
Introduction
Primary hyperparathyroidism (PHPT) manifests with symptoms from many organs and systems, mainly kidney stones or osteoporotic fractures. Therefore, patients with PHPT are usually first diagnosed by non-endocrinologists (including urologists, nephrologists, orthopedists, osteoporosis specialists and rheumatologists), who should know its symptoms, diagnostic criteria, and current evaluation.
Advances in knowledge about epidemiology, genetics, diagnosis, clinical presentitions and other aspects of PHPT have led to publishing in 2022 new international guidelines regarding its evaluation and treatment [1].
Primary hyperparathyroidism is one of the most frequent endocrine diseases and the most common cause of hypercalcemia in the general population. Age-adjusted prevalence in the United States is estimated to be 233 per 100,000 women and 85 per 100,000 men [2]. The reported incidence and prevalence of PHPT increased in well-developed countries, where automated chemistry panels that include serum calcium concentration measurements and osteoporosis screening are common. For these reasons, most PHPT patients in these countries are asymptomatic or oligosymptomatic. In countries where screening is not routine, PHPT remains a less frequent and more symptomatic disease [3].
It is well established that the classical, severe course of PHPT is associated with increased mortality, however, the impact of mild forms of the disease on survival is uncertain. Some data indicate increased mortality from cardiovascular disease or cancer even in PHPT patients with only mildly elevated serum calcium [4, 5]. Hyperparathyroidism is usually caused by a single benign parathyroid adenoma (85%), less often by multiple parathyroid gland disease (hyperplasia and/or multiple adenomas, 15%) and only in less than 1% of cases by parathyroid carcinoma.
More than 10% of PHPT cases are genetic. Mutations of 10 genes (MEN, RET, CDKN1B, CDKN1A, CDKN2B, CDKN2C, CDC73, CASR, GNA11, AP2S1) are now known to cause PHPT [6].
Hereditary PHPT may be an isolated endocrinopathy such as familial isolated hyperparathyroidism (FIHP), familial hypocalciuric hypercalcemia (FHH) or neonatal severe hyperparathyroidism (NSHPT) or a part of complex syndromes such as multiple endocrine neoplasia type 1, 2A, and 4 (MEN1, MEN2A, MEN4) or hyperparathyroidism-jaw tumor syndrome (HPT-JT). Patients with genetic forms of PHPT are generally younger than those with sporadic PHPT and have usually synchronic or metachronic multigland disease.
Parathormone (PTH) is a major hormone regulating calcium homeostasis. Even small increase in the serum ionized calcium concentration activates the calcium sensing receptor (CaSR) on the parathyroid cell surface, which inhibits PTH secretion, PTH gene expression and parathyroid cell proliferation. Clonal overgrowth of parathyroid tissue with reduced expression of CaSR leads to increased PTH gene expression and inadequately high secretion of PTH [6]. This hormone stimulates renal calcium reabsorption, suppress renal phosphate reabsorption, increases bone resorption and, indirectly, intestinal calcium absorption by stimulation of 25(OH)D hydroxylation to 1,25(OH)2D in the proximal renal tubule.
Hypersecretion of PTH in PHPT leads to hypercalcemia, which is usually mild, but occasionally may be even life-threatening condition. On the other hand, routine PTH measurements in patients with osteoporosis have shown that there is also a normocalcemic variant of PHPT. Serum phosphate is usually in low normal range, but in some patients hypophosphatemia also occurs. Primary hyperparathyroidism leads to an increased bone loss mainly at cortically enriched skeletal sites, but also to more subtle changes in trabecular bone. Renal overproduction of 1,25(OH)2D and increased intestinal calcium absorption accounts in part for hypercalciuria, increasing the risk of nephrolithiasis and nephrocalcinosis. Parathormone directly inhibits proximal tubular bicarbonate reabsorption, leading to mild metabolic acidosis (proximal tubular acidosis) [6].
What is crucial for this article is to note that PHPT often causes musculoskeletal symptoms, therefore knowledge about this disease is essential not only for endocrinologists, but also for rheumatologists.
Clinical manifestations
The clinical picture of PHPT depends on disease severity and chronicity. There are three clinical phenotypes of PHPT:
Symptomatic hypercalcemic – with overt skeletal and/or renal complications.
Asymptomatic hypercalcemic – with typical biochemical abnormalities but without overt symptoms or signs.
Normocalcemic (with or without skeletal or renal complications) [1].
The most common phenotype – asymptomatic PHPT is usually observed in patients with mild hypercalcemia (albumin-adjusted serum calcium < 1 mg/dl above upper limit of normal) [3]. In this group of PHPT patients asymptomatic nephrolithiasis, nephrocalcinosis, osteoporosis and silent vertebral fractures may be found during investigation.
The classical manifestations of PHPT (bone pain, proximal myopathy, osteoporotic fractures, bone cysts, brown tumors, nephrolithiasis and/or nephrocalcinosis) typically, but not always, occurs in those with higher calcium concentrations. It has been clearly established that PHPT causes skeletal and renal manifestations and that they are at least partially reversed with parathyroidectomy [1, 7, 8].
There are also many nonclassical symptoms described in association studies, which may occur in patients with mild, moderate and severe hypercalcemia [9, 10]. They include gastrointestinal, cardiovascular, neurobehavioral and neurocognitive features (Table I).
Table I
Classical and nonclassical symptoms of primary hyperparathyroidism
| Classical PHPT symptoms | Non-classical PHPT symptoms | ||||
|---|---|---|---|---|---|
| Renal | Skeletal | Gastrointestinal | Cardiovascular | Neuromuscular | Neurobehavioural |
However, randomized clinical trials have not shown a long-term benefit of parathyroidectomy versus observation on the vast majority of these manifestations [7].
Some of these symptoms are the consequence of hypercalcemia (e.g. polyuria, polydipsia, dehydration, nausea, vomiting, constipation, anorexia, headache, altered cognitive function and mental status, acute kidney injury or nephrocalcinosis), while others are exclusively caused directly by PTH (skeletal manifestations) [11, 12].
Rarely PHPT, may cause a life-threatening emergency – hypercalcemic crisis, which usually develops, when serum calcium is > 14 mg/dl, and manifests with oliguria or even anuria, somnolence and coma [13].
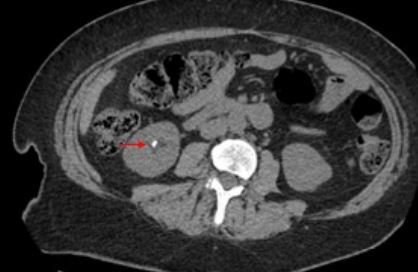
Nephrolithiasis (Fig. 1) is the most common clinical expression of PHPT with a prevalence ranging from 5 to 55% depending on PHPT severity and kidney imaging techniques [7, 8, 14–18]. Risk factors include hypercalciuria, hypomagnesuria, urinary calcium to magnesium ratio and genetic factors (CaSR polymorphism) [19, 20]. Chronic renal impairment with GFR reduction < 60 ml/min is also quite common in PHPT, affecting 12–20.6% of patients [21, 22]. When occurs, it alters calcium metabolism increasing serum phosphate and PTH, and decreasing calcium and 1,25(OH)2 vitamin D.
Fig. 1
Computed tomography (CT) of the abdomen (axial scans) from a 44-year-old woman with primary hyperparathyroidism and bilateral nephrolithiasis. Red arrows indicate kidney stones.
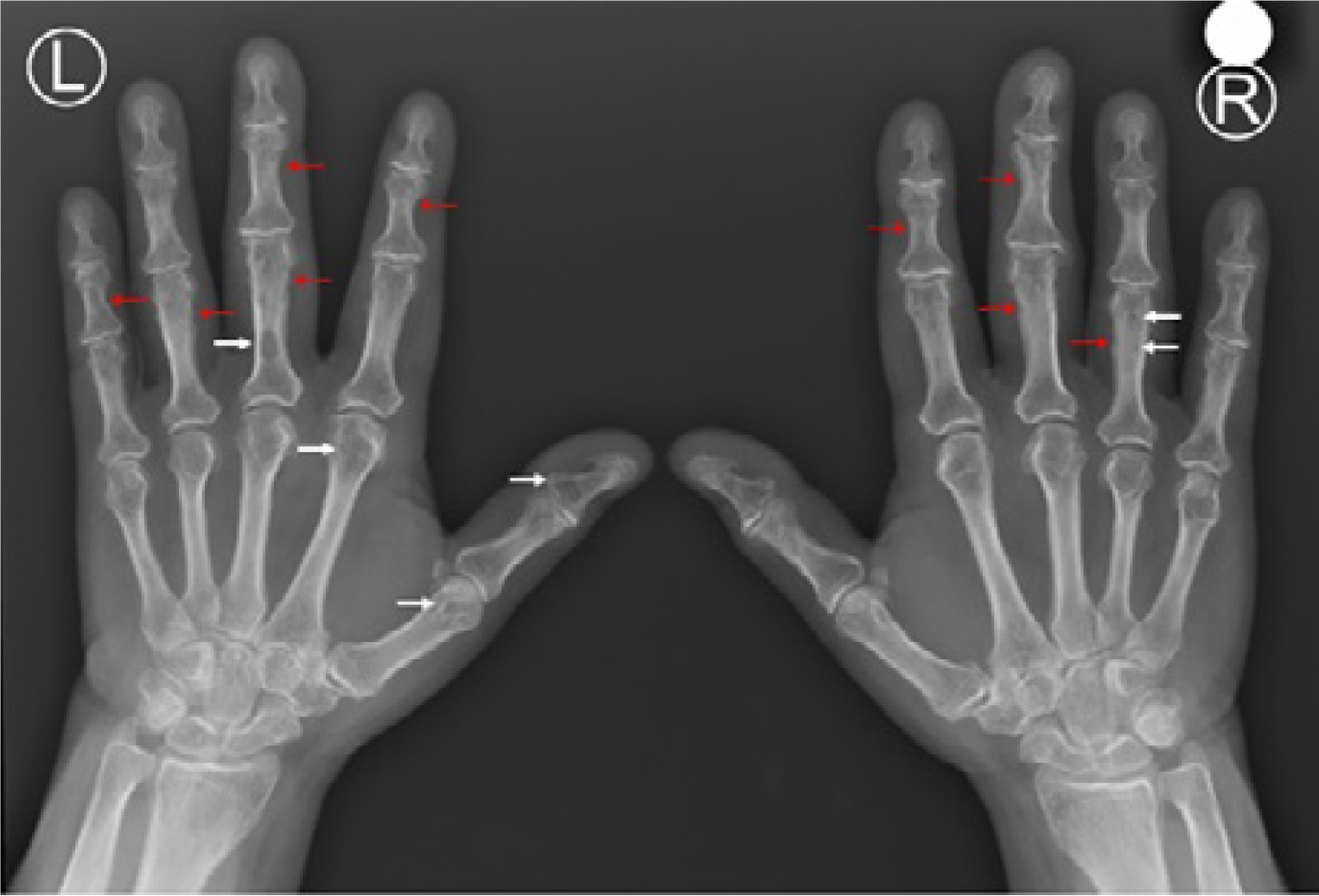
Osteoporosis is found in 50–65% PHPT patients [17, 18]. In PHPT bone remodeling increases and bone turnover markers are elevated [23]. Cortical bone becomes more porous and thinned due to “trabecularization” of endocortical surface. This is clearly visible on hand radiographs as thinned and feathery cortical bone, sometimes accompanied by subperiosteal resorption with acro-osteolysis, as well as bone cysts (Fig. 2).
Fig. 2
Plain X-ray of hands of 70-year old woman with primary hyperparathyroidism showing subperiostal bone resorption along the radial aspects of the proximal and middle phalanges (red arrows) and bone cysts (white arrows).
Bone mineral density (BMD) measured by dual-energy X-ray absorptiometry (DXA) is reduced, even in mild forms of PHPT, to the greatest extent at the cortical site (1/3 distal radius) and least at the trabecular site (lumbar spine) [9, 17, 18]. However, occasionally an opposite pattern may be observed, particularly in postmenopausal women [24]. Trabecular bone score (TBS) and high-resolution peripheral quantitative computed tomography (HRpQCT) analyses indicate, that PHPT leads to deterioration of cortical and also trabecular bone [25, 26]. Osteitis fibrosa cystica with brown tumors formation is a classic but rare manifestation of PHPT occurring in 2–5% patients.
Patients with PHPT have twice the risk of all fractures (odds ratio 2.01, 95% confidence interval, 1.61–2.50) with an increased fracture risk at the forearm and vertebrae [27].
Rarely, PHPT is associated with calcium pyrophosphate crystal deposition disease (CPPD), which causes calcification of the fibrocartilage and joint capsule (chondrocalcinosis), mainly of the knees. Pseudogout attacks are seen in these patients when calcium levels decrease after successful parathyroidectomy [28].
It is well known, that severe PHPT is associated with increased mortality [29]. Some studies also detected increased mortality in PHPT with even mild hypercalcemia with a relative risk (RR) of 1.2–1.6, when compared to a control population [4, 30, 31]. The main reported cause of deaths was cardiovascular disease and, in a lesser extent, cancer [4, 5, 30–33].
Diagnosis
Primary hyperparathyroidism diagnosis is based on identifying an elevated serum calcium concentration adjusted for albumin in the presence of an elevated or inappropriately high intact PTH level twice at least 2 weeks apart [1].
Normocalcemic PHPT is diagnosed when elevated intact PTH along with normal adjusted total calcium and normal ionized calcium concentration is present on at least two occasions over 3-6 months after ruling out all causes of secondary hyperparathyroidism (including stage ≥ 3 chronic kidney disease, vitamin D deficiency, calcium malabsorption, bisphosphonate or denosumab use). The serum calcium concentration in secondary hyperparathyroidism states is usually normal or low and when low, normocalcemic PHPT is ruled out [1].
Differential diagnosis of PHPT includes:
familial hypocalciuric hypercalcemia (FHH),
thiazide diuretics and/or lithium use,
ectopic secretion of PTH (very rare),
tertiary hyperparathyroidism.
The greatest diagnostic challenge in differential diagnosis of PHPT is caused by FHH, a genetically heterogenous disease due to inactivating mutations of calcium-sensing receptor (CaSR), which causes a shift of the Ca–PTH set point to the right. Familial hypocalciuric hypercalcemia manifests in early childhood as mild hypercalcemia with decreased renal calcium excretion. In FHH the calcium to creatinine clearance ratio is typically < 0.01, while in PHPT it is > 0.02. However about 40% of patients with either disease have values between 0.01 and 0.02. Moreover, up to 20% of PHPT patients have Ca/Cr Cl ratios < 0.01 and up to 10% of FHH patients have ratios > 0.02 [34–37]. In patients with FHH cases of kidney stones have also been rarely reported [34].
The differential diagnosis is important, as in FHH organ damage is not observed and parathyroidectomy does not cure hypercalcemia, so it is contraindicated [1].
Both thiazides and lithium increase renal calcium reabsorption and can be associated with hypercalcemia and increased serum PTH concentration, which may persist after medication withdrawal [1].
Tertiary hyperparathyroidism is characterized by excessive PTH secretion in patients with longstanding secondary hyperparathyroidism (caused for example by uncontrolled renal insufficiency or malabsorption syndromes such as active celiac disease, extensive bowel resection or gastric bypass surgery), which leads to hypercalcemia. It is usually identified by the clinical context.
Hypercalcemia of malignancy due to osteolytic metastases, tumors synthesizing parathyroid hormone-related protein (PTHrP) or 1,25(OH)2D as well as hypercalcemia observed in granulomatous diseases producing 1,25(OH)2D such as: sarcoidosis and tuberculosis are all characterized by suppression of PTH and, therefore, easily distinguishable from PHPT.
Evaluation of patients with primary hyperparathyroidism
According to current guidelines the following tests should be performed to diagnose PHPT and to assess its severity:
total serum calcium adjusted to albumin level, phosphorus, intact PTH, 25(OH)D, creatinine and ionized calcium if normocalcemic PHPT is considered,
dual X-ray absorptiometry (DXA) at lumbar spine, hip and distal 1/3 radius,
vertebral X-rays or vertebral fracture assessment (VFA) by lateral DXA scanning, TBS if available,
creatinine clearance (preferred) or estimated glomerular filtration (eGFR),
24-hour urinary calcium excretion, biochemical risk factors for stones,
imaging for nephrolithiasis/nephrocalcinosis (abdominal X-ray, ultrasonography or computed tomography which is a “gold standard”) [1].
There is a lack of evidence to justify routine evaluation of non-classical manifestations of PHPT [1, 38–42].
The key decision to be made is whether the patient should undergo parathyroidectomy, which is the only curative treatment of PHPT. It can be considered in every patient, provided there are no contraindications and the patient and physician agree on that choice. Surgery is recommended in all symptomatic PHPT patients and in those asymptomatic ones who meet one of the criteria listed in Table II [1].
Table II
Guidelines for surgery in asymptomatic patients with primary hyperparathyroidism according to the Fifth International Workshop (2022)
| Criteria for surgical treatment of patients with PHPT (one of following) | |
|---|---|
| Serum calcium | Total serum calcium adjusted to albumin > 1 mg/dl (0.25 mmol/l) above the upper limit of normal |
| Skeletal | Vertebral fracture by X-ray, CT, MRI or VFA |
| Bone mineral density (BMD): T-score ≤ –2.5 at L1–L4, total hip/neck or radius 33%* | |
| Renal | Creatinine clearance or eGFR < 60 ml/min** |
| Nephrocalcinosis/nephrolithiasis by X-ray, USG, CT, other method | |
| Hypercalciuria (in women > 250 mg/24 h, in men > 300 mg/24 h) | |
| Age | < 50 years |
There are no guidelines for surgery in normocalcemic PHPT due to limited data [1].
Impaired neurocognitive functions, poor quality of life or cardiovascular symptoms cannot be an indication for parathyroidectomy, as there is a lack of unequivocal data indicating improvement of these manifestations after surgery [38–42].
Patients with a mild subclinical PHPT without organ complications, in whom surgical treatment is not planned, may be further observed without pharmacological treatment [1].
Genetic testing should be considered for patients:
< 30 years old,
with multi-glandular disease,
with family history of hypercalcemia and/or syndromes associated with PHPT.
The results of genetic tests may have a significant impact on further clinical management. For example, in FHH patients surgical treatment is contraindicated, whereas in hyperparathyroidism-jaw tumor syndrome (HPT-JT), early parathyroidectomy with bilateral neck exploration is recommended due to the increased risk of parathyroid carcinoma. In MEN1 and MEN2 syndromes a bilateral exploration and total or subtotal parathyroidectomy is needed because of the presence of multiglandular disease in these patients, and, therefore, frequent recurrences after less radical management. Moreover, confirmation of the mutation in the proband allows early identification of his family members who may or may not be at risk [1].
Pre-operative imaging
Imaging of abnormal parathyroid tissue is recommended only for PHPT patients who are going to have parathyroid surgery. The diagnosis of PHPT and indications for surgical treatment are completely independent of the results of localization studies and those results should not influence the decision about parathyroidectomy [1].
Preoperative localization of enlarged parathyroid allows surgeon to perform minimally invasive selective parathyroidectomy. The potential benefits of a selective approach are shorter surgery time, lower tissue damage and risk of complications, and as a result shorter hospitalization time and reduction of costs. However, there is still a lack of publications directly comparing the effectiveness and safety of minimally invasive methods with the traditional bilateral neck exploration [1].
The main parathyroid tumor imaging modalities are neck ultrasound and 99mTc-sestamibi subtraction scintigraphy. Meta-analysis which included over 20,000 patients with PHPT reported sensitivity of neck ultrasound to be 79% in detecting single parathyroid adenoma, but it was much lower when two or all four parathyroid glands were enlarged (16% and 35%, respectively) [43].
The results of ultrasound imaging are frequently false negative in patients with small or ectopic adenomas, a coexisting large multinodular goiter or after neck surgery. Tc-MIBI SPECT/CT allows one to localize ectopic parathyroid nodules and its sensitivity in a single gland disease reaches 88%, but it is also significantly lower in multiple gland enlargement [44].
The third main method currently recommended is contrast enhanced 4D-computed tomography (4D-CT) however, it is a method with limited availability in many countries [1, 45]. Positron emission tomography (PET) with 18F-fluorocholine or 11C-methionine may be considered as second-choice method. Both of them have a proven very high sensitivity in the location of pathological parathyroid glands, but they are not widely recommended due to limited availability and high costs [45].
Non-surgical management of primary hyperparathyroidism
Monitoring includes at least annual measurements of calcium, 25(OH)D and creatinine with estimated eGFR (or preferably creatinine clearance measurement). PTH measurements are of lesser value in assessing the severity of the disease, but are reasonable if clinically indicated. Bone mineral density of the lumbar spine, hip and distal 1/3 radius should be measured every 1 or 2 years using the same DXA instrument. There is no need for routine periodic monitoring of 24-hour urinary calcium excretion, repeated abdominal ultrasound, CT or X-ray or for assessment of the spine X-ray/VFA or TBS. Nevertheless, it is recommended in the case of clinical indications [1]. The monitored patient may become a candidate for parathyroid surgery over time.
Parathyroidectomy should be reconsidered in the following cases:
serum calcium becomes consistently > 1 mg/dl (> 0.25 mmol/l) above the upper limit of normal,
a nontraumatic fracture or
a significant reduction in BMD (above the least significant change) to a T-score ≤ –2.5 at L1–L4, total hip/neck or radius 33% or
a kidney stone or
a significant reduction in creatinine clearance (> 3 ml/min on average over 1–2-year period) to < 60 ml/min if there are other changes indicating progression [1].
It is not recommended to restrict dietary calcium sources or to eliminate supplemental calcium in PHPT, as it may stimulate parathyroid glands to grow and secrete PTH without a measurable effect on serum calcium levels. Therefore, the calcium intake recommended for the general population (800 mg daily for women < 50 years and men < 70 years, and 1,000 mg daily for women > 50 years and men > 70 years) is considered to be appropriate also for subjects with PHPT. Current guidelines suggest vitamin D supplementation to keep the concentration of 25(OH)D > 30 ng/ml and below the laboratory upper limit of normal (usually < 50 ng/ml) [1].
In patients with hypercalcemia or low BMD who do not undergo parathyroidectomy, available pharmacological options may be offered.
The calcium sensing receptor agonist (calcimimetic) – cinacalcet is recommended to reduce serum calcium levels. However, it only modestly reduces PTH and does not influence bone mineral density.
The authors of the current guidelines suggest cinacalcet treatment in PHPT subjects with calcium levels > 1 mg/dl (> 0.25 mmol/l) above the upper limit of normal [1]. Unfortunately, the drug causes numerous side effects (e.g. anorexia, nausea, vomiting, diarrhea, dizziness) that sometimes lead to its discontinuation. Moreover, few patients can afford that long-term therapy as it is relatively expensive and not refunded in some countries, including Poland.
In the absence of contraindications, bisphosphonates (e.g. alendronate) or human monoclonal antibody that binds the RANKL (denosumab) can be used to improve BMD and to reduce bone turnover in PHPT.
If there is a need to reduce calcium levels and improve BMD, a combination of cinacalcet and denosumab/bisphosphonate may be considered [1].
Conclusions
Primary hyperparathyroidism is associated with multiple symptoms, but classical manifestations are from skeletal system and kidneys.
Significant hypercalcemia, young age, musculoskeletal and kidney symptoms are absolute indications to parathyroidectmy.
Mild subclinical PHPT without organ complications may be observed without pharmacological treatment.
The new international guidelines provide evidence-based recommendations for the evaluation and treatment of PHPT patients. The parathyroid surgery as the main and proper curative method of PHPT treatment was confirmed, however other medical treatment methods and proposed adequate management strategies also were discussed.