Linstedt AD. A Sensor of Protein O-Glycosylation Based on Sequential Processing in the Golgi Apparatus. Traffic (2013) 14(1):47–56. doi: 10.1111/tra.12019
231. Magesh S, Moriya S, Suzuki T, Miyagi T, Ishida H, Kiso M. Design, Synthesis, and Biological Evaluation of Human Sialidase Inhibitors. Part 1: Selective Inhibitors of Lysosomal Sialidase (Neu1). Bioorg Med Chem Lett (2008) 18(2):532–7. doi: 10.1016/j.bmcl.2007.11.084
232. Verceles AC, Bhat P, Nagaria Z, Martin D, Patel H, Ntem-Mensah A, et al. Muc1 Ectodomain Is a Flagellin-Targeting Decoy Receptor and Biomarker Operative During Pseudomonas Aeruginosa Lung Infection. Sci Rep (2021) 11(1):22725. doi: 10.1038/s41598-021-02242-x
233. Hyun SW, Imamura A, Ishida H, Piepenbrink KH, Goldblum SE, Lillehoj EP. The Sialidase Neu1 Directly Interacts With the Juxtamembranous Segment of the Cytoplasmic Domain of Mucin-1 to Inhibit Downstream Pi3k-Akt Signaling. J Biol Chem (2021) 297(5):101337. doi: 10.1016/j.jbc.2021.101337
234. Yang WH, Heithoff DM, Aziz PV, Haslund-Gourley B, Westman JS, Narisawa S, et al. Accelerated Aging and Clearance of Host Anti-Inflammatory Enzymes by Discrete Pathogens Fuels Sepsis. Cell Host Microbe (2018) 24(4):500–13 e5. doi: 10.1016/j.chom.2018.09.011
235. Yang WH, Heithoff DM, Aziz PV, Sperandio M, Nizet V, Mahan MJ, et al. Recurrent Infection Progressively Disables Host Protection Against Intestinal Inflammation. Science (2017) 358(6370):eaao5610. doi: 10.1126/science.aao5610
236. Yang WH, Westman JS, Heithoff DM, Sperandio M, Cho JW, Mahan MJ, et al. Neu3 Neuraminidase Induction Triggers Intestinal Inflammation and Colitis in a Model of Recurrent Human Food-Poisoning. Proc Natl Acad Sci USA (2021) 118(29):e2100937118. doi: 10.1073/pnas.2100937118
237. Chen QQ, Ma G, Liu JF, Cai YY, Zhang JY, Wei TT, et al. Neuraminidase 1 Is a Driver of Experimental Cardiac Hypertrophy. Eur Heart J (2021) 42(36):3770–82. doi: 10.1093/eurheartj/ehab347
238. Thulasiraman P, Kerr K, McAlister K, Hardisty S, Wistner A, McCullough I. Neuraminidase 1 Regulates Proliferation, Apoptosis and the Expression of Cadherins in Mammary Carcinoma Cells. Mol Cell Biochem (2019) 462(1-2):207–15. doi: 10.1007/s11010-019-03623-7
239. Kapishon V, Allison S, Whitney RA, Cunningham MF, Szewczuk MR, Neufeld RJ. Oseltamivir-Conjugated Polymeric Micelles Prepared by Raft Living Radical Polymerization as a New Active Tumor Targeting Drug Delivery Platform. Biomater Sci (2016) 4(3):511–21. doi: 10.1039/c5bm00519a
240. Formiga RO, Amaral FC, Souza CF, Mendes D, Wanderley CWS, Lorenzini CB, et al. Neuraminidase Inhibitors Rewire Neutrophil Function In Vivo in Murine Sepsis and Ex Vivo in Covid-19. bioRxiv (2021) 2020:11.12.379115. doi: 10.1101/2020.11.12.379115
241. Teixeira A, David L, Reis CA, Costa J, Sobrinho-Simoes M. Expression of Mucins (Muc1, Muc2, Muc5ac, and Muc6) and Type 1 Lewis Antigens in Cases With and Without Helicobacter Pylori Colonization in Metaplastic Glands of the Human Stomach. J Pathol (2002) 197(1):37–43. doi: 10.1002/path.1083
242. Linden S, Nordman H, Hedenbro J, Hurtig M, Boren T, Carlstedt I. Strain- and Blood Group-Dependent Binding of Helicobacter Pylori to Human Gastric Muc5ac Glycoforms. Gastroenterology (2002) 123(6):1923–30. doi: 10.1053/gast.2002.37076
243. Linden S, Mahdavi J, Hedenbro J, Boren T, Carlstedt I. Effects of Ph on Helicobacter Pylori Binding to Human Gastric Mucins: Identification of Binding to Non-Muc5ac Mucins. Biochem J (2004) 384(Pt 2):263–70. doi: 10.1042/BJ20040402
244. McGuckin MA, Every AL, Skene CD, Linden SK, Chionh YT, Swierczak A, et al. Muc1 Mucin Limits Both Helicobacter Pylori Colonization of the Murine Gastric Mucosa and Associated Gastritis. Gastroenterology (2007) 133(4):1210–8. doi: 10.1053/j.gastro.2007.07.003
245. Costa NR, Mendes N, Marcos NT, Reis CA, Caffrey T, Hollingsworth MA, et al. Relevance of Muc1 Mucin Variable Number of Tandem Repeats Polymorphism in H Pylori Adhesion to Gastric Epithelial Cells. World J Gastroenterol (2008) 14(9):1411–4. doi: 10.3748/wjg.14.1411
246. Radziejewska I, Borzym-Kluczyk M, Leszczynska K. Are Lewis B and H Type 1 on Helicobacter Pylori Involved in Binding of Bacteria to Muc1 Mucin? Adv Clin Exp Med (2013) 22(3):347–53.
247. Koning N, Kessen SF, van der Voorn JP, Appelmelk BJ, Jeurink PV, Knippels LM, et al. Human Milk Blocks Dc-Sign-Pathogen Interaction Via Muc1. Front Immunol (2015) 6:112. doi: 10.3389/fimmu.2015.00112
248. Radziejewska I, Borzym-Kluczyk M, Leszczynska K. Luteolin Alters Muc1 Extracellular Domain, St Antigen, Adam-17, Il-8, Il-10 and Nf-Kappab Expression in Helicobacter Pylori-Infected Gastric Cancer Crl-1739 Cells: A Preliminary Study. BioMed Rep (2021) 14(2):19. doi: 10.3892/br.2020.1395
249. Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TG, Lillehoj EP. Muc1 Cell Surface Mucin Attenuates Epithelial Inflammation in Response to a Common Mucosal Pathogen. J Biol Chem (2010) 285(27):20547–57. doi: 10.1074/jbc.M110.121319
250. Guang W, Twaddell WS, Lillehoj EP. Molecular Interactions Between Muc1 Epithelial Mucin, Beta-Catenin, and Caga Proteins. Front Immunol (2012) 3:105. doi: 10.3389/fimmu.2012.00105
251. Lillehoj EP, Guang W, Ding H, Czinn SJ, Blanchard TG. Helicobacter Pylori and Gastric Inflammation: Role of Muc1 Mucin. J Pediatr Biochem (2012) 2(3):125–32. doi: 10.3233/JPB-2012-00058
252. Park YS, Guang W, Blanchard TG, Kim KC, Lillehoj EP. Suppression of Il-8 Production in Gastric Epithelial Cells by Muc1 Mucin and Peroxisome Proliferator-Associated Receptor-Gamma. Am J Physiol Gastrointest Liver Physiol (2012) 303(6):G765–74. doi: 10.1152/ajpgi.00023.2012
253. Sheng YH, Triyana S, Wang R, Das I, Gerloff K, Florin TH, et al. Muc1 and Muc13 Differentially Regulate Epithelial Inflammation in Response to Inflammatory and Infectious Stimuli. Mucosal Immunol (2013) 6(3):557–68. doi: 10.1038/mi.2012.98
254. Guang W, Czinn SJ, Blanchard TG, Kim KC, Lillehoj EP. Genetic Regulation of Muc1 Expression by Helicobacter Pylori in Gastric Cancer Cells. Biochem Biophys Res Commun (2014) 445(1):145–50. doi: 10.1016/j.bbrc.2014.01.142
255. Ng GZ, Menheniott TR, Every AL, Stent A, Judd LM, Chionh YT, et al. The Muc1 Mucin Protects Against Helicobacter Pylori Pathogenesis in Mice by Regulation of the Nlrp3 Inflammasome. Gut (2016) 65(7):1087–99. doi: 10.1136/gutjnl-2014-307175
256. Sheng YH, Ng GZ, Summers KM, Every AL, Price G, Hasnain SZ, et al. Influence of the Muc1 Cell Surface Mucin on Gastric Mucosal Gene Expression Profiles in Response to Helicobacter Pylori Infection in Mice. Front Cell Infect Microbiol (2020) 10:343. doi: 10.3389/fcimb.2020.00343
257. Kardon R, Price RE, Julian J, Lagow E, Tseng SC, Gendler SJ, et al. Bacterial Conjunctivitis in Muc1 Null Mice. Invest Ophthalmol Vis Sci (1999) 40(7):1328–35.
258. DeSouza MM, Surveyor GA, Price RE, Julian J, Kardon R, Zhou X, et al. Muc1/Episialin: A Critical Barrier in the Female Reproductive Tract. J Reprod Immunol (1999) 45(2):127–58. doi: 10.1016/s0165-0378(99)00046-7
259. McAuley JL, Linden SK, Png CW, King RM, Pennington HL, Gendler SJ, et al. Muc1 Cell Surface Mucin Is a Critical Element of the Mucosal Barrier to Infection. J Clin Invest (2007) 117(8):2313–24. doi: 10.1172/JCI26705
260. Sando L, Pearson R, Gray C, Parker P, Hawken R, Thomson PC, et al. Bovine Muc1 Is a Highly Polymorphic Gene Encoding an Extensively Glycosylated Mucin That Binds Bacteria. J Dairy Sci (2009) 92(10):5276–91. doi: 10.3168/jds.2009-2216
261. Parker P, Sando L, Pearson R, Kongsuwan K, Tellam RL, Smith S. Bovine Muc1 Inhibits Binding of Enteric Bacteria to Caco-2 Cells. Glycoconj J (2010) 27(1):89–97. doi: 10.1007/s10719-009-9269-2
262. Kyo Y, Kato K, Park YS, Gajghate S, Umehara T, Lillehoj EP, et al. Antiinflammatory Role of Muc1 Mucin During Infection With Nontypeable Haemophilus Influenzae. Am J Respir Cell Mol Biol (2012) 46(2):149–56. doi: 10.1165/rcmb.2011-0142OC
263. Ng GZ, Sutton P. The Muc1 Mucin Specifically Inhibits Activation of the Nlrp3 Inflammasome. Genes Immun (2016) 17(3):203–6. doi: 10.1038/gene.2016.10
264. Boll EJ, Ayala-Lujan J, Szabady RL, Louissaint C, Smith RZ, Krogfelt KA, et al. Enteroaggregative Escherichia Coli Adherence Fimbriae Drive Inflammatory Cell Recruitment Via Interactions With Epithelial Muc1. mBio (2017) 8(3):e00717–17. doi: 10.1128/mBio.00717-17
265. Dhar P, Ng GZ, Dunne EM, Sutton P. Mucin 1 Protects Against Severe Streptococcus Pneumoniae Infection. Virulence (2017) 8(8):1631–42. doi: 10.1080/21505594.2017.1341021
266. Li X, Bleumink-Pluym NMC, Luijkx Y, Wubbolts RW, van Putten JPM, Strijbis K. Muc1 Is a Receptor for the Salmonella Siie Adhesin That Enables Apical Invasion Into Enterocytes. PLoS Pathog (2019) 15(2):e1007566. doi: 10.1371/journal.ppat.1007566
267. Li X, Wubbolts RW, Bleumink-Pluym NMC, van Putten JPM, Strijbis K. The Transmembrane Mucin Muc1 Facilitates Beta1-Integrin-Mediated Bacterial Invasion. mBio (2021) 12(2):e03491–20. doi: 10.1128/mBio.03491-20
268. Pech M, Weckmann M, Konig IR, Franke A, Heinsen FA, Oliver B, et al. Rhinovirus Infections Change DNA Methylation and Mrna Expression in Children With Asthma. PLoS One (2018) 13(11):e0205275. doi: 10.1371/journal.pone.0205275
269. Kong F, Li N, Tu T, Tao Y, Bi Y, Yuan D, et al. Hepatitis B Virus Core Protein Promotes the Expression of Neuraminidase 1 to Facilitate Hepatocarcinogenesis. Lab Invest (2020) 100(12):1602–17. doi: 10.1038/s41374-020-0465-9
270. Murray J, Todd KV, Bakre A, Orr-Burks N, Jones L, Wu W, et al. A Universal Mammalian Vaccine Cell Line Substrate. PLoS One (2017) 12(11):e0188333. doi: 10.1371/journal.pone.0188333
271. Anastasia L, Holguera J, Bianchi A, D’Avila F, Papini N, Tringali C, et al. Over-Expression of Mammalian Sialidase Neu3 Reduces Newcastle Disease Virus Entry and Propagation in Cos7 Cells. Biochim Biophys Acta (2008) 1780(3):504–12. doi: 10.1016/j.bbagen.2007.11.011
272. Lu W, Liu X, Wang T, Liu F, Zhu A, Lin Y, et al. Elevated Muc1 and Muc5ac Mucin Protein Levels in Airway Mucus of Critical Ill Covid-19 Patients. J Med Virol (2021) 93(2):582–4. doi: 10.1002/jmv.26406
273. Kim D, Maharjan S, Kim J, Park S, Park JA, Park BK, et al. Muc1-C Influences Cell Survival in Lung Adenocarcinoma Calu-3 Cells After Sars-Cov-2 Infection. BMB Rep (2021) 54(8):425–30. doi: 10.5483/BMBRep.2021.54.8.018
274. Habte HH, de Beer C, Lotz ZE, Tyler MG, Kahn D, Mall AS. Inhibition of Human Immunodeficiency Virus Type 1 Activity by Purified Human Breast Milk Mucin (Muc1) in an Inhibition Assay. Neonatology (2008) 93(3):162–70. doi: 10.1159/000108414
275. Saeland E, de Jong MA, Nabatov AA, Kalay H, Geijtenbeek TB, van Kooyk Y. Muc1 in Human Milk Blocks Transmission of Human Immunodeficiency Virus From Dendritic Cells to T Cells. Mol Immunol (2009) 46(11-12):2309–16. doi: 10.1016/j.molimm.2009.03.025
276. Li Y, Dinwiddie DL, Harrod KS, Jiang Y, Kim KC. Anti-Inflammatory Effect of Muc1 During Respiratory Syncytial Virus Infection of Lung Epithelial Cells In Vitro. Am J Physiol Lung Cell Mol Physiol (2010) 298(4):L558–63. doi: 10.1152/ajplung.00225.2009
277. Nguyen Y, Procario MC, Ashley SL, O’Neal WK, Pickles RJ, Weinberg JB. Limited Effects of Muc1 Deficiency on Mouse Adenovirus Type 1 Respiratory Infection. Virus Res (2011) 160(1-2):351–9. doi: 10.1016/j.virusres.2011.07.012
278. Mthembu Y, Lotz Z, Tyler M, de Beer C, Rodrigues J, Schoeman L, et al. Purified Human Breast Milk Muc1 and Muc4 Inhibit Human Immunodeficiency Virus. Neonatology (2014) 105(3):211–7. doi: 10.1159/000357201
279. McAuley JL, Corcilius L, Tan HX, Payne RJ, McGuckin MA, Brown LE. The Cell Surface Mucin Muc1 Limits the Severity of Influenza a Virus Infection. Mucosal Immunol (2017) 10(6):1581–93. doi: 10.1038/mi.2017.16
280. Li K, Cao P, McCaw JM. Modelling the Effect of Muc1 on Influenza Virus Infection Kinetics and Macrophage Dynamics. Viruses (2021) 13(5):850. doi: 10.3390/v13050850
281. Karmakar J, Mandal C. Interplay Between Sialic Acids, Siglec-E, and Neu1 Regulates Myd88- and Trif-Dependent Pathways for Tlr4-Activation During Leishmania Donovani Infection. Front Immunol (2021) 12:626110. doi: 10.3389/fimmu.2021.626110
282. Takeda K, Akira S. Toll-Like Receptors in Innate Immunity. Int Immunol (2005) 17(1):1–14. doi: 10.1093/intimm/dxh186
283. Sundararaj K, Rodgers J, Angel P, Wolf B, Nowling TK. The Role of Neuraminidase in Tlr4-Mapk Signalling and the Release of Cytokines by Lupus Serum-Stimulated Mesangial Cells. Immunology (2021) 162(4):418–33. doi: 10.1111/imm.13294
284. Landolfi NF, Leone J, Womack JE, Cook RG. Activation of T Lymphocytes Results in an Increase in H-2-Encoded Neuraminidase. Immunogenetics (1985) 22(2):159–67. doi: 10.1007/BF00563513
285. Landolfi NF, Cook RG. Activated T-Lymphocytes Express Class I Molecules Which Are Hyposialylated Compared to Other Lymphocyte Populations. Mol Immunol (1986) 23(3):297–309. doi: 10.1016/0161-5890(86)90057-x
286. Chen XP, Enioutina EY, Daynes RA. The Control of Il-4 Gene Expression in Activated Murine T Lymphocytes: A Novel Role for Neu-1 Sialidase. J Immunol (1997) 158(7):3070–80.
287. Bilyy R, Stoika R. Search for Novel Cell Surface Markers of Apoptotic Cells. Autoimmunity (2007) 40(4):249–53. doi: 10.1080/08916930701358867
288. Wang P, Zhang J, Bian H, Wu P, Kuvelkar R, Kung TT, et al. Induction of Lysosomal and Plasma Membrane-Bound Sialidases in Human T-Cells Via T-Cell Receptor. Biochem J (2004) 380(Pt 2):425–33. doi: 10.1042/BJ20031896
289. Kaminuma O, Katoh S, Miyagi T, Watanabe N, Kitamura N, Nishimura T, et al. Contribution of Neuraminidase 3 to the Differentiation of Induced Regulatory T Cells. Genes Cells (2018) 23(2):112–6. doi: 10.1111/gtc.12553
290. Wei Y, Jing J, Peng Z, Liu X, Wang X. Acacetin Ameliorates Insulin Resistance in Obesity Mice Through Regulating Treg/Th17 Balance Via Mir-23b-3p/Neu1 Axis. BMC Endocr Disord (2021) 21(1):57. doi: 10.1186/s12902-021-00688-8
291. Sundararaj K, Rodgers JI, Marimuthu S, Siskind LJ, Bruner E, Nowling TK. Neuraminidase Activity Mediates Il-6 Production by Activated Lupus-Prone Mesangial Cells. Am J Physiol Renal Physiol (2018) 314(4):F630–F42. doi: 10.1152/ajprenal.00421.2017
292. Sehnert B, Mietz J, Rzepka R, Buchholz S, Maul-Pavicic A, Schaffer S, et al. Neuraminidase Inhibitor Zanamivir Ameliorates Collagen-Induced Arthritis. Int J Mol Sci (2021) 22(3):1428. doi: 10.3390/ijms22031428
293. Haxho F, Haq S, Szewczuk MR. Biased G Protein-Coupled Receptor Agonism Mediates Neu1 Sialidase and Matrix Metalloproteinase-9 Crosstalk to Induce Transactivation of Insulin Receptor Signaling. Cell Signal (2018) 43:71–84. doi: 10.1016/j.cellsig.2017.12.006
294. Sasaki A, Hata K, Suzuki S, Sawada M, Wada T, Yamaguchi K, et al. Overexpression of Plasma Membrane-Associated Sialidase Attenuates Insulin Signaling in Transgenic Mice. J Biol Chem (2003) 278(30):27896–902. doi: 10.1074/jbc.M212200200
295. Yoshizumi S, Suzuki S, Hirai M, Hinokio Y, Yamada T, Yamada T, et al. Increased Hepatic Expression of Ganglioside-Specific Sialidase, Neu3, Improves Insulin Sensitivity and Glucose Tolerance in Mice. Metabolism (2007) 56(3):420–9. doi: 10.1016/j.metabol.2006.10.027
296. Lipina C, Nardi F, Grace H, Hundal HS. Neu3 Sialidase as a Marker of Insulin Sensitivity: Regulation by Fatty Acids. Cell Signal (2015) 27(9):1742–50. doi: 10.1016/j.cellsig.2015.05.010
297. Miklavcic JJ, Hart TD, Lees GM, Shoemaker GK, Schnabl KL, Larsen BM, et al. Increased Catabolism and Decreased Unsaturation of Ganglioside in Patients With Inflammatory Bowel Disease. World J Gastroenterol (2015) 21(35):10080–90. doi: 10.3748/wjg.v21.i35.10080
298. Bhagavatula S, Banerjee P, Sood A, Midha V, Thelma BK, Senapati S. Multiple Allelic Associations From Genes Involved in Energy Metabolism Were Identified in Celiac Disease. J Biosci (2021) 46:61. doi: 10.1007/s12038-021-00184-0
299. Kim S, Becker J, Bechheim M, Kaiser V, Noursadeghi M, Fricker N, et al. Characterizing the Genetic Basis of Innate Immune Response in Tlr4-Activated Human Monocytes. Nat Commun (2014) 5:5236. doi: 10.1038/ncomms6236
300. Allendorf DH, Puigdellivol M, Brown GC. Activated Microglia Desialylate Their Surface, Stimulating Complement Receptor 3-Mediated Phagocytosis of Neurons. Glia (2020) 68(5):989–98. doi: 10.1002/glia.23757
301. Nomura K, Vilalta A, Allendorf DH, Hornik TC, Brown GC. Activated Microglia Desialylate and Phagocytose Cells Via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J Immunol (2017) 198(12):4792–801. doi: 10.4049/jimmunol.1502532
302. Hickey MJ, Kubes P. Intravascular Immunity: The Host-Pathogen Encounter in Blood Vessels. Nat Rev Immunol (2009) 9(5):364–75. doi: 10.1038/nri2532
303. Cross AS, Wright DG. Mobilization of Sialidase From Intracellular Stores to the Surface of Human Neutrophils and Its Role in Stimulated Adhesion Responses of These Cells. J Clin Invest (1991) 88(6):2067–76. doi: 10.1172/JCI115536
304. Cross AS, Sakarya S, Rifat S, Held TK, Drysdale BE, Grange PA, et al. Recruitment of Murine Neutrophils In Vivo Through Endogenous Sialidase Activity. J Biol Chem (2003) 278(6):4112–20. doi: 10.1074/jbc.M207591200
305. Feng C, Li J, Snyder G, Huang W, Goldblum SE, Chen WH, et al. Antibody Against Microbial Neuraminidases Recognizes Human Sialidase 3 (Neu3): The Neuraminidase/Sialidase Superfamily Revisited. mBio (2017) 8(3):e00078–17. doi: 10.1128/mBio.00078-17
306. Sakarya S, Rifat S, Zhou J, Bannerman DD, Stamatos NM, Cross AS, et al. Mobilization of Neutrophil Sialidase Activity Desialylates the Pulmonary Vascular Endothelial Surface and Increases Resting Neutrophil Adhesion to and Migration Across the Endothelium. Glycobiology (2004) 14(6):481–94. doi: 10.1093/glycob/cwh065
307. Stamatos NM, Liang F, Nan X, Landry K, Cross AS, Wang LX, et al. Differential Expression of Endogenous Sialidases of Human Monocytes During Cellular Differentiation Into Macrophages. FEBS J (2005) 272(10):2545–56. doi: 10.1111/j.1742-4658.2005.04679.x
308. Sieve I, Ricke-Hoch M, Kasten M, Battmer K, Stapel B, Falk CS, et al. A Positive Feedback Loop Between Il-1beta, Lps and Neu1 May Promote Atherosclerosis by Enhancing a Pro-Inflammatory State in Monocytes and Macrophages. Vascul Pharmacol (2018) 103-105:16–28. doi: 10.1016/j.vph.2018.01.005
309. Gayral S, Garnotel R, Castaing-Berthou A, Blaise S, Fougerat A, Berge E, et al. Elastin-Derived Peptides Potentiate Atherosclerosis Through the Immune Neu1-Pi3kgamma Pathway. Cardiovasc Res (2014) 102(1):118–27. doi: 10.1093/cvr/cvt336
310. White EJ, Gyulay G, Lhotak S, Szewczyk MM, Chong T, Fuller MT, et al. Sialidase Down-Regulation Reduces Non-Hdl Cholesterol, Inhibits Leukocyte Transmigration, and Attenuates Atherosclerosis in Apoe Knockout Mice. J Biol Chem (2018) 293(38):14689–706. doi: 10.1074/jbc.RA118.004589
311. Heimerl M, Sieve I, Ricke-Hoch M, Erschow S, Battmer K, Scherr M, et al. Neuraminidase-1 Promotes Heart Failure After Ischemia/Reperfusion Injury by Affecting Cardiomyocytes and Invading Monocytes/Macrophages. Basic Res Cardiol (2020) 115(6):62. doi: 10.1007/s00395-020-00821-z
312. Tembely D, Henry A, Vanalderwiert L, Toussaint K, Bennasroune A, Blaise S, et al. The Elastin Receptor Complex: An Emerging Therapeutic Target Against Age-Related Vascular Diseases. Front Endocrinol (2022) 13:815356. doi: 10.3389/fendo.2022.815356
313. Potier M, Lu Shun Yan D, Womack JE. Neuraminidase Deficiency in the Mouse. FEBS Lett (1979) 108(2):345–8. doi: 10.1016/0014-5793(79)80560-8
314. Rottier RJ, Bonten E, d’Azzo A. A Point Mutation in the Neu-1 Locus Causes the Neuraminidase Defect in the Sm/J Mouse. Hum Mol Genet (1998) 7(2):313–21. doi: 10.1093/hmg/7.2.313
315. Taira S, Nariuchi H. Possible Role of Neuraminidase in Activated T Cells in the Recognition of Allogeneic Ia. J Immunol (1988) 141(2):440–6.
316. Wipfler D, Srinivasan GV, Sadick H, Kniep B, Arming S, Willhauck-Fleckenstein M, et al. Differentially Regulated Expression of 9-O-Acetyl Gd3 (Cd60b) and 7-O-Acetyl-Gd3 (Cd60c) During Differentiation and Maturation of Human T and B Lymphocytes. Glycobiology (2011) 21(9):1161–72. doi: 10.1093/glycob/cwr050
317. Kijimoto-Ochiai S, Koda T, Suwama T, Matsukawa H, Fujii M, Tomobe K, et al. Low Expression of Neu2 Sialidase in the Thymus of Sm/J Mice-Existence of Neuraminidase Positive Cells “Neu-Medullocyte” in the Murine Thymus. Glycoconj J (2008) 25(8):787–96. doi: 10.1007/s10719-008-9126-8
318. Kijimoto-Ochiai S, Doi N, Matsukawa H, Fujii M, Tomobe K. Localization of Sialidase-Positive Cells Expressing Mac-1 and Immunoglobulin in the Mouse Thymus. Glycoconj J (2004) 20(6):375–84. doi: 10.1023/B:GLYC.0000033994.99464.ce
319. Kijimoto-Ochiai S, Kamimura K, Koda T. Neu-Medullocytes, Sialidase-Positive B Cells in the Thymus, Express Autoimmune Regulator (Aire). Sci Rep (2019) 9(1):858. doi: 10.1038/s41598-018-37225-y
320. Koda T, Kijimoto-Ochiai S, Uemura S, Inokuchi J. Specific Expression of Neu2 Type B in Mouse Thymus and the Existence of a Membrane-Bound Form in Cos Cells. Biochem Biophys Res Commun (2009) 387(4):729–35. doi: 10.1016/j.bbrc.2009.07.100
321. Chen XP, Ding X, Daynes RA. Ganglioside Control Over Il-4 Priming and Cytokine Production in Activated T Cells. Cytokine (2000) 12(7):972–85. doi: 10.1006/cyto.1999.0596
322. Azuma Y, Taniguchi A, Matsumoto K. Decrease in Cell Surface Sialic Acid in Etoposide-Treated Jurkat Cells and the Role of Cell Surface Sialidase. Glycoconj J (2000) 17(5):301–6. doi: 10.1023/a:1007165403771
323. Azuma Y, Sato H, Higai K, Matsumoto K. Enhanced Expression of Membrane-Associated Sialidase Neu3 Decreases Gd3 and Increases Gm3 on the Surface of Jurkat Cells During Etoposide-Induced Apoptosis. Biol Pharm Bull (2007) 30(9):1680–4. doi: 10.1248/bpb.30.1680
324. Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, et al. Decrease of Sialic Acid Residues as an Eat-Me Signal on the Surface of Apoptotic Lymphocytes. J Cell Sci (2010) 123(Pt 19):3347–56. doi: 10.1242/jcs.066696
325. Bilyy RO, Shkandina T, Tomin A, Munoz LE, Franz S, Antonyuk V, et al. Macrophages Discriminate Glycosylation Patterns of Apoptotic Cell-Derived Microparticles. J Biol Chem (2012) 287(1):496–503. doi: 10.1074/jbc.M111.273144
326. Katoh S, Kaminuma O, Hiroi T, Mori A, Ohtomo T, Maeda S, et al. Cd44 Is Critical for Airway Accumulation of Antigen-Specific Th2, But Not Th1, Cells Induced by Antigen Challenge in Mice. Eur J Immunol (2011) 41(11):3198–207. doi: 10.1002/eji.201141521
327. Lu LQ, Liao W. Screening and Functional Pathway Analysis of Genes Associated With Pediatric Allergic Asthma Using a DNA Microarray. Mol Med Rep (2015) 11(6):4197–203. doi: 10.3892/mmr.2015.3277
328. Hewison M. Vitamin D. And Innate and Adaptive Immunity. Vitam Horm (2011) 86:23–62. doi: 10.1016/B978-0-12-386960-9.00002-2
329. Yamamoto N, Homma S, Millman I. Identification of the Serum Factor Required for In Vitro Activation of Macrophages. Role of Vitamin D3-Binding Protein (Group Specific Component, Gc) in Lysophospholipid Activation of Mouse Peritoneal Macrophages. J Immunol (1991) 147(1):273–80.
330. Yamamoto N, Naraparaju VR. Role of Vitamin D3-Binding Protein in Activation of Mouse Macrophages. J Immunol (1996) 157(4):1744–9.
331. Yamamoto N, Naraparaju VR. Vitamin D3-Binding Protein as a Precursor for Macrophage Activating Factor in the Inflammation-Primed Macrophage Activation Cascade in Rats. Cell Immunol (1996) 170(2):161–7. doi: 10.1006/cimm.1996.0148
332. Naraparaju VR, Yamamoto N. Roles of Beta-Galactosidase of B Lymphocytes and Sialidase of T Lymphocytes in Inflammation-Primed Activation of Macrophages. Immunol Lett (1994) 43(3):143–8. doi: 10.1016/0165-2478(94)90214-3
333. Yamamoto N, Kumashiro R. Conversion of Vitamin D3 Binding Protein (Group-Specific Component) to a Macrophage Activating Factor by the Stepwise Action of Beta-Galactosidase of B Cells and Sialidase of T Cells. J Immunol (1993) 151(5):2794–802.
334. Agrawal B, Gupta N, Konowalchuk JD. Muc1 Mucin: A Putative Regulatory (Checkpoint) Molecule of T Cells. Front Immunol (2018) 9:2391. doi: 10.3389/fimmu.2018.02391
335. Nowling TK, Gilkeson GS. Mechanisms of Tissue Injury in Lupus Nephritis. Arthritis Res Ther (2011) 13(6):250. doi: 10.1186/ar3528
336. Nowling TK, Rodgers J, Thiyagarajan T, Wolf B, Bruner E, Sundararaj K, et al. Targeting Glycosphingolipid Metabolism as a Potential Therapeutic Approach for Treating Disease in Female Mrl/Lpr Lupus Mice. PLoS One (2020) 15(3):e0230499. doi: 10.1371/journal.pone.0230499
337. Sundararaj KP, Thiyagarajan T, Molano I, Basher F, Powers TW, Drake RR, et al. Fli1 Levels Impact Cxcr3 Expression and Renal Infiltration of T Cells and Renal Glycosphingolipid Metabolism in the Mrl/Lpr Lupus Mouse Strain. J Immunol (2015) 195(12):5551–60. doi: 10.4049/jimmunol.1500961
338. Richard EM, Thiyagarajan T, Bunni MA, Basher F, Roddy PO, Siskind LJ, et al. Reducing Fli1 Levels in the Mrl/Lpr Lupus Mouse Model Impacts T Cell Function by Modulating Glycosphingolipid Metabolism. PLoS One (2013) 8(9):e75175. doi: 10.1371/journal.pone.0075175
339. Nowling TK, Mather AR, Thiyagarajan T, Hernandez-Corbacho MJ, Powers TW, Jones EE, et al. Renal Glycosphingolipid Metabolism Is Dysfunctional in Lupus Nephritis. J Am Soc Nephrol (2015) 26(6):1402–13. doi: 10.1681/ASN.2014050508
340. Rodgers J, Sundararaj K, Bruner E, Wolf B, Nowling TK. The Role of Neuraminidase 1 (Neu1) in Cytokine Release by Primary Mouse Mesangial Cells and Disease Outcomes in Murine Lupus Nephritis. Autoimmunity (2021) 54(3):163–75. doi: 10.1080/08916934.2021.1897978
341. Mao Z, Tan Y, Yu F, Zhao M. Discovery of Neu1 as a Candidatedone. Renal Biomarker for Proliferative Lupus Nephritis Chronicity. Lupus Sci Med (2021) 8(1):e000569. doi: 10.1136/lupus-2021-000569
342. Liou LB, Huang CC. Sialyltransferase and Neuraminidase Levels/Ratios and Sialic Acid Levels in Peripheral Blood B Cells Correlate With Measures of Disease Activity in Patients With Systemic Lupus Erythematosus and Rheumatoid Arthritis: A Pilot Study. PLoS One (2016) 11(3):e0151669. doi: 10.1371/journal.pone.0151669
343. Yang Z, Liang Y, Li C, Zhong R. Serum Ca125 Elevation Is Independently Associated With Serositis in Sle Patients. Clin Exp Rheumatol (2012) 30(1):93–8.
344. Meena DS, Kumar B, Gopalakrishnan M, Kachhwaha A, Kumar S, Sureka B, et al. Pseudo-Pseudo Meigs’ Syndrome (Ppms) in Chronic Lupus Peritonitis: A Case Report With Review of Literature. Mod Rheumatol Case Rep (2021) 5(2):300–5. doi: 10.1080/24725625.2021.1916160
345. Quintero-Munoz E, Gomez Pineda MA, Araque Parra C, Vallejo Castillo CA, Ortega Marrugo V, Bonilla Jassir J, et al. Is There Any Relationship Between Massive Ascites and Elevated Ca-125 in Systemic Lupus Erythematosus? Case Report and Review of the Literature. Mod Rheumatol Case Rep (2021) 5(2):292–9. doi: 10.1080/24725625.2021.1909213
346. Tansir G, Kumar P, Pius A, Sunny SK, Soneja M. Pseudo-Pseudo Meigs’ Syndrome: A Rare Presentation of Systemic Lupus Erythematosus. Reumatismo (2019) 71(2):108–12. doi: 10.4081/reumatismo.2019.1140
347. Higuchi T, Orita T, Nakanishi S, Katsuya K, Watanabe H, Yamasaki Y, et al. Molecular Cloning, Genomic Structure, and Expression Analysis of Muc20, a Novel Mucin Protein, Up-Regulated in Injured Kidney. J Biol Chem (2004) 279(3):1968–79. doi: 10.1074/jbc.M304558200
348. de Geest N, Bonten E, Mann L, de Sousa-Hitzler J, Hahn C, d’Azzo A. Systemic and Neurologic Abnormalities Distinguish the Lysosomal Disorders Sialidosis and Galactosialidosis in Mice. Hum Mol Genet (2002) 11(12):1455–64. doi: 10.1093/hmg/11.12.1455
349. Radu AF, Bungau SG. Management of Rheumatoid Arthritis: An Overview. Cells (2021) 10(11):2857. doi: 10.3390/cells10112857
350. Liou LB, Jang SS. Alpha-2,3-Sialyltransferase 1 and Neuraminidase-3 From Monocytes in Patients With Rheumatoid Arthritis Correlate With Disease Activity Measures: A Pilot Study. J Chin Med Assoc (2019) 82(3):179–85. doi: 10.1097/JCMA.0000000000000027
351. Marchand NW, Kishore GS, Carubelli R. Neuraminidase Activity in the Blood and Liver of Arthritic Rats. Exp Mol Pathol (1978) 29(3):273–80. doi: 10.1016/0014-4800(78)90070-9
352. Correa I, Plunkett T, Vlad A, Mungul A, Candelora-Kettel J, Burchell JM, et al. Form and Pattern of Muc1 Expression on T Cells Activated In Vivo or In Vitro Suggests a Function in T-Cell Migration. Immunology (2003) 108(1):32–41. doi: 10.1046/j.1365-2567.2003.01562.x
353. Wang Y, Chen S, Zheng S, Lin J, Hu S, Zhuang J, et al. The Role of Lung Ultrasound B-Lines and Serum Kl-6 in the Screening and Follow-Up of Rheumatoid Arthritis Patients for an Identification of Interstitial Lung Disease: Review of the Literature, Proposal for a Preliminary Algorithm, and Clinical Application to Cases. Arthritis Res Ther (2021) 23(1):212. doi: 10.1186/s13075-021-02586-9
354. Volin MV, Shahrara S, Haines GK 3rd, Woods JM, Koch AE. Expression of Mucin 3 and Mucin 5ac in Arthritic Synovial Tissue. Arthritis Rheum (2008) 58(1):46–52. doi: 10.1002/art.23174
355. Morisset J, Lee JS. New Trajectories in the Treatment of Interstitial Lung Disease: Treat the Disease or Treat the Underlying Pattern? Curr Opin Pulm Med (2019) 25(5):442–9. doi: 10.1097/MCP.0000000000000600
356. Flowers SA, Ali L, Lane CS, Olin M, Karlsson NG. Selected Reaction Monitoring to Differentiate and Relatively Quantitate Isomers of Sulfated and Unsulfated Core 1 O-Glycans From Salivary Muc7 Protein in Rheumatoid Arthritis. Mol Cell Proteomics (2013) 12(4):921–31. doi: 10.1074/mcp.M113.028878
357. Pickl WF, Majdic O, Fischer GF, Petzelbauer P, Fae I, Waclavicek M, et al. Muc18/Mcam (Cd146), an Activation Antigen of Human T Lymphocytes. J Immunol (1997) 158(5):2107–15.
358. Neidhart M, Wehrli R, Bruhlmann P, Michel BA, Gay RE, Gay S. Synovial Fluid Cd146 (Muc18), a Marker for Synovial Membrane Angiogenesis in Rheumatoid Arthritis. Arthritis Rheum (1999) 42(4):622–30. doi: 10.1002/1529-0131(199904)42:4<622::AID-ANR4>3.0.CO;2-Y
359. Richardson SJ, Pugliese A. 100 Years of Insulin: Pancreas Pathology in Type 1 Diabetes: An Evolving Story. J Endocrinol (2021) 252(2):R41–57. doi: 10.1530/JOE-21-0358
360. Natori Y, Ohkura N, Nasui M, Atsumi G, Kihara-Negishi F. Acidic Sialidase Activity Is Aberrant in Obese and Diabetic Mice. Biol Pharm Bull (2013) 36(6):1027–31. doi: 10.1248/bpb.b12-00995
361. Inim MD, Ibrahim MA, Isah MB, Onyike E. Variations in the Mrna Expression Level of Udp-Glcnac Epimerase/Mannac Kinase and Neuraminidase 1 Genes in Organs of Type 2 Diabetic Animals. Glycoconj J (2021) 38(1):129–34. doi: 10.1007/s10719-021-09979-7
362. Minami A, Fujita Y, Shimba S, Shiratori M, Kaneko YK, Sawatani T, et al. The Sialidase Inhibitor 2,3-Dehydro-2-Deoxy-N-Acetylneuraminic Acid Is a Glucose-Dependent Potentiator of Insulin Secretion. Sci Rep (2020) 10(1):5198. doi: 10.1038/s41598-020-62203-8
363. Xia QS, Lu FE, Wu F, Huang ZY, Dong H, Xu LJ, et al. New Role for Ceramide in Hypoxia and Insulin Resistance. World J Gastroenterol (2020) 26(18):2177–86. doi: 10.3748/wjg.v26.i18.2177
364. Albaghdadi AJ, Kan FW. Endometrial Receptivity Defects and Impaired Implantation in Diabetic Nod Mice. Biol Reprod (2012) 87(2):30. doi: 10.1095/biolreprod.112.100016
365. Di G, Qi X, Zhao X, Zhang S, Zhou Q. Efficacy of Sodium Hyaluronate in Murine Diabetic Ocular Surface Diseases. Cornea (2017) 36(9):1133–8. doi: 10.1097/ICO.0000000000001271
366. Mednieks MI, Szczepanski A, Clark B, Hand AR. Protein Expression in Salivary Glands of Rats With Streptozotocin Diabetes. Int J Exp Pathol (2009) 90(4):412–22. doi: 10.1111/j.1365-2613.2009.00662.x
367. Bi J, Huang Y, Liu Y. Effect of Nop2 Knockdown on Colon Cancer Cell Proliferation, Migration, and Invasion. Transl Cancer Res (2019) 8(6):2274–83. doi: 10.21037/tcr.2019.09.46
368. Baumgart DC, Carding SR. Inflammatory Bowel Disease: Cause and Immunobiology. Lancet (2007) 369(9573):1627–40. doi: 10.1016/S0140-6736(07)60750-8
369. Tuin A, Poelstra K, de Jager-Krikken A, Bok L, Raaben W, Velders MP, et al. Role of Alkaline Phosphatase in Colitis in Man and Rats. Gut (2009) 58(3):379–87. doi: 10.1136/gut.2007.128868
370. Parlato M, Charbit-Henrion F, Pan J, Romano C, Duclaux-Loras R, Le Du MH, et al. Human Alpi Deficiency Causes Inflammatory Bowel Disease and Highlights a Key Mechanism of Gut Homeostasis. EMBO Mol Med (2018) 10(4):e8483. doi: 10.15252/emmm.201708483
371. Chen Y, He Y, Zhan X, Chen D, Feng P, Yan Y, et al. Sialic Acid as a Suitable Marker of Clinical Disease Activity in Patients With Crohn’s Disease. Lab Med (2022) lmac006. doi: 10.1093/labmed/lmac006
372. Baba R, Yashiro K, Nagasako K, Obata H. Significance of Serum Sialic Acid in Patients With Crohn’s Disease. Gastroenterol Jpn (1992) 27(5):604–10. doi: 10.1007/BF02774974
373. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat Genet (2010) 42(12):1118–25. doi: 10.1038/ng.717
374. Furr AE, Ranganathan S, Finn OJ. Aberrant Expression of Muc1 Mucin in Pediatric Inflammatory Bowel Disease. Pediatr Dev Pathol (2010) 13(1):24–31. doi: 10.2350/08-06-0479.1
375. Begue B, Verdier J, Rieux-Laucat F, Goulet O, Morali A, Canioni D, et al. Defective Il10 Signaling Defining a Subgroup of Patients With Inflammatory Bowel Disease. Am J Gastroenterol (2011) 106(8):1544–55. doi: 10.1038/ajg.2011.112
376. Miro L, Amat C, Rosell-Cardona C, Campbell JM, Polo J, Perez-Bosque A, et al. Dietary Supplementation With Spray-Dried Porcine Plasma Attenuates Colon Inflammation in a Genetic Mouse Model of Inflammatory Bowel Disease. Int J Mol Sci (2020) 21(18):6760. doi: 10.3390/ijms21186760
377. Moehle C, Ackermann N, Langmann T, Aslanidis C, Kel A, Kel-Margoulis O, et al. Aberrant Intestinal Expression and Allelic Variants of Mucin Genes Associated With Inflammatory Bowel Disease. J Mol Med (Berl) (2006) 84(12):1055–66. doi: 10.1007/s00109-006-0100-2
378. Nishida A, Lau CW, Zhang M, Andoh A, Shi HN, Mizoguchi E, et al. The Membrane-Bound Mucin Muc1 Regulates T Helper 17-Cell Responses and Colitis in Mice. Gastroenterology (2012) 142(4):865–74 e2. doi: 10.1053/j.gastro.2011.12.036
379. Beatty PL, Plevy SE, Sepulveda AR, Finn OJ. Cutting Edge: Transgenic Expression of Human Muc1 in Il-10-/- Mice Accelerates Inflammatory Bowel Disease and Progression to Colon Cancer. J Immunol (2007) 179(2):735–9. doi: 10.4049/jimmunol.179.2.735
380. Beatty P, Ranganathan S, Finn OJ. Prevention of Colitis-Associated Colon Cancer Using a Vaccine to Target Abnormal Expression of the Muc1 Tumor Antigen. Oncoimmunology (2012) 1(3):263–70. doi: 10.4161/onci.18950
381. Beatty PL, Narayanan S, Gariepy J, Ranganathan S, Finn OJ. Vaccine Against Muc1 Antigen Expressed in Inflammatory Bowel Disease and Cancer Lessens Colonic Inflammation and Prevents Progression to Colitis-Associated Colon Cancer. Cancer Prev Res (Phila) (2010) 3(4):438–46. doi: 10.1158/1940-6207.CAPR-09-0194
382. Poh TW, Madsen CS, Gorman JE, Marler RJ, Leighton JA, Cohen PA, et al. Downregulation of Hematopoietic Muc1 During Experimental Colitis Increases Tumor-Promoting Myeloid-Derived Suppressor Cells. Clin Cancer Res (2013) 19(18):5039–52. doi: 10.1158/1078-0432.CCR-13-0278
383. Khemiri M, Doghri R, Mrad K, Friedrich K, Oueslati R. Mucin-1 Expression and Localization in Epithelial Cells Shows Characteristic and Distinct Patterns in Inflammatory Bowel Diseases and Colorectal Cancer. Int J Clin Exp Pathol (2019) 12(5):1731–7.
384. Li W, Zhang N, Jin C, Long MD, Rajabi H, Yasumizu Y, et al. Muc1-C Drives Stemness in Progression of Colitis to Colorectal Cancer. JCI Insight (2020) 5(12):e137112. doi: 10.1172/jci.insight.137112
385. Liu Y, Yu X, Zhao J, Zhang H, Zhai Q, Chen W. The Role of Muc2 Mucin in Intestinal Homeostasis and the Impact of Dietary Components on Muc2 Expression. Int J Biol Macromol (2020) 164:884–91. doi: 10.1016/j.ijbiomac.2020.07.191
386. Engler DB, Leonardi I, Hartung ML, Kyburz A, Spath S, Becher B, et al. Helicobacter Pylori-Specific Protection Against Inflammatory Bowel Disease Requires the Nlrp3 Inflammasome and Il-18. Inflammation Bowel Dis (2015) 21(4):854–61. doi: 10.1097/MIB.0000000000000318
387. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That Muc2 Is Critical for Colonic Protection. Gastroenterology (2006) 131(1):117–29. doi: 10.1053/j.gastro.2006.04.020
388. Estaki M, Morck DW, Ghosh S, Quin C, Pither J, Barnett JA, et al. Physical Activity Shapes the Intestinal Microbiome and Immunity of Healthy Mice But Has No Protective Effects Against Colitis in Muc2(-/-) Mice. mSystems (2020) 5(5):e00515–20. doi: 10.1128/mSystems.00515-20
389. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant Mucin Assembly in Mice Causes Endoplasmic Reticulum Stress and Spontaneous Inflammation Resembling Ulcerative Colitis. PLoS Med (2008) 5(3):e54. doi: 10.1371/journal.pmed.0050054
390. Kyo K, Muto T, Nagawa H, Lathrop GM, Nakamura Y. Associations of Distinct Variants of the Intestinal Mucin Gene Muc3a With Ulcerative Colitis and Crohn’s Disease. J Hum Genet (2001) 46(1):5–20. doi: 10.1007/s100380170118
391. Kyo K, Parkes M, Takei Y, Nishimori H, Vyas P, Satsangi J, et al. Association of Ulcerative Colitis With Rare Vntr Alleles of the Human Intestinal Mucin Gene, Muc3. Hum Mol Genet (1999) 8(2):307–11. doi: 10.1093/hmg/8.2.307
392. Buisine MP, Desreumaux P, Leteurtre E, Copin MC, Colombel JF, Porchet N, et al. Mucin Gene Expression in Intestinal Epithelial Cells in Crohn’s Disease. Gut (2001) 49(4):544–51. doi: 10.1136/gut.49.4.544
393. Yamamoto-Furusho JK, Ascano-Gutierrez I, Furuzawa-Carballeda J, Fonseca-Camarillo G. Differential Expression of Muc12, Muc16, and Muc20 in Patients With Active and Remission Ulcerative Colitis. Mediators Inflamm (2015) 2015:659018. doi: 10.1155/2015/659018
394. Takagawa T, Kitani A, Fuss I, Levine B, Brant SR, Peter I, et al. An Increase in Lrrk2 Suppresses Autophagy and Enhances Dectin-1-Induced Immunity in a Mouse Model of Colitis. Sci Transl Med (2018) 10(444):eaan8162. doi: 10.1126/scitranslmed.aan8162
Keywords: post-translational modification, sialidase, sialic acid, fibrosis, inflammation, infection, autoimmunity
Citation: Lillehoj EP, Luzina IG and Atamas SP (2022) Mammalian Neuraminidases in Immune-Mediated Diseases: Mucins and Beyond. Front. Immunol. 13:883079. doi: 10.3389/fimmu.2022.883079
Received: 24 February 2022; Accepted: 21 March 2022;
Published: 11 April 2022.
Edited by:
Gaby Palmer, Université de Genève, SwitzerlandReviewed by:
Tamara Nowling, Medical University of South Carolina, United StatesEugenio Monti, University of Brescia, Italy
Copyright © 2022 Lillehoj, Luzina and Atamas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erik P. Lillehoj, elillehoj@som.umaryland.edu