

Downloads
Download



This work is licensed under a Creative Commons Attribution 4.0 International License.
Review
Transforming Growth Factor β Signaling Pathway as a Potential Drug Target in Treating Aortic Diseases
Zijie Liu 1,2, Tianyu Song 3, and Liping Xie 1,2,3, *
1 Key Laboratory of Targeted Intervention of Cardiovascular Disease, Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, Nanjing Medical University, Nanjing 211166, China
2 School of Basic Medical Sciences, Nanjing Medical University, Nanjing 211166, China
3 Key Laboratory of Cardiovascular and Cerebrovascular Medicine, Nanjing Medical University, Nanjing 211166, China
* Correspondence: lipingxie@njmu.edu.cn
Received: 16 October 2023
Accepted: 20 November 2023
Published: 18 March 2024
Abstract: The transforming growth factor β (TGF-β) signaling pathway is crucial for preserving the structural homeostasis of the aorta and promoting aortic development. This pathway encompasses both SMAD-dependent canonical pathway and SMAD-independent non-canonical signaling pathway. Heritable thoracic aortic aneurysms and dissection are highly correlated with genetic alterations in TGF-β canonical signaling-related genes. However, depending on the stage of the disease, the TGF-β signaling pathway can have either inhibitory or aggravation effects, making its roles in aortic disease complex and occasionally contradictory. This review aims to elucidate the biological mechanisms underlying the TGF-β signaling pathway in the most common aortic diseases, namely acute aortic syndromes and aortic aneurysms, and to evaluate the potential clinical application of TGF-β-targeting therapies in aortic diseases.
Keywords:
TGF-β signaling pathway aortic aneurysm acute aortic syndromes aortic dissection1. Introduction
Aortic diseases are primarily categorized into thoracic aortic aneurysm (TAA), abdominal aortic aneurysm (AAA) and acute aortic syndromes (AAS) [1]. Aortic dissection, intramural hematoma and penetrating atherosclerotic ulcer (PAU) are the most typical AAS, each posing a risk of rupture. Aortic aneurysm is characterized by a localized dilatation of the aorta, exceeding 1.5 times the normal diameter of corresponding aortic segments in age- and sex-matched healthy individual [2]. Among AAS, aortic dissection is the most common, involving a tear in the inner layer of the aortic wall. This tear allows blood to penetrate the aortic media, splitting the intima in two longitudinally and forming a dissection flap that divides the true lumen from a newly formed false lumen [3]. Open surgical repair or endovascular surgery are recommended to prevent rupture of large aortic aneurysm and dissection [4]. However, a large number of patients with persistent aortic degeneration require further pharmacotherapy [5]. To date, despite the fact that statins and antihypertensive agents could improve the overall cardiovascular risk profile, there are no effective drugs to block the expansion of aortic aneurysms and the occurrence of aortic dissections [6].
The etiology underlying aortic aneurysms and AAS is multifactorial. Factors such as genetic changes affecting cellular function and vascular structure, inflammation, endothelial dysfunction, vascular smooth muscle cells (VSMCs) phenotypic switching, extracellular matrix (ECM) destruction and the formation of intraluminal thrombi are all considered causes of sporadic aortic diseases [7]. Yet, the molecular mechanisms underlying aortic diseases remain elusive. Thus, in-depth study in the pathological characteristics and potential molecular mechanisms of aortic aneurysm and aortic dissection and identifying key intervention targets are of great importance for early prevention and drug treatment.
The transforming growth factor β (TGF-β) signaling pathway plays a vital role in regulating the structure and function of the aorta. Genome-wide association analysis (GWAS) has indicated that the single nucleotide polymorphisms (SNPs) of multiple genes in the TGF-β pathway (such as LTBP4 and HIPK3) are closely related to aortic dilation [8]. For example, some SNPs in LTBP4 lead to unstable TGF-β receptors [9], and others in HIPK3 might decrease the noncanonical TGF-β signaling [10]. But in general, these SNPs are loss-of-function mutations. Syndromic thoracic aortic aneurysm and dissection (TAAD), including Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS) and Shprintzen-Goldberg syndrome (SGS) are usually caused by sequence variants associated to the TGF-β signaling system [11]. Also, early studies suggested that excessive TGF-β pathway activation is a pathogenic factor in TAAD, with dramatically enhanced TGF-β signaling observed in the aorta of patients with familial aortic dissection and MFS [12-14]. However, the precise role of TGF-β signaling in aortic diseases, particularly whether it exacerbates or mitigates aortic dissection and aneurysms, remains a subject of debate. The main focus of this review is to present and discuss the current molecular knowledge of the effect of the TGF-β signaling pathway on aortic aneurysms and aortic dissection.
2. Overview of TGF-β Signaling Pathway
The activins, bone morphogenetic proteins (BMPs), growth differentiation factors (GDFs), müllerian inhibiting substance (MIS), nodal and TGF-βs are all members of the TGF-β superfamily. These cytokines, depending on the intrinsic properties of the target cells and the surrounding environment, can exhibit autocrine, paracrine, or endocrine functions. While different members of TGF-β superfamily might share similar functions, they can also exhibit antagonistic effects on one another [15]. TGF-β is synthesized as a proprotein that consists of a latency-associated peptide (LAP) at the N-terminus and a mature cytokine at the C-terminus, which is cut in the Golgi apparatus and secreted in a non-covalent form, known as the small latent complex (SLC), also called LAP-TGF-β. This complex can further bind to latent TGF-β binding proteins (LTBPs) to form the large latent complex (LLC), rendering TGF-β inactive. LTBPs crosslink with matrix proteins, such as fibrillin, to ensure that LLC is deposited and stored in the ECM after secretion [16,17]. TGF-β activation is regulated by integrin proteins, particularly αvβ6 and αvβ8. These integrins bind to the arginine-glycine-aspartic acid (RGD) motif in the LAP segment, leading to a physical deformation of the latent complex under traction or stress, which then releases active TGF-β [18]. Interestingly, various proteinases, especially matrix metalloproteinases (MMPs), are also involved in the activation of the TGF-β complex. The conformational change induced by integrin proteins makes the complex more prone to proteolytic cleavage and latent TGF-β become activated. The most widely recognized mechanism of latent TGF-β activation via integrins involves direct activation through traction forces generated between cells and the extracellular matrix [19,20].
The TGF-β signaling pathway participates in various biological processes, including the regulation of inflammation, cell proliferation, apoptosis, migration, adhesion, ECM generation and cytoskeleton construction. Therefore, it plays a momentous role in the process of embryonic development, cell fate determination and tissue homeostasis repair. Generally, the TGF-β signaling pathway can be classified into two distinct categories: the canonical TGF-β (SMAD-dependent) signaling pathway and the noncanonical TGF-β (SMAD-independent) signaling pathway [21]. The SMADs family, as transcription factors, can directly mediate transmission of signals from the membrane to the nucleus. The canonical TGF-β signaling pathway, being SMAD-dependent, is essential for aortic development and maintaining aortic wall stability [22]. In the noncanonical TGF-β pathway, the TGF-β receptor complex transmits signals through various alternative pathways, including JNK, TGF-β-activated kinase 1 (TAK1), extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (p38 MAPK) and nuclear factor κB (NF-κB) signaling pathways [23].
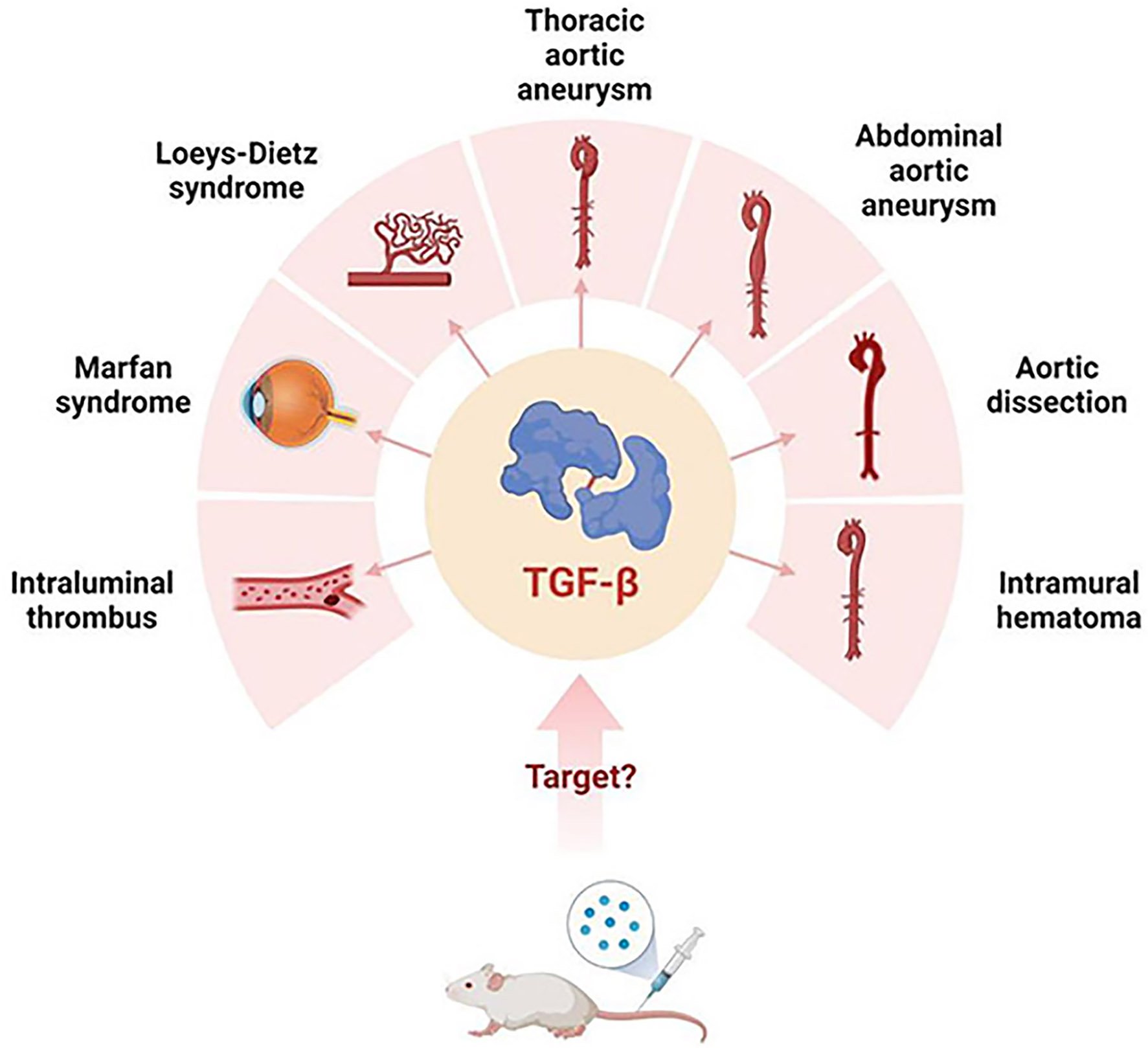
Figure 1. TGF-β’s possibly related aortic diseases..
3. The Role of Canonical TGF-β Signaling Pathway in Aortic Diseases
In recent years, a growing body of evidence has suggested the dysregulation of TGF-β signaling in various aortic diseases. Multiple comprehensive reviews have analyzed and discussed the activation of TGF-β signaling in the progression of TAAD. These studies concluded that TGF-β signaling pathway is essential for vascular function: excessive activation may contribute to the development of aneurysms and dissection, while over-inhibition of TGF-β signaling may lead to the overactivation of alternative bypass pathways, thereby inducing the occurrence of aortopathy [11,22,24,25]. The activation of the canonical TGF-β signaling pathway in aortopathy relies on the binding of TGF-β to the serine/threonine kinase homodimer TGF-β type II receptors (TβRII), which promotes phosphorylation of the intracellular domain of TGF-β type I receptors (TβRI). This results in the recruitment and phosphorylation of SMAD2/3, which bind to SMAD4 and form the heterodimer complex. The complex then translocates into the nucleus and binds to SMAD response elements to induce or repress the expression of various genes encoding ECM, anti-proteases, LTBPs, or others [26]. Here, we will summary the effect of canonical TGF-β signaling pathway on the development of aortic aneurysm and aortic dissection.
3.1. Canonical TGF-β Signaling Pathway-Related Genes and TAAD
Knocking out genes such as Tgfbr1 or Tgfbr2, which encode TβRI and TβRII, can impede the process of normal vascular morphogenesis [27] and hinder the differentiation of smooth muscle cells (SMCs), ultimately resulting in fetal death [28]. To circumvent embryonic lethality, several studies have demonstrated observable aortic pathological changes through the induction of loss of Tgfbr2 and other genes in mice at different ages [29-32]. Postnatal deletion of Tgfbr2 in SMCs leads to aortic thickening, dilatation, and thoracic aorta dissection in both C57BL/6 and Fbn1C1039G/+ (MFS model) mice. The inactivation of TβRII results in a decrease in canonical SMAD signaling, while simultaneously causing an increase in MAPK signaling [30]. SMAD2 phosphorylation is unchanged in aortic SMCs of youthful MFS mice, but SMC-specific deletion of Tgfbr2 in Fbn1C1039G/+ mice remarkably decreases the level of SMAD2 phosphorylation and aggravates the aortic lesions [31]. Chen et al. showed that inducible SMC-specific Tgfbr2 knockout significantly decreased the expression of phosphorylated SMAD2 (p-SMAD2) in aortic SMCs, inhibited TGF-β1-induced SMAD2/3 phosphorylation, and eventually resulted in the development of aortic aneurysms in Apoe-/- mice [33]. Additionally, a previous study reported that postnatal deletion of Tgfbr2 in SMCs caused severe aortopathy in all areas of the aorta, including aortic dilation, elastolysis, dissection, matrix accumulation, wall thickening and ulceration [32]. These studies strongly suggest that physiological TβRII signaling is crucial for maintaining postnatal aortic health.
Smads deletions can cause aortic aneurysm as well. Pluijm et al. identified that Smad3 deficiency in mice caused spontaneous dilation of the ascending aorta and an increased incidence of thoracic aortic aneurysm. Smad3-/- aortas exhibited elevated levels of nuclear p-SMAD2 and p-ERK, indicative of upregulated TGF-β receptor activation, yet without activating transcription of downstream TGF-β-targeted genes [34]. In calcium chloride-induced AAA mouse model, Smad3 deficiency was found to promote AAA formation, characterized by thickened abdominal aortic wall, fragmented elastic fiber and reorganized collagen fibers, and vascular remodeling [35]. These findings provide support for the pivotal functions of SMAD3 in maintaining the integrity of the vessel wall during the development of aortopathy. Aside from Smad3, deletion of Smad4 in SMCs promotes the development of both thoracic and abdominal aortic aneurysms along with reduced canonical TGF-β signaling. The phenomenon is mainly attributed to the upregulation of MMP-12, which is one of the proteases essential for elastin degradation [36]. Another study also reported that inducible Smad4 deletion in SMCs specifically provoked aortic aneurysms, suggesting that TGF-β signaling disruption triggers interleukin-1β as a hazard signal for the onset of aortic diseases [37].
3.2. TGF-β Neutralizing Antibodies and Aortic Diseases
Based on the activation of TGF-β signaling in the pathogenesis of aortopathy, the therapeutic efficacy of TGF-β neutralization was observed when the intervention was initiated at P28, P45, or P49 of age in MFS mice [14,38]. However, treatment with TGF-β neutralizing antibody (1D11) from P16 onward exacerbated arterial disease [39]. Apart from that, the application of 1D11 increased the incidence of aortic aneurysm rupture [40]. The detrimental impact of TGF-β neutralizing antibodies on AAA has also been observed. Since the elevated level of TGF-β1 had been detected in the aneurysmal tissue from AAA patients and mice [41], TGF-β neutralizing antibodies were utilized. Unexpectedly, it exacerbated angiotensin II (Ang II) or elastase-induced AAA [42]. In a similar context, after intraperitoneal injection of another TGF-β neutralizing antibody, 2G7, in Ang II micro-pump-induced AAA mice, the serum levels of TGF-β1 and TGF-β3 decrease, and the adventitia of abdominal aorta further expand [43]. Based on these data, it has been proposed that the detrimental effect of TGF-β neutralizing antibodies is associated with the recruitment of macrophages, monocyte invasiveness, activation of MMP-12, and promotion of matrix degradation, which can be partially alleviated by Mmp12 knockout or acute monocyte depletion. In other words, these tactics are unable to significantly change aneurysm incidence, yet they notably alleviate the severity of aneurysms [44]. Moreover, using neutralizing antibodies to inhibit TGF-β resulted in an enhanced infiltration of leukocytes in both the aortic wall and intraluminal thrombus, thereby leading to persistent dilation of the aortic lumen and aggravation of the ECM degradation. Therefore, the early inhibition of IL-1β or the suppression of monocyte-dependent responses could significantly mitigate the severity of AAA [45]. TGF-β neutralizing antibodies can also promote the apoptosis of VSMCs, resulting in substantial loss of medial smooth muscle cells [46], potentially increasing the risk of aortic dissection and rupture. Cyclosporine A (CsA), known to induce TGF-β1 production, maintains vascular elastic fiber integrity, reduces inflammation, increases α-SMA-positive cell content and eventually prevents AAA. However, these protective effects of CsA are disappeared after the administration of TGF-β neutralizing antibody [47]. Intriguingly, there were significant differences in aortic pathological damage caused by systematic inhibition of TGF-β with neutralizing antibody and SMC-specific knockout of Tgfbr2. The severity of AAA increases after TGF-β neutralizing antibody administration, but the severity of TAA appeared unaffected. In contrast, SMC-specific Tgfbr2 knockout prevented further expansion of the abdominal aortic adventitia but increased the incidence of intramural hematoma in the thoracic aortic wall considerably [43]. Hence, identifying the distinction of the two TGF-β inhibition methods and understanding the specific mechanisms of TGFβ1, 2 and 3 is beneficial for future research in this field.
3.3. TGF-β Overexpression and AAA
Early studies found that local overexpression of TGF-β in the aorta, using adenovirus-mediated delivery, delayed the expansion of AAA. Dai et al. reported that overexpressing active TGF-β1 in already-formed experimental AAA by endovascular gene delivery could reduce the accumulation of monocytes, macrophages and T cells, inhibit MMPs activity, increase collagen and elastic proteins level, and promote the proliferation of VSMCs [48]. Additionally, heart-specific overexpression of TGF-β1 upregulated plasma concentration of TGF-β1 and cardiac phosphorylated SMAD2, and prevented aortic dilation [49]. Bai et al. illustrated that in a calcium chloride-induced AAA mouse model, injection of TGF-β1 hydrogel into the vessel wall elevated SMAD2 phosphorylation, and reduced pathological damage to the aneurysm. Most crucial of all, hydrogels could be effectively and easily delivered between the medium and adventitia in swine aortas. This observation suggested that this technique held promise as a potential drug delivery method and therapeutic option [50]. In addition, in a pseudoaneurysm model induced by angioplasty, administration of TGF-β nanoparticles increases the expression of TGF-β1 and phosphorylation SMAD2, limiting the formation of pseudoaneurysms. Conversely, inhibition of TβRI signaling with SB431542 increases the incidence of pseudoaneurysms, indicating that TGF-β signaling deficiency is closely related to the formation of large pseudoaneurysms [51]. Kojima et al. explored AAA alleviation by overexpressing TGF-β through efferocytosis-stimulating therapy, and demonstrated that administration of the CD47 antibody (MIAP410) into AAA mice promoted the clearance of apoptotic cells, upregulated TGF-β and SMAD3 expression in the vascular media, reduced the accumulation of free apoptotic bodies and inflammatory aggregates in the aortic lesion, and relieved aortic dilation [52]. If proven safe and effective, this method could represent a novel non-surgical approach for treating aortic disease. Furthermore, since the safety of stem cell therapy for AAA has been confirmed, and a multicenter, double-blind, phase III clinical trial has illustrated that local injection of chondrocytes with high expression of TGF-β could remarkably alleviate arthritis, exploring stem cell therapy with high expression of TGF-β might offer new intervention for AAA [53,54].
3.4. Underlying Mechanisms of Improving AAD via TGF-β/SMAD Pathway
TGF-β signaling is of utmost importance in maintaining the equilibrium between ECM synthesis and degradation [55]. TGF-β promotes the synthesis of various elastic proteins by SMCs and stimulates the synthesis of collagen by fibroblasts. Furthermore, TGF-β inhibits MMP-9 secretion by monocytes or macrophages, preventing excessive degradation of ECM under the condition of AAA, thus maintaining vascular structural stability [56]. SMC-specific deletion of Tgfbr2 in Apoe-/- mice on a hypercholesterolemic diet promotes extensive reprogramming of contractile medial SMCs to mesenchymal stem cell (MSC)-like cells and leads to development of aortic aneurysms, which is mainly controlled by krüppel-like factor 4 (KLF4) [33]. Postnatal deletion of Tgfbr2 in SMCs decreases the expression of contractile-associated proteins smooth muscle protein 22 (SM22) and smooth muscle myosin heavy chain (SMMHC), inability to maintain contractile phenotype, and increases proliferation capacity, which may potentially increase the susceptibility of the vessel wall to complications such as dissection and dilatation [30]. However, Dichek DA et al. reported that postnatal SMC-specific Tgfbr2 deletion did not alter contractile protein abundance, but resulted in severe endothelial dysfunction and increased contractility of the aorta [32,57]. The authors suggested that this paradox may be due to different aortic mRNA collection sites (total aortic mRNA versus aortic medial mRNA). The TGFBR1A230T mutation, which has been newly identified as a mutation associated with LDS, impairs contractile transcript and protein levels, and function of cardiovascular progenitor cell (CPC)-VSMCs via decreasing SMAD3 phosphorylation [58]. Meanwhile, SMAD3 deficiency in CPC-VSMCs notably disrupts canonical TGF-β signaling, thus reducing contractile function and downregulating the expression of contractile-related genes, such as SM α-actin, myosin heavy chain 11, calponin-1 [58,59]. Analogously, the deficiency of microRNA-21 in SMAD3 heterozygous (Smad3+/- ) mice increases the expression of SMAD7 and suppresses the activation of canonical TGF-β signaling, thereby promoting VSMC phenotype switching from the contractile phenotype to the synthetic phenotype and accelerating aneurysm and dissection [60]. Another study showed that the matrix protein cellular communication network factor 2 (CCN2) has the ability to interact with the TGF-β1-TGF-β receptor complex, thereby enhancing TGF-β1-mediated SMAD3 phosphorylation and improving the contractile function of SMCs. Additionally, the deletion of CCN2 triggered SMC reprogramming, thus exacerbating the development of Ang II-induced AAA [61]. GDF11, belonging to the TGF-β superfamily, can hamper SMC phenotype switching and keep its contractile state via inhibition of MMP activity and activation of the canonical TGF-β signaling pathway. Consequently, GDF11 serves as a protective factor against the development of thoracic aortic aneurysms [62]. Interestingly, the mechanisms of SMC phenotype switching in a disease context differ from those during the differentiation of SMCs from CPCs. CPC-VSMCs maintain the contractile phenotype mainly by relying on SMAD3-dependent TGF-β signalling, whereas many other factors besides SMAD3 play a role in phenotype switching of SMCs in a disease context. Besides inducing medial degeneration, disruption of the TGF-β signaling brings on a large number of monocytes and macrophages to accumulate outside the blood vessels, thus promoting inflammation infiltration and accelerating vascular damage. The specific deletion of Smad4 in SMCs induces the recruitment of monocytes and triggers inflammation in the adventitia through the activation of the IL-1β/C-C motif chemokine ligand 2 (CCL2)/C-C motif chemokine receptor 2 (CCR2) axis. Inhibiting this axis by knockout of IL-1β receptor 1 or Ccr2, or using an anti-IL-1β antibody (canakinumab), blocks pro-inflammatory monocyte infiltration and alleviates the aortic pathology and aneurysm dilation caused by the inactivation of Smad4 in SMCs [38]. Similarly, in patients with TAAD resulting from rs12455792 variant of SMAD4, macrophage recruitment and M1 type inflammatory response increase in the aortic tissue [63]. These results suggest that SMC phenotype switching, endothelial dysfunction and monocyte inflammation, which were induced by disruption of TGF-β signaling, promote the development of aortic diseases.
4. The Role of Noncanonical TGF-β Signaling Pathway in Aortic Diseases
Apart from the canonical SMAD pathway, TGF-β family cytokines also activate non-SMAD signaling pathways, including TAK1, ERK, JNK, p38 MAPK and NF-κB signaling pathways. Typically, canonical TGF-β signaling is weakened while noncanonical TGF-β signaling is activated. The loss of SMAD4 leads to diminished canonical TGF-β signaling, concurrent activation of the JNK pathway, tissue proteinase S (CTSS) and MMPs, resulting in increased elastin degradation [36]. Likewise, the SMC-specific knockout of Tgfbr2 leads to the downregulation of SMAD2 phosphorylation and the upregulation of p38 phosphorylation in the thoracic aorta. Also, the MAPK pathway is abnormally activated [30]. Unlike the canonical TGF-β signaling pathway, noncanonical TGF-β signaling is believed to promote the progression of aortic dilation, aneurysm and dissection. In MFS mice, the selective inhibition of ERK1/2 has been shown to alleviate the progression of aneurysms, while Smad4 deletion exacerbates aortic diseases. Moreover, Smad4 deficiency increases the activation of JNK1, and a JNK antagonist ameliorates aortic growth in MFS mice [64]. One possible explanation is that the ERK pathway inhibits the transcription of SMAD-mediated smooth muscle contraction-related genes and promotes the expression of cell cycle and apoptosis-related genes by Notch 3 in MFS [65]. All of these studies suggest that the canonical and noncanonical TGF-β signaling pathways exert divergent regulatory effects on aortic diseases.
4.1. MAPK Signaling Pathway
The MAPK family includes three key kinases: ERK, JNK, and p38 [66]. ERK1/2 activation participates in non-canonical TGF-β signalling, which is excessively increased in MFS, and has been recognized as a critical factor in the cultivation of aortic pathology [64]. Increase of phosphorylated ERK and activation of the ERK pathway can be detected in the aortas of AAA patients, MFS mice, elastase and Ang II-induced AAA mice. Such activation of the ERK pathway promotes the production and release of MMPs and plasminogen activator, which degrade ECM and accelerate aortic dilation [39,67-70]. Notably, Erk1-/- mice exhibit resistance to AAA development, and inhibiting ERK activation blocks AAA formation [68,69]. Besides, the specific inhibition of TGF-β-mediated ERK signaling activation by losartan has been shown to delay aortic aneurysm expansion, while, enalapril, another agent capable of attenuating TGF-β signaling in the aorta, demonstrates less efficacy [71]. Nettersheim et al. reported that nitro-oleic acid reduces aortic dilation and wall stiffening in MFS via inhibition of aortic ERK1/2 phosphorylation without altering TGF-β1 or TβRI/II expression [72]. Additionally, some drugs, such as enzastaurin, hydralazine and cycloastragenol significantly reduce aortic root growth in MFS mice or Ang II-treated Apoe-/- mice by inhibiting ERK activation [73]. Women diagnosed with MFS face a high risk of experiencing aortic dissection during pregnancy. Habashi et al. proposed that oxytocin mediated the increased risk of aortic dissection during pregnancy through phosphorylation of ERK. Therefore, treatment with oxytocin receptor antagonist, ERK kinase inhibitor (trametinib) or hydralazine substantially decrease pregnancy-associated aortic disease [74]. These studies suggest that the activation of ERK pathway promotes the occurrence and development of aortic dilation and dissection. Therefore, targeting TGF-β1-mediated ERK signaling could represent a more efficacious therapeutic strategies for aortic diseases.
High levels of phosphorylated JNK have been observed in human AAA tissues [75]. Activated JNK signaling is known to stimulate the secretion of MMPs by monocytes and macrophages, especially MMP-2, which could exacerbate aortic ECM degradation. In addition, activated JNK constricts the biosynthesis of ECM through downregulating ECM synthetic enzymes to hinder collagen deposition, thus participating in aortic structural damage [76,77]. The activity of JNK is regulated by various signaling molecules. For example, the loss of ephrin-B2 in smooth muscle cells enhances the internalization of platelet-derived growth factor receptor β, subsequently activating the JNK pathway. This activation leads to increased aortic root diameter and vessel wall defects [78]. Also, a recent study demonstrated that prostaglandin E receptor 4 (EP4) in SMCs increased IL-6 secretion, triggered inflammatory cell infiltration via activating the TAK1-NF-κB/JNK/p38 pathway, leading to exacerbating AAA [79]. Furthermore, knockout of microRNA-33 in macrophages inactivates JNK, reduces the expression of MMP-9, suppresses matrix degradation and attenuates AAA [80]. Comparative studies indicate that administration of JNK inhibitor, SP600125, could alleviate Ang II- and calcium chloride-induced aortic dilation, medial thinning and elastic plate rupture [76,81]. All these studies demonstrate that JNK-targeted therapy has the potential to offer alternative therapeutic approaches for the treatment of AAA.
It has been reported that an increase of the p38 MAPK signaling activity aggravates the progression of aortic diseases as well. Granata et al. used an MFS patient-derived human induced pluripotent stem cell (hiPSC) model to recapitulate the major phenotypes of MFS vascular pathology and identified a key role for p38 MAPK in disease development. They found that p38 MAPK inhibition by SB203580 increased fibrillin-1 deposition and promoted SMC proliferation, while inhibition of canonical SMAD signaling or ERK1/2 had no significant effect, highlighting a crucial role of p38 MAPK in MFS [82]. Similarly, SMC-specific deletion of Mapk14 (p38α), which is the primary isoform of p38 family in SMCs, has been shown to improve AAA formation, as well as vascular senescence and inflammation [83]. Another study indicates that sodium-glucose cotransporter 2 (SGLT-2) inhibitor, empagliflozin, significantly inhibits Ang II-induced AAA formation, independently of blood pressure effects via inactivation of p38 MAPK signaling pathway [84]. All of these studies indicate the beneficial effect of p38MAPK inhibition on aortic aneurysm and dissection.
4.2. NF-κB Signaling Pathway
It is known that under stimuli, such as reactive oxygen species (ROS), the NF-κB pathway is activated, which subsequently triggers a series of inflammatory reactions, including the increased expression of MMPs and monocyte chemoattractant protein 1 (MCP-1). During the formation of AAA, the process of pathological vascular remodeling is primarily triggered by the infiltration of macrophage. The epigenetic enzyme JMJD3 facilitates NF-κB-mediated transcription of inflammatory genes and enhances monocyte and macrophage infiltration into the aortic wall. Inhibition of JMJD3 prevents AAA formation and decreases macrophage inflammation [85]. Abnormal amino acid metabolism has also been associated with vascular diseases. One study found that the level of 3-hydroxyanthranilic acid (3-HAA) rises in the plasma and aorta of AAA. 3-HAA activates MMP2 exceptionally by increasing the phosphorylation of NF-κB, thereby promoting AAA formation [86]. In addition, some studies have identified an association between membrane attack complex (MAC) and AAA. MAC can activate NF-κB and increase the expression of MMP-2 and MMP-9, thus accelerating the development of AAA [87]. Furthermore, evidence suggests that the activation of mesenchymal cells is closely related to vascular inflammation and the formation of AAA. The absence of NF-κB signaling in mesenchymal cells can appreciably inhibit monocytes recruitment and vascular inflammation, thereby blocking Ang II-induced AAA formation [88]. In addition, Liu et al. has found that eosinophils could block the activation of NF-κB and polarization of macrophages and monocytes to protect against AAA [89]. These studies collectively demonstrate that NF-κB-mediated vascular inflammation plays a pivotal role in the progression of AAA.
Apart from promoting the infiltration of inflammatory cells, the TGF-β1/ROS/NF-κB signaling pathway also induces SMC senescence and phenotype switching. In aortic tissue harvested from the MFS patients, TGF-β1 has been found to induce VSMC senescence by activating NF-κB signaling through the generation of ROS. This effect can be suppressed by NF-κB inhibitor (SC-514) [90]. Likely, the histone deacetylase SIRT1 has been observed to blocks vascular cell senescence and AAA formation via impeding the binding of NF-κB to the MCP-1 promoter [91]. The NF-κB pathway is known to promote SMC phenotypic switching, as evidenced by the downregulation of SM22α. Inhibition of NF-κB or ROS has been shown to partially mitigate the promoting effect of SM22α deficiency on AAA formation and reduce the maximal diameter of the aorta [92]. Besides, the loss of α-actin in SMCs leads to NF-κB-dependent abnormal dilation of the mouse aortic root [93]. Liang et al. reported that Ang II promotes SMC proliferation, migration, apoptosis, and MMPs’ expression. However, these phenomena are alleviated by inhibiting the NF-κB signaling pathway [94]. In conclusion, TGF-β1/ROS/NF-κB signaling pathway induces monocytes and macrophages inflammation and SMC dysfunction, thus promoting aortic diseases.
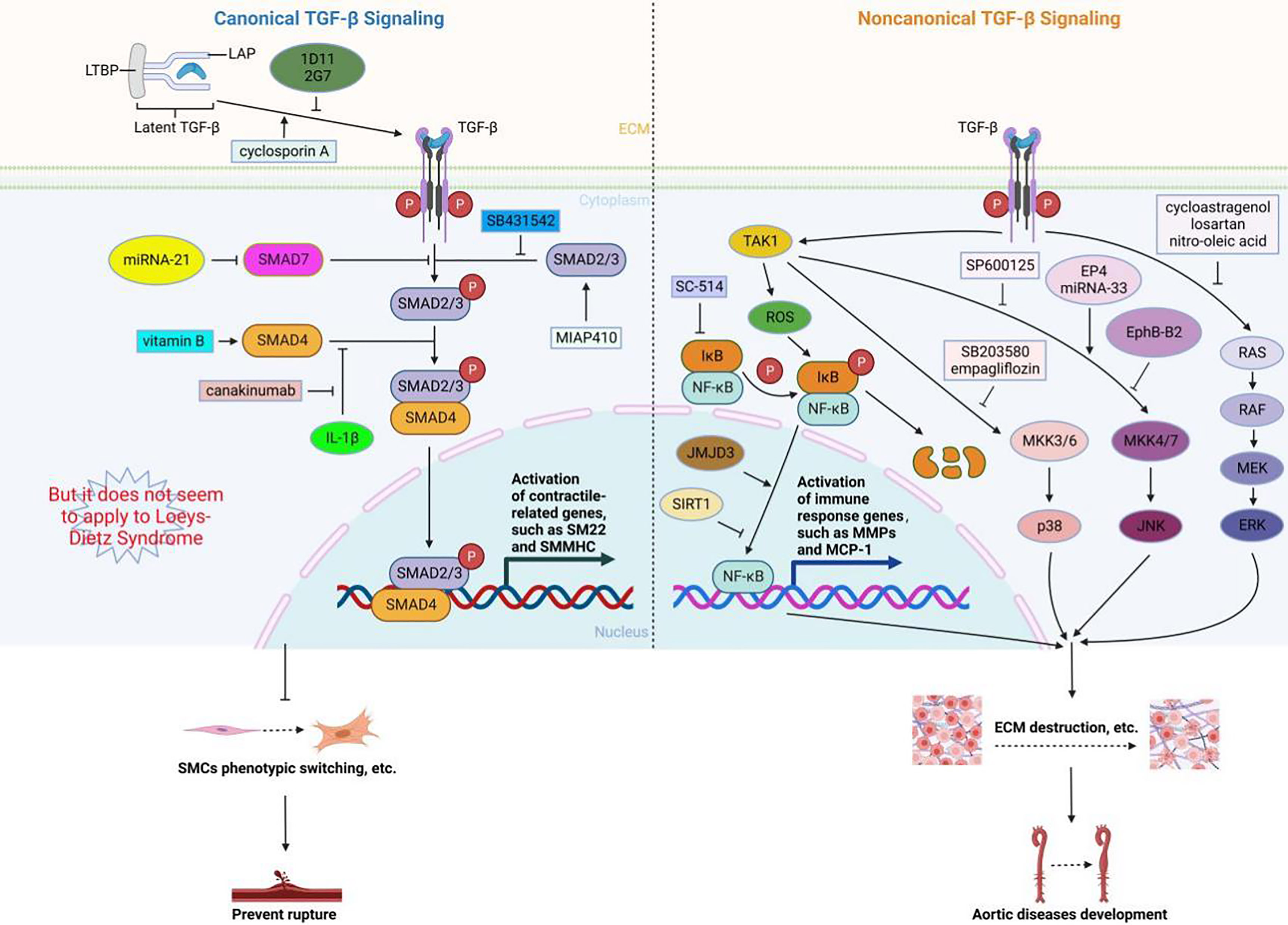
Figure 2. The role of canonical and noncanonical TGF-β signaling pathways in aortic diseases..
5. TGF-β Signaling Pathway and Heritable TAAD
Pathogenic variants in numerous genes, including those within the TGF-β signaling pathway, as well as genes related to extracellular matrix protein, and vascular smooth muscle contraction components or cytoskeleton genes, have been discovered to be associated with an increased risk of heritable TAAD [95,96]. Sequence variants related to the TGF-β signaling pathway have been identified as the underlying cause of syndromic TAAD, encompassing conditions such as MFS, LDS, and SGS. In this review, we will focus on current molecular knowledge and the effect of TGF-β signaling pathway on MFS and LDS.
5.1. Marfan Syndrome (MFS)
MFS is a genetic disorder affecting the connective tissue, primarily manifesting in cardiovascular abnormalities such as aortic aneurysm and dissection, ocular complications like ectopia lentis, pulmonary issues including pneumothorax, and skeletal system disorders characterized by elongated limbs and fingers, scoliosis, and pectus deformities. As a hereditary disease, MFS is primarily caused by heterozygous mutations in the fibrillin-1 gene (FBN1). Fibrillin-1 monomers undergo self-assembly to form microfibrils with a diameter of 10 nm. These microfibrils, constituting part of the extracellular matrix in the aortic wall, play a key role in the regulation of TGF-β bioavailability in the extracellular matrix through interacting with latent TGF-β-binding proteins [97]. Thus, mutations in the fibrilin-1 gene not only leads to structural weakness in connective tissue, but also results in an increase in TGF-β signaling. Both of these factors contribute to the complex pathogenesis observed in MFS. Deletion or mutation in Fbn1 increases the activation of TGF-β signaling in murine models of MFS [13,14,98]. Although the role of TGF-β in MFS pathogenesis is still debated, early investigations have demonstrated that the overactivation of TGF-β signaling exerted a causal effect on progressive aortic root enlargement and the formation of aortic aneurysm in mice with non-lethal MFS (Fbn1 C1039G/+ mice). This conclusion was derived from the observation that TGF-β antagonists, such as TGF-β-neutralizing antibody or the angiotensin II type 1 receptor (AT1) blocker, could effectively inhibit the development of aneurysms [14]. Another report also showed that treatment with anti-TGF-β antibody in Fbn1C1039G/+ mice at 4 weeks old could prevent TAA progression [38]. By investigating the effects of TGF-β on the development and advancement of aortic aneurysm in mice with progressively severe MFS (Fbn1mgR/mgR ), Ramirez et al. revealed that the impact of TGF-β on the formation and progression of aortic aneurysm varied, with some cases showing a protective effect while others demonstrated a detrimental effect. The researchers reached the conclusion that TGF-β hypersignaling played a secondary role in driving the progression of aneurysms in individuals with MFS. This conclusion was based on the observation that blocking TGF-β in young MFS animals during the early stages of the disease (2 weeks old/P16) worsened the aneurysm, whereas treatment administered at later stages proved to be beneficial [39]. Similarly, loss of physiological TGF-β signaling induced by disruption of Tgfbr2 gene exacerbates the degree of TAA and aortic dissection in Fbn1C1039G/+ mice [32]. In addition, Galunisertib, a highly effective small-molecule inhibitor of TβRI, fails to mitigate the disrupted architecture of the medial wall in MFS mice [99]. These findings suggest that the impairment of basal TGF-β signaling can lead to the development of aortic dilatation and dissection.
The underlying causes for the divergent outcomes observed in aortopathy following TGF-β inhibition remain unclear. However, a growing body of research suggests that the noncanonical TGF-β/ERK signaling pathway plays a significant role in driving the progression of aortic aneurysms in mice with MFS [64,71]. Both ERK 1/2 and SMAD2/3 are activated in MFS mice, and nitro-oleic acid or recombinant angiogenic factor with G patch and FHA domains 1 (AGGF1) protein attenuates TAA, accompanied by the inhibition of ERK1/2 and SMAD2/3 phosphorylation [72,100]. Selective inhibition of ERK1/2 activation ameliorates aortic dilation but has no effect on SMAD2 activation. Inhibition of canonical SMAD4 exacerbates the aortopathy and causes premature mortality in mice with MFS [65]. Park JH et al. reported MFS mice treated with galunisertib or losartan completely blocked SMAD2 phosphorylation. But only losartan exhibited remarkable inhibition of aortopathy and a marked decrease in ERK1/2 phosphorylation [99]. A preventive treatment with TGF-β inhibitor peptide (P144) before the onset of the aortic aneurysm (at the age of 4 weeks in MFS mice) fully prevented the formation of the aneurysm and reduces the nuclear translocation of p-ERK1/2, but did not significantly inhibit p-SMAD2 activation [101]. Vitamin B mixture has been shown to restore canonical TGF-β signaling and mitigates aortic dilation in MFS mice [102]. Given the pleiotropic effects of TGF-β in the aortic wall and various tissues, direct inhibition of TGF-β may not be suitable for MFS and could even have harmful effects. Therefore, alternative targets need to be identified. Lim et al. proposed that IL-11 induced by activation of TGF-β1/TβRII/SMAD2 signaling pathway may potentially play a vital role in noncanonical TGF-β/ERK-mediated aortic dilatation and dissection. They concluded that specifically targeting IL-11-induced ERK activation, either through genetic means or with a neutralizing antibody, rather than the overall TGF-β signaling pathway, had uniquely benefit as this approach effectively limited aortopathy in MFS mouse model and without influencing SMAD4-targeted gene transcription [103-105]. Therefore, the IL-11 emerges as a potential therapeutic target for aortopathy in individuals with MFS.
5.2. Loeys-Dietz Syndrome (LDS)
Loeys-Dietz syndrome (LDS) is characterized by the presence of aortic and branch vessel aneurysms and dissections, distinctive facial features such as hypertelorism, arterial tortuosity, and bifid uvula. Individuals with this syndrome are particularly susceptible to arterial dissection or rupture at a young age [106]. LDS is usually induced by pathogenic variants in multiple genes, including TGF-β ligands (TGFB2, TGFB3), receptors (TGFBR1, TGFBR2), or intracellular signaling mediators SMAD3 and SMAD2 [2,107-112]. Almost all sequence variants related to LDS indicate that development of aortic disease can be linked to the deficiency of TGF-β signaling. However, pathogenic variants in TGFBR1 or TGFBR2 have been shown to upregulate the level p-SMAD2 in the aortic wall [110]. Consistently, SMAD2 and ERK phosphorylation in aortic media are evidently increased in knock-in mice with loss-of-function mutations (Tgfbr1M318R/+ and Tgfbr2G357W/+ ) at end-stage aneurysmal samples. Furthermore, overall activation of TGF-β signature is also observed in the aortic tissues with mutations in SMAD3 or TGFB2/3 [108,111,113]. A recent study identified a novel TGFBR1 variant (TGFBR1A230T ) causing LDS, revealing that the TGFBR1A230T mutation reduced phosphorylated SMAD3 but does not affect SMAD2 or ERK activation [58]. Several reviews have summarized extensive investigations and proposed some hypotheses to explain this paradox [26,114-117]. Nevertheless, further investigation is required to provide a conclusive explanation for the variations in TGF-β signaling in relation to the development of aneurysms.
6. Conclusions
TGF-β signaling dysregulation plays a vital role in the occurrence and development of aortic diseases. Pathogenic sequence variants in TGF-β signaling-related genes are implicated in hereditary TAAD. In general, SMAD-dependent canonical TGF-β signaling pathways not only promote aortic development and maintain aortic structural homeostasis under normal physiological conditions, but also reduce the incidence of aortic diseases under pathological conditions by inhibiting aortic dilation. The protective mechanisms include promoting extracellular matrix synthesis, activating regulatory T cells, inhibiting inflammation and matrix degradation, and improving VSMCs phenotype switching. On the contrary, TGF-β, through noncanonical signaling pathways, exacerbates aortic injury and the development of aortic aneurysm and dissection. In the clinical setting, the primary approaches targeting TGF-β involving inhibiting TGF-β receptor signal transduction, attenuating its activation and expression, and blocking TGF-β binding to its receptor [118]. However, there are significant challenges in advancing these treatments clinically. For instance, individuals treated with Fresolimumab, a TGF-β-blocking antibody, have experienced the development of acanthoma [119]. Given the complexity of TGF-β’s role in aortic diseases, precise and targeted therapies need to be developed. Exploring effective and safe intervention methods to activate canonical TGF-β signals or inhibit non-canonical TGF-β signals can provide potential new treatment strategies for aortic diseases.
Author Contributions: Conceptualization, Liping Xie and Tianyu Song; writing-original draft preparation, Zijie Liu and Tianyu Song; writing-review and editing, Liping Xie and Zijie Liu; supervision and project administration, Liping Xie.All authors have read and agreed to the published version of the manuscript.
Funding: This research funded by the National Natural Science Foundation of China (82070278).
Institutional Review Board Statement: Not applicable.
Informed Consent Statement: Not applicable.
Data Availability Statement: Not applicable.
Acknowledgments: The authors would like to thank the associate editor and the reviewers for their useful comments, suggestions and criticism that improved this paper. Additionally, we extend our gratitude to Prof. Xin Wang for her invaluable assistance in this manuscript.
Conflicts of Interest: The authors declare no conflict of interest.

References
- Bossone, E.; Eagle, K.A. Epidemiology and management of aortic disease: aortic aneurysms and acute aortic syndromes. Nat. Rev. Cardiol. 2021, 18, 331‒348. DOI: https://doi.org/10.1038/s41569-020-00472-6
- Writing Committee, Members; Isselbacher, E.M.; Preventza, O.; et al. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2022, 80, e223‒e393.
- Juang, D.; Braverman, A.C.; Eagle, K. Cardiology patient pages. Aortic dissection. Circulation 2008, 118, e507‒e510. DOI: https://doi.org/10.1161/CIRCULATIONAHA.108.799908
- Schanzer, A.; Oderich, G.S. Management of Abdominal Aortic Aneurysms. N. Engl. J. Med. 2021, 385, 1690‒1698. DOI: https://doi.org/10.1056/NEJMcp2108504
- Isselbacher, E.M. Thoracic and abdominal aortic aneurysms. Circulation 2005, 111, 816‒828. DOI: https://doi.org/10.1161/01.CIR.0000154569.08857.7A
- Gao, J.; Cao, H.; Hu, G.; et al. The mechanism and therapy of aortic aneurysms. Signal Transduct. Target. Ther. 2023, 8, 55. DOI: https://doi.org/10.1038/s41392-023-01325-7
- Li, J.; Pan, C.; Zhang, S.; et al. Decoding the Genomics of Abdominal Aortic Aneurysm. Cell 2018, 174, 1361‒1372. DOI: https://doi.org/10.1016/j.cell.2018.07.021
- Francis, C.M.; Futschik, M.E.; Huang, J.; et al. Genome-wide associations of aortic distensibility suggest causality for aortic aneurysms and brain white matter hyperintensities. Nat. Commun. 2022, 13, 4505. DOI: https://doi.org/10.1038/s41467-022-32219-x
- Su, C.T.; Huang, J.W.; Chiang, C.K. Latent transforming growth factor binding protein 4 regulates transforming growth factor beta receptor stability. Hum. Mol. Genet. 2015, 24, 4024‒4036. DOI: https://doi.org/10.1093/hmg/ddv139
- Lan, H.C.; Li, H.J.; Lin, G.; et al. Cyclic AMP stimulates SF-1-dependent CYP11A1 expression through homeodomain-interacting protein kinase 3-mediated Jun N-terminal kinase and c-Jun phosphorylation. Mol. Cell. Biol. 2007, 27, 2027‒2036. DOI: https://doi.org/10.1128/MCB.02253-06
- Takeda, N.; Hara, H.; Fujiwara, T.; et al. TGF-β Signaling-Related Genes and Thoracic Aortic Aneurysms and Dissections. Int. J. Mol. Sci. 2018, 19, 2125. DOI: https://doi.org/10.3390/ijms19072125
- Ng, C.M.; Cheng, A.; Myers, L.A.; et al. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Invest. 2004, 114, 1586‒1592. DOI: https://doi.org/10.1172/JCI22715
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; et al. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 3, 407‒411. DOI: https://doi.org/10.1038/ng1116
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117‒121. DOI: https://doi.org/10.1126/science.1124287
- Chen, W.; Ten, Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723‒740. DOI: https://doi.org/10.1038/nri.2016.112
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; et al. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44‒53. DOI: https://doi.org/10.1016/j.matbio.2015.05.005
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325‒338. DOI: https://doi.org/10.1038/nrneph.2016.48
- Nolte, M.A.; Margadant, C. Controlling Immunity and Inflammation through Integrin-Dependent Regulation of TGF-β. Trends. Cell. Biol. 2020, 30, 833. DOI: https://doi.org/10.1016/j.tcb.2020.08.001
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. DOI: https://doi.org/10.1101/cshperspect.a021907
- Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell. Biol. 2008, 40, 1068‒1078. DOI: https://doi.org/10.1016/j.biocel.2007.11.026
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577‒584. DOI: https://doi.org/10.1038/nature02006
- Chen, J.; Chang, R. Association of TGF-β Canonical Signaling-Related Core Genes with Aortic Aneurysms and Aortic Dissections. Front. Pharmacol. 2022, 13, 888563. DOI: https://doi.org/10.3389/fphar.2022.888563
- Tie, Y.; Tang, F.; Peng, D.; et al. TGF-beta signal transduction: biology, function and therapy for diseases. Mol. Biomed. 2022, 3, 45. DOI: https://doi.org/10.1186/s43556-022-00109-9
- Tingting, T.; Wenjing, F.; Qian, Z.; et al. The TGF-β pathway plays a key role in aortic aneurysms. Clin. Chim. Acta. 2020, 501, 222‒228. DOI: https://doi.org/10.1016/j.cca.2019.10.042
- van Dorst, D.C.H.; de Wagenaar, N.P.; van der Pluijm, I.; et al. Transforming Growth Factor-β and the Renin-Angiotensin System in Syndromic Thoracic Aortic Aneurysms: Implications for Treatment. Cardiovasc. Drugs Ther. 2021, 35, 1233‒1252. DOI: https://doi.org/10.1007/s10557-020-07116-4
- Shi, Y.; Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685‒700. DOI: https://doi.org/10.1016/S0092-8674(03)00432-X
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends. Cell. Biol. 2010, 20, 556‒567. DOI: https://doi.org/10.1016/j.tcb.2010.06.006
- Goumans, M.J.; Liu, Z.; ten, Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell. Res. 2009, 19, 116‒127. DOI: https://doi.org/10.1038/cr.2008.326
- Schlecht, A.; Leimbeck, S.V.; Jägle, H.; et al. Deletion of Endothelial Transforming Growth Factor-β Signaling Leads to Choroidal Neovascularization. Am. J. Pathol. 2017, 187, 2570‒2589. DOI: https://doi.org/10.1016/j.ajpath.2017.06.018
- Li, W.; Li, Q.; Jiao, Y.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest. 2014, 124, 755‒767. DOI: https://doi.org/10.1172/JCI69942
- Wei, H.; Hu, J.H.; Angelov, S.N.; et al. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor β Signaling. J. Am. Heart. Assoc., 2017, 6, e004968. DOI: https://doi.org/10.1161/JAHA.116.004968
- Hu, J.H.; Wei, H.; Jaffe, M.; et al. Postnatal Deletion of the Type II Transforming Growth Factor-β Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2647‒2656. DOI: https://doi.org/10.1161/ATVBAHA.115.306573
- Chen, P.Y.; Qin, L.; Li, G.; et al. Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 2020, 26, 542‒557 DOI: https://doi.org/10.1016/j.stem.2020.02.013
- van der Pluijm, I.; van Vliet, N.; von der Thusen, J.H.; et al. Defective Connective Tissue Remodeling in Smad3 Mice Leads to Accelerated Aneurysmal Growth Through Disturbed Downstream TGF-β Signaling. EbioMedicine 2016, 12, 280‒294. DOI: https://doi.org/10.1016/j.ebiom.2016.09.006
- Dai, X.; Shen, J.; Annam, N.P.; et al. SMAD3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model. Sci. Rep. 2015, 5, 10180. DOI: https://doi.org/10.1038/srep10180
- Zhang, P.; Hou, S.; Chen, J.; et al. Smad4 Deficiency in Smooth Muscle Cells Initiates the Formation of Aortic Aneurysm. Circ. Res. 2016, 118, 388‒399. DOI: https://doi.org/10.1161/CIRCRESAHA.115.308040
- Da, Ros F.; Carnevale, R.; Cifelli, G.; et al. Targeting Interleukin-1β Protects from Aortic Aneurysms Induced by Disrupted Transforming Growth Factor β Signaling. Immunity. 2017, 47, 959‒973. DOI: https://doi.org/10.1016/j.immuni.2017.10.016
- Huang, K.; Wang, Y.; Siu, K.L.; et al. Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics. Redox. Biol. 2021, 38, 101757. DOI: https://doi.org/10.1016/j.redox.2020.101757
- Cook, J.R.; Clayton, N.P.; Carta, L.; et al. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 911‒917. DOI: https://doi.org/10.1161/ATVBAHA.114.305150
- Chen, X.; Rateri, D.L.; Howatt, D.A.; et al. TGF-β Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS One 2016, 11, e0153811. DOI: https://doi.org/10.1371/journal.pone.0153811
- Doyle, A.J.; Redmond, E.M.; Gillespie, D.L.; et al. Differential expression of Hedgehog/Notch and transforming growth factor-β in human abdominal aortic aneurysms. J. Vasc. Surg. 2015, 62, 464‒470. DOI: https://doi.org/10.1016/j.jvs.2014.02.053
- Rush, C.; Nyara, M.; Moxon, J.V.; et al. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm. BMC Genomics 2009, 10, 298. DOI: https://doi.org/10.1186/1471-2164-10-298
- Angelov, S.N.; Hu, J.H.; Wei, H.; et al. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102‒2113. DOI: https://doi.org/10.1161/ATVBAHA.117.309401
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Invest. 2010, 120, 422‒432. DOI: https://doi.org/10.1172/JCI38136
- Lareyre, F.; Clément, M.; Raffort, J.; et al. TGFβ (Transforming Growth Factor-β) Blockade Induces a Human-Like Disease in a Nondissecting Mouse Model of Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2171‒2181. DOI: https://doi.org/10.1161/ATVBAHA.117.309999
- Meng, X.; Yang, J.; Zhang, K.; et al. Regulatory T cells prevent angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E knockout mice. Hypertension 2014, 64, 875‒882. DOI: https://doi.org/10.1161/HYPERTENSIONAHA.114.03950
- Dai, J.; Michineau S.; Franck, G.; et al. Long term stabilization of expanding aortic aneurysms by a short course of cyclosporine A through transforming growth factor-beta induction. PLoS One 2011, 6, e28903. DOI: https://doi.org/10.1371/journal.pone.0028903
- Dai, J.; Losy, F.; Guinault, A.M.; et al. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation 2005, 112, 1008‒1015. DOI: https://doi.org/10.1161/CIRCULATIONAHA.104.523357
- Frutkin, A.D.; Otsuka, G.; Stempien-Otero, A.; et al. TGF-[beta]1 limits plaque growth, stabilizes plaque structure, and prevents aortic dilation in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1251‒1257. DOI: https://doi.org/10.1161/ATVBAHA.109.186593
- Bai, H.; Sun, P.; Wei, S.; et al. A novel intramural TGF β 1 hydrogel delivery method to decrease murine abdominal aortic aneurysm and rat aortic pseudoaneurysm formation and progression. Biomed. Pharmacother. 2021, 137, 111296. DOI: https://doi.org/10.1016/j.biopha.2021.111296
- Bai, H.; Lee, J.S.; Hu, H.; et al. Transforming Growth Factor-β1 Inhibits Pseudoaneurysm Formation After Aortic Patch Angioplasty. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 195‒205. DOI: https://doi.org/10.1161/ATVBAHA.117.310372
- Kojima, Y.; Werner, N.; Ye, J.; et al. Proefferocytic Therapy Promotes Transforming Growth Factor-β Signaling and Prevents Aneurysm Formation. Circulation 2018, 137, 750‒753. DOI: https://doi.org/10.1161/CIRCULATIONAHA.117.030389
- Golledge, J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225‒242. DOI: https://doi.org/10.1038/s41569-018-0114-9
- Kim, M.K.; Ha, C.W.; In, Y.; et al. A Multicenter, Double-Blind, Phase III Clinical Trial to Evaluate the Efficacy and Safety of a Cell and Gene Therapy in Knee Osteoarthritis Patients. Hum. Gene. Ther. Clin. Dev. 2018, 29, 48‒59. DOI: https://doi.org/10.1089/humc.2017.249
- Massagué, J.; Sheppard, D. TGF-β signaling in health and disease. Cell 2023, 186, 4007‒4037. DOI: https://doi.org/10.1016/j.cell.2023.07.036
- Li, Z.; Kong, W. Cellular signaling in Abdominal Aortic Aneurysm. Cell. Signal. 2020, 70, 109575. DOI: https://doi.org/10.1016/j.cellsig.2020.109575
- Zhu, J.; Angelov, S.; Alp Yildirim, I.; et al. Loss of Transforming Growth Factor Beta Signaling in Aortic Smooth Muscle Cells Causes Endothelial Dysfunction and Aortic Hypercontractility. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1956‒1971. DOI: https://doi.org/10.1161/ATVBAHA.121.315878
- Zhou, D.; Feng, H.; Yang, Y.; et al. hiPSC Modeling of Lineage-Specific Smooth Muscle Cell Defects Caused by TGFBR1A230T Variant, and Its Therapeutic Implications for Loeys-Dietz Syndrome. Circulation 2021, 144, 1145‒1159. DOI: https://doi.org/10.1161/CIRCULATIONAHA.121.054744
- Gong, J.; Zhou, D.; Jiang, L.; et al. In Vitro Lineage-Specific Differentiation of Vascular Smooth Muscle Cells in Response to SMAD3 Deficiency: Implications for SMAD3-Related Thoracic Aortic Aneurysm. Arterioscler. Thromb. Vasc Biol., 2020, 40, 1651‒1663. DOI: https://doi.org/10.1161/ATVBAHA.120.313033
- Huang, X.; Yue, Z.; Wu, J.; et al. MicroRNA-21 Knockout Exacerbates Angiotensin II-Induced Thoracic Aortic Aneurysm and Dissection in Mice With Abnormal Transforming Growth Factor-β-SMAD3 Signaling. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1086‒1101. DOI: https://doi.org/10.1161/ATVBAHA.117.310694
- Wang, Y.; Liu, X.; Xu, Q.; et al. CCN2 deficiency in smooth muscle cells triggers cell reprogramming and aggravates aneurysm development. JCI Insight, 2023, 8, e162987. DOI: https://doi.org/10.1172/jci.insight.162987
- Ren, K.; Li, B.; Liu, Z.; et al. GDF11 prevents the formation of thoracic aortic dissection in mice: Promotion of contractile transition of aortic SMCs. J. Cell. Mol. Med. 2021, 25, 4623‒4636. DOI: https://doi.org/10.1111/jcmm.16312
- Wang, Y.; Yin, P.; Chen, Y.H.; et al. A functional variant of SMAD4 enhances macrophage recruitment and inflammatory response via TGF-β signal activation in Thoracic aortic aneurysm and dissection. Aging 2018, 10, 3683‒3701. DOI: https://doi.org/10.18632/aging.101662
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011, 332, 358‒361. DOI: https://doi.org/10.1126/science.1192149
- Pedroza, A.J.; Koyano, T.; Trojan, J.; et al. Divergent effects of canonical and non-canonical TGF-β signalling on mixed contractile-synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J. Cell. Mol. Med. 2020, 24, 2369‒2383. DOI: https://doi.org/10.1111/jcmm.14921
- Sinkala, M.; Nkhoma, P.; Mulder, N.; et al. Integrated molecular characterisation of the MAPK pathways in human cancers reveals pharmacologically vulnerable mutations and gene dependencies. Commun. Biol. 2021, 4, 9. DOI: https://doi.org/10.1038/s42003-020-01552-6
- Groeneveld, M.E.; van Burink, M.V.; Begieneman, M.P.; et al. Activation of extracellular signal-related kinase in abdominal aortic aneurysm. Eur. J. Clin. Invest. 2016, 46, 440‒447. DOI: https://doi.org/10.1111/eci.12618
- Ghosh, A.; DiMusto, P.D.; Ehrlichman, L.K.; et al. The role of extracellular signal-related kinase during abdominal aortic aneurysm formation. J. Am. Coll. Surg. 2012, 215, 668‒680. DOI: https://doi.org/10.1016/j.jamcollsurg.2012.06.414
- Zhang, Y.; Naggar, J.C.; Welzig, C.M.; et al. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1764‒1771. DOI: https://doi.org/10.1161/ATVBAHA.109.192609
- Xiong, W.; Meisinger, T.; Knispel, R.; et al. MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ. Res. 2012, 110, e92‒e101. DOI: https://doi.org/10.1161/CIRCRESAHA.112.268268
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011, 332, 361‒365. DOI: https://doi.org/10.1126/science.1192152
- Nettersheim, F.S.; Lemties, J.; Braumann, S.; et al. Nitro-oleic acid reduces thoracic aortic aneurysm progression in a mouse model of Marfan syndrome. Cardiovasc. Res. 2022, 118, 2211‒2225. DOI: https://doi.org/10.1093/cvr/cvab256
- Doyle, J.J.; Doyle, A.J.; Wilson, N.K.; et al. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife 2015, 4, e08648. DOI: https://doi.org/10.7554/eLife.08648
- Habashi, J.P.; MacFarlane, E.G.; Bagirzadeh, R.; et al. Oxytocin antagonism prevents pregnancy-associated aortic dissection in a mouse model of Marfan syndrome. Sci. Transl. Med. 2019, 11, eaat4822. DOI: https://doi.org/10.1126/scitranslmed.aat4822
- Kugo, H.; Sukketsiri, W.; Iwamoto, K.; et al. Low glucose and serum levels cause an increased inflammatory factor in 3T3-L1 cell through Akt, MAPKs and NF-кB activation. Adipocyte 2021, 10, 232‒241. DOI: https://doi.org/10.1080/21623945.2021.1914420
- Yoshimura, K.; Aoki, H.; Ikeda, Y.; et al. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat. Med. 2005, 11, 1330‒1338. DOI: https://doi.org/10.1038/nm1335
- Zhang, C.; Zhang, M.X.; Shen, Y.H.; et al. TNF-alpha suppresses prolyl-4-hydroxylase alpha1 expression via the ASK1-JNK-NonO pathway. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1760‒1767. DOI: https://doi.org/10.1161/ATVBAHA.107.144881
- Nakayama, A.; Nakayama, M.; Turner, C.J.; et al. Ephrin-B2 controls PDGFRβ internalization and signaling. Genes Dev. 2013, 27, 2576‒2589. DOI: https://doi.org/10.1101/gad.224089.113
- Hiromi, T.; Yokoyama, U.; Kurotaki, D.; et al. Excessive EP4 Signaling in Smooth Muscle Cells Induces Abdominal Aortic Aneurysm by Amplifying Inflammation. Arterioscler, Thromb, Vasc, Biol. 2020, 40, 1559‒1573. DOI: https://doi.org/10.1161/ATVBAHA.120.314297
- Nakao, T.; Horie, T.; Baba, O.; et al. Genetic Ablation of MicroRNA-33 Attenuates Inflammation and Abdominal Aortic Aneurysm Formation via Several Anti-Inflammatory Pathways. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2161‒2170. DOI: https://doi.org/10.1161/ATVBAHA.117.309768
- Guo, Z.Z.; Cao, Q.A.; Li, Z.Z.; et al. SP600125 Attenuates Nicotine-Related Aortic Aneurysm Formation by Inhibiting Matrix Metalloproteinase Production and CC Chemokine-Mediated Macrophage Migration. Mediators. Inflamm. 2016, 2016, 9142425. DOI: https://doi.org/10.1155/2016/9142425
- Granata, A.; Serrano, F.; Bernard, W.G.; et al. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat. Genet. 2017, 49, 97‒109. DOI: https://doi.org/10.1038/ng.3723
- Gao, P.; Gao, P.; Zhao, J.; et al. MKL1 cooperates with p38MAPK to promote vascular senescence, inflammation, and abdominal aortic aneurysm. Redox. Biol. 2021, 41, 101903. DOI: https://doi.org/10.1016/j.redox.2021.101903
- Ortega, R.; Collado, A.; Selles, F.; et al. SGLT-2 (Sodium-Glucose Cotransporter 2) Inhibition Reduces Ang II (Angiotensin II)-Induced Dissecting Abdominal Aortic Aneurysm in ApoE (Apolipoprotein E) Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1614‒1628. DOI: https://doi.org/10.1161/ATVBAHA.119.312659
- Davis, F.M.; Tsoi, L.C.; Melvin, W.J.; et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J. Exp. Med. 2021, 218, e20201839. DOI: https://doi.org/10.1084/jem.20201839
- Wang, Q.; Ding, Y.; Song, P.; et al. Tryptophan-Derived 3-Hydroxyanthranilic Acid Contributes to Angiotensin II-Induced Abdominal Aortic Aneurysm Formation in Mice In Vivo. Circulation 2017, 136, 2271‒2283. DOI: https://doi.org/10.1161/CIRCULATIONAHA.117.030972
- Wu, G.; Chen, T.; Shahsafaei, A.; et al. Complement regulator CD59 protects against angiotensin II-induced abdominal aortic aneurysms in mice. Circulation 2010, 121, 1338‒1346. DOI: https://doi.org/10.1161/CIRCULATIONAHA.108.844589
- Ijaz, T.; Sun, H.; Pinchuk, I.V.; et al. Deletion of NF-κB/RelA in Angiotensin II-Sensitive Mesenchymal Cells Blocks Aortic Vascular Inflammation and Abdominal Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1881‒1890. DOI: https://doi.org/10.1161/ATVBAHA.117.309863
- Liu, C.L.; Liu, X.; Zhang, Y.; et al. Eosinophils Protect Mice from Angiotensin-II Perfusion-Induced Abdominal Aortic Aneurysm. Circ. Res., 2021, 128, 188‒202. DOI: https://doi.org/10.1161/CIRCRESAHA.120.318182
- You, W.; Hong, Y.; He, H.; et al. TGF-β mediates aortic smooth muscle cell senescence in Marfan syndrome. Aging 2019, 11, 3574‒3584. DOI: https://doi.org/10.18632/aging.101998
- Chen, H.Z.; Wang, F.; Gao, P.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076‒1088. DOI: https://doi.org/10.1161/CIRCRESAHA.116.308895
- Zhong, L.; He, X.; Si, X.; et al. SM22α (Smooth Muscle 22α) Prevents Aortic Aneurysm Formation by Inhibiting Smooth Muscle Cell Phenotypic Switching Through Suppressing Reactive Oxygen Species/NF-κB (Nuclear Factor-κB). Arterioscler. Thromb. Vasc. Biol., 2019, 39, e10‒e25. DOI: https://doi.org/10.1161/ATVBAHA.118.311917
- Chen, J.; Peters, A.; Papke, C.L.; et al. Loss of Smooth Muscle α-Actin Leads to NF-κB-Dependent Increased Sensitivity to Angiotensin II in Smooth Muscle Cells and Aortic Enlargement. Circ. Res. 2017, 120, 1903‒1915. DOI: https://doi.org/10.1161/CIRCRESAHA.117.310563
- Liang, E.S.; Bai, W.W.; Wang, H.; et al. PARP-1 (Poly[ADP-Ribose] Polymerase 1) Inhibition Protects From Ang II (Angiotensin II)-Induced Abdominal Aortic Aneurysm in Mice. Hypertension 2018, 72, 1189‒1199. DOI: https://doi.org/10.1161/HYPERTENSIONAHA.118.11184
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516‒2528. DOI: https://doi.org/10.1161/CIRCULATIONAHA.116.009762
- Braverman, A.C. Heritable thoracic aortic aneurysm disease: recognizing phenotypes, exploring genotypes. J. Am. Coll. Cardiol. 2015, 65, 1337‒1339. DOI: https://doi.org/10.1016/j.jacc.2014.12.056
- Fletcher, A.J.; Syed, M.B.J.; Aitman, T.J.; et al. Inherited Thoracic Aortic Disease: New Insights and Translational Targets. Circulation 2020, 141, 1570‒1587. DOI: https://doi.org/10.1161/CIRCULATIONAHA.119.043756
- Carta, L.; Smaldone, S.; Zilberberg, L.; et al. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J. Biol. Chem. 2009, 284, 5630‒5636. DOI: https://doi.org/10.1074/jbc.M806962200
- Park, J.H.; Kim, M.S.; Ham, S.; et al. Transforming Growth Factor β Receptor Type I Inhibitor, Galunisertib, Has No Beneficial Effects on Aneurysmal Pathological Changes in Marfan Mice. Biomol. Ther. 2020, 28, 98‒103. DOI: https://doi.org/10.4062/biomolther.2019.042
- Da, X.; Li, Z.; Huang, X.; et al. AGGF1 therapy inhibits thoracic aortic aneurysms by enhancing integrin α7-mediated inhibition of TGF-β1 maturation and ERK1/2 signaling. Nat. Commun. 2023, 14, 2265. DOI: https://doi.org/10.1038/s41467-023-37809-x
- Arce, C.; Rodríguez-Rovira, I.; De Rycke, K.; et al. Anti-TGFβ (Transforming Growth Factor β) Therapy with Betaglycan-Derived P144 Peptide Gene Delivery Prevents the Formation of Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e440‒e452. DOI: https://doi.org/10.1161/ATVBAHA.121.316496
- Huang, T.H.; Chang, H.H.; Guo, Y.R.; et al. Vitamin B Mitigates Thoracic Aortic Dilation in Marfan Syndrome Mice by Restoring the Canonical TGF-β Pathway. Int. J. Mol. Sci. 2021, 22, 11737. DOI: https://doi.org/10.3390/ijms222111737
- Lim, W.W.; Dong, J.; Ng, B.; et al. Inhibition of IL11 Signaling Reduces Aortic Pathology in Murine Marfan Syndrome. Circ. Res. 2022, 130, 728‒740. DOI: https://doi.org/10.1161/CIRCRESAHA.121.320381
- Ng, B.; Dong, J.; D’Agostino, G.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. DOI: https://doi.org/10.1126/scitranslmed.aaw1237
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110‒115.
- MacCarrick, G.; Black, J.H.3rd.; Bowdin, S.; et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet. Med. 2014, 16, 576‒587. DOI: https://doi.org/10.1038/gim.2014.11
- Lindsay, M.E.; Schepers, D.; Bolar, N.A.; et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat. Genet. 2012, 44, 922‒927. DOI: https://doi.org/10.1038/ng.2349
- Bertoli-Avella, A.M.; Gillis, E.; Morisaki, H.; et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J. Am. Coll. Cardiol. 2015, 65, 1324‒1336.
- Loeys, B.L.; Chen, J.; Neptune, E.R.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275‒281. DOI: https://doi.org/10.1038/ng1511
- Loeys, B.L.; Schwarze, U.; Holm, T.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788‒798. DOI: https://doi.org/10.1056/NEJMoa055695
- van de Laar, I.M.; Oldenburg, R.A.; Pals, G.; et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat. Genet. 2011, 43, 121‒126. DOI: https://doi.org/10.1038/ng.744
- Cannaerts, E.; Kempers, M.; Maugeri, A.; et al. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: further delineation of the phenotype. J. Med. Genet. 2019, 56, 220‒227. DOI: https://doi.org/10.1136/jmedgenet-2018-105304
- Boileau, C.; Guo, D.C.; Hanna, N.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916‒921. DOI: https://doi.org/10.1038/ng.2348
- Akhurst, R.J. The paradoxical TGF-β vasculopathies. Nat. Genet. 2012, 44, 838‒839. DOI: https://doi.org/10.1038/ng.2366
- Schepers, D.; Tortora, G.; Morisaki, H.; et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum. Mutat. 2018, 39, 621‒634. DOI: https://doi.org/10.1002/humu.23407
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; et al. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. DOI: https://doi.org/10.3390/biom10020182
- Velchev, J.D.; Van Laer, L.; Luyckx, I.; et al. Loeys-Dietz Syndrome. Adv. Exp. Med. Biol. 2021, 1348, 251‒264. DOI: https://doi.org/10.1007/978-3-030-80614-9_11
- Liu, S.; Ren, J.; Ten Dijke, P. Targeting TGFβ signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 8. DOI: https://doi.org/10.1038/s41392-020-00436-9
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437‒446. DOI: https://doi.org/10.1007/s00262-015-1653-0


