Operationally Simple Enantioselective Silane Reduction of Ketones by the [Ir(OMe)(cod)]2/Azolium Catalytic System ()
1. Introduction
Transition metal complexes of N-heterocyclic carbenes (NHCs) have been extensively researched in organometallic chemistry and homogeneous catalysis. Strong interactions between the metal and NHCs prevent the decomposition of the complex to free and inactive metal under catalytic conditions. Lin et al. achieved a decisive breakthrough in the synthesis of metal complexes bearing NHCs; they reported the preparation of silver NHCs by allowing azolium salts to react with Ag2O [1] . This strategy, known as the Ag2O method, offers a direct approach for synthesizing other metal/carbene systems via transmetalation. Thus far, a wide variety of transition metal complexes bearing NHCs have been developed [2] . Using the Ag2O method, Crabtree et al. prepared IrX(NHC)(cod)-type Ir complexes (cod = 1,5-cyclooctadiene, X = halogen) [3] . In 2004, Peris et al. reported the first catalytic application of metal complexes bearing NHCs; in this study, the hydrosilylation reaction of alkynes was catalyzed by bimetallic rhodium or iridium complexes bearing tripodal NHC ligands [4] . Liu et al. developed iridium pyridinyl NHC complexes for the catalytic transfer hydrogenation (TH) reaction [5] . Similarly, Herrmann and Kühn synthesized a series of IrX(NHC)(cod) complexes to study their catalytic activities in TH reactions. [6] . Currently, a large number of IrX(NHC)(cod)-type Ir complexes are continuously being investigated for homogeneous catalysis [7] [8] [9] . In 2006, Herrmann et al. developed well-defined chiral IrX(NHC)(cod) complexes using the Ag2O method [10] . The IrCl(NHC)(cod) complexes derived from chiral heterocyclic diamine 2,2’-bipiperidine were used to catalyze the asymmetric hydrosilylation and TH reactions of acetophenone. More recently, Yoshida and Yanagisawa demonstrated the highly enantioselective TH reaction of ketones, which was catalyzed by an Ir complex possessing a chiral bicyclic NHC ligand system [11] [12] .
We developed a series of chiral IrCl(NHC)(cod) complexes via the transmetalation reaction of a hydroxyamide-functionalized NHC/Ag complex with [IrCl(cod)]2 using the Ag2O method (Figure 1) [13] . Interestingly, the Ir-catalyzed enantioselective silane reduction (ESR) reaction of ketones was successfully carried out in the presence of a small amount of AgBF4 at room temperature without temperature control [14] . Subsequently, we investigated the ESR reaction using [Ir(cod)2]BF4 (1) as an Ir catalyst precursor without AgBF4 additive (Figure 1) [15] . Consequently, a simple combination of 1 and chiral hydroxyamide-functionalized benzimidazolium salt 2 promoted the catalytic ESR reaction of ketones. However, the complex 1 is unstable and sensitive toward air and moisture. These drawbacks limit the use of the 1/2 catalytic system for practical synthetic purposes. To overcome these limitations, we envisioned that [Ir(OMe)(cod)]2 (3) could be used as an Ir catalyst precursor (Figure 1). We assumed that the reaction of 2 with 3 would furnish the corresponding NHC/Ir complex, in which the methoxy group of 3 could serve as an internal base that deprotonates the C-H bond of 2 at the C2 position. Importantly, the commercially available Ir complex 3 is relatively inexpensive and easy to manipulate because of its benchtop stability [16] . The ESR reaction of ketone using the 2/3 catalytic system can be performed at room temperature under benchtop conditions. Hence, the present protocol is suitable for the practical synthesis of optically active alcohols.
![]()
Figure 1. Development of enantioselective Ir-catalyzed silane reductions.
To the best of our knowledge, only three previous studies have synthesized IrX(NHC)(cod) complexes by reacting azolium halides with the dinuclear alkoxy complex of Ir; these representative examples are shown in Figure 2. Jiménez et al. demonstrated that the reaction of 1-(2-methoxybenzyl)-3-methyl-1H-benzimidazol-3-ium bromide with 3 yields the corresponding monodentate NHC/Ir complex. In addition, the catalytic TH reaction of cyclohexanone was successfully performed in the presence of KOH in 2-propanol at 80˚C [17] . Voutchkova-Kostal et al. reported that the reaction of 1-methyl-3-(4-sulfophenyl)-1H-imidazolium
![]()
Figure 2. Previous studies that have reported the synthesis of IrX(NHC)(cod) complexes by reacting azolium salt with well-defined [Ir(OMe)(cod)]2 or in situ-generated [Ir(OEt)(cod)]2 from [IrCl(cod)]2 and NaH in EtOH.
with [Ir(OEt)(cod)]2, which was generated in situ from the reaction of [IrCl(cod)]2 with NaH in EtOH, produced the corresponding water-soluble NHC/Ir complex with sulfonate-functionalized wingtips [18] . These Ir complexes promoted the conversion of glycerol into lactic acid in water in the presence of KOH under microwave conditions at 150˚C. More recently, Pérez-Torrent et al. synthesized a series of Ir complexes bearing a hemilabile NHC ligand by treating coumarin-functionalized azolium chlorides with 3 [19] . The Ir complex bearing hemilabile coumarin-functionalized NHC catalyzed the hydrosilylation reaction of terminal alkynes with PhMe2SiH. However, to the best our knowledge, the synthesis of a chiral IrX(NHC)(cod) complex using 3 as an Ir precursor has not been reported so far.
Herein, we synthesized the IrCl(NHC)(cod) complex 4 by reacting 2 with 3. The well-defined NHC/Ir complex 4 exhibited good catalytic activity for the ESR reaction of ketones at room temperature. In this reaction, we employed our previously developed pre-mixing reaction procedure [20] . Additionally, we designed and implemented an operationally simple protocol for the ESR reaction, which was catalyzed by an in situ-generated Ir species derived from 2 and 3. This study reports an extremely useful method for synthesizing optically active alcohols, which can be performed on a benchtop under ambient conditions.
2. Results and Discussion
2.1. Preparation of 4 from the Reaction of 2 with 3 and Its Application in the ESR Reaction
Azolium salt 2 was reacted with 3 (0.5 equiv.) at room temperature for 16 h, to produce the corresponding IrCl(NHC)(cod) complex 4 as a yellow solid in 93% yield (Equation (1)). The formation of the carbene species strongly suggests that the OMe group of 3 acts as a base and deprotonates the C-H bond at azolium ring (Figure 1). Indeed, no reaction occurred when 2 was treated with [IrCl(cod)]2 (5) under these reaction conditions. The use of 5 instead of 3 as an Ir catalyst precursor will be explained in Section 2.3.
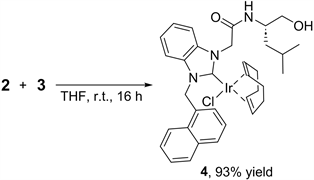 (1)
(1)
Complex 4 was characterized using NMR spectroscopy and elemental analysis. Unfortunately, complex 4 failed to yield satisfactory crystals for an X-ray crystal structure. Notably, the 13C-NMR spectrum of 4 indicated that it exists as a mixture of two NHC/Ir complexes. Enders et al. observed the hindered rotation of the carbene-metal bond in an NHC/Rh complex, which contained a bulky cyclooctadiene ligand [21] [22] . As 4 contains a chiral carbon center on the side arm of the NHC ligand, the hindered rotation of carbene-Ir bond results in the formation of a mixture of two diastereotopic rotamers. These two rotamers could not be separated by conventional chromatographic techniques. In the 1H-NMR spectrum of 4, the characteristic downfield signals for the NCHN+ protons of 2 disappeared. Complex 4 also exhibits 13C chemical shifts at both δ 192.5 and 192.3 ppm, which are comparable to those of other reported NHC/Ir(I) complexes [23] [24] . The 13C chemical shifts indicate that Ccarbene is substantially deshielded. In addition to 4, we synthesized two other IrCl(NHC)(cod) complexes, 4-Et and 4-Me, from benzimidazolium salts bearing both N-methyl and N-hydroxyamide wingtips derived from (S)-2-amino-1-butanol and (S)-alaninol, respectively. These complexes, 4-Et and 4-Me, also exist as a mixture of two NHC/Ir isomers, and characteristic carbene signals at approximately δ 191-192 ppm were observed in their 13C-NMR spectra. Notably, the obtained iridium complexes were completely stable in air and moisture environments.
The asymmetric catalytic property of 4 was examined by applying it in the reduction reaction of ketones with silanes at room temperature (Figure 3). The ESR reaction of propiophenone (6) with (EtO)2MeSiH was performed using our recently developed pre-mixing reaction procedure [20] . After stirring the mixture
![]()
Figure 3. ESR reaction of 6 with (EtO)2MeSiH catalyzed by 4 using the pre-mixing reaction procedure.
of 4 (8 mol% wrt 6) and (EtO)2MeSiH (4.5 equiv. wrt 6) in THF at room temperature for 20 h, 6 was reduced in MeOH in the presence of a small amount of K2CO3. Consequently, (S)-1-phenyl-1-propanol (6a) was obtained in 86% yield with 88% ee. This result indicates that the newly prepared Ir complex 4 is very stable and is a suitable catalyst for the ESR reaction under ambient conditions.
2.2. ESR Reaction Catalyzed by the in Situ-Generated Ir Species Derived from 2 and 3
Encouraged by the success of the well-defined IrX(NHC)(cod) complex 4, we continued our investigation on the ESR reaction of ketones. Next, we used the in situ-generated Ir species derived from 2 and 3 as a catalyst precursor. The in situ-generated metal complexes offer several distinct advantages over well-defined metal complexes [25] . In this approach, there was no need to prepare the NHC/metal catalyst in advance.
The catalytic ESR reaction was performed using the 2/3 combined catalytic system and the pre-mixing reaction procedure. The operational simplicity of this protocol enables the synthesis of various optically active alcohols (Table 1). All alcohol products listed in Table 1 have the (S)-configuration. First, 2, 3, and (EtO)2MeSiH were premixed, and 6 was reacted with the resulting mixture in MeOH in the presence of a small amount of K2CO3 at room temperature for 2 h. This reaction produced 6a in 86% yield with 92% ee (Entry 1). Similarly, butyrophenone (7), valerophenone (8), and benzyl phenyl ketone (9) were successfully reduced to the corresponding optically active alcohols 7a-9a with excellent enantioselectivities (Entries 2-4). Alkyl aryl ketones with branched alkyl groups can also be reduced using this reaction. The reduction of isobutyrophenone (11) and cyclohexyl phenyl ketone (12) yielded 11a and 12a, respectively, with 90% ee (Entries 6 and 7).
Although p-methoxypropiophenone (13), which contained an electron-donating substituent, was reduced to produce the alcohol 13a in 53% yield, higher product yield (85%) was obtained in the reduction of p-chloropropiophenone (14), which contained an electron-withdrawing substituent (Table 1, Entry 8 vs. Entry 9). These results suggest that the electrophilicity of the ketone affected the catalytic activity of the 2/3 catalytic system. Similar results were obtained in the
![]()
Table 1. Enantioselective reduction of various ketones with (EtO)2MeSiH catalyzed by an in situ-generated NHC/Ir speciesa.
a2 (0.03 mmol), 3 (0.015 mmol), and (EtO)2MeSiH (2.25 mmol) were added to THF (2 mL). After stirring the mixture at room temperature for 20 h, ketone (0.5 mmol), K2CO3 (5 mg) and MeOH (2 mL) were added. Then, the resulting mixture was reacted at room temperature for 2 h. bIsolated yield. cDetermined by GC or LC using a chiral stationary phase. dKetone was reacted with (EtO)2MeSiH for 6 h.
reduction reactions of p-methoxyacetophenone (16) and p-chloroacetophenone (18), which formed the corresponding alcohols 16a and 18a in 61% and 80% yields, respectively (Entry 11 vs. Entry 13). The product 16a was obtained in moderate yield (61%) when p-methoxyacetophenone (16) was reacted with (EtO)2MeSiH. However, m-methoxyacetophenone (17) was reduced smoothly to furnish 17a in good yield (90%) with 85% ee (Entry 11 vs. Entry 12). In general, the meta-OMe group acts as an electron-withdrawing group, whereas the para-OMe group acts as an electron-donating group. Therefore, the higher yield of 17a may have been caused by the higher electrophilicity of the carbonyl group in 17 (Entry 12). In contrast to the reduction of p-chloroacetophenone (18) to form 18a in 80% yield with 88% ee under the standard conditions, o-chloroacetophenone (19) was converted into 19a in moderate yield (61%) with low enantioselectivity (41% ee); this probably occurred because of the steric effect (Entry 13 vs. Entry 14). A poor product yield (24%) was observed in the reduction reaction of p-(dimethylamino)acetophenone (20) (Entry 15). This may have occurred because the dimethylamino group acts as a catalyst poison for the 2/3 catalytic system.
The in situ-generated 2/3 catalytic system was also suitable for the ESR reduction of 2-acetylnaphthalene (21), which afforded (S)-1-(2-naphthyl)ethanol (21a) in 96% yield with 83% ee (Table 1, Entry 16). However, the reduction of 1-acetylnaphthalene (22) furnished the alcohol 22a in a moderate yield and enantioselectivity (Entry 17). These results indicate that the reactivity of the NHC/Ir catalyst was sterically affected by the arene moiety on the substrate (Entry 16 vs. Entry 17). A satisfactory ee value (85%) was obtained in the reduction of 2-acetylthiophene (23); however, the product yield was slightly lower (Entry 18). Diaryl ketones such as 2,4’-dichlorobenzophenone (26) produced the corresponding diarylmethanol derivative 26a with moderate yield (47%) and enantioselectivity (60% ee) (Entry 21). However, 4-chromanone (27) was reduced to 4-chromanol (27a) with poor stereoselectivity (Entry 22).
2.3. ESR Reaction Catalyzed by in Situ-Generated Ir Species Derived from 2 and 26 or 5
The good performance of the 2/3 catalytic system prompted us to investigate the ability of other metal catalyst precursors for the ESR reaction of 6 using the pre-mixing reaction procedure (Table 2). To compare the relative abilities of the catalysts, the results of the ESR reaction catalyzed by the 2/3 catalytic system are also listed in Table 2 as Entries 1 and 2.
First, we selected the commercially available Ir(acac)(cod) (acac = acetylacetonate) (28) complex by assuming that the CH3C(O)CHC(O)CH3⁻ (acac) moiety of 28 may serve as an internal base to deprotonate the C-H bond of 2 [26] [27] [28] . Notably, complex 28 exhibited a high catalytic activity for the ESR reaction of 6. When 6 was treated with (EtO)2MeSiH in the presence of the 2/28 catalytic system in MeOH at room temperature, 6a was obtained in 90% yield with 85% ee (Table 2, Entry 4). To our surprise, the combination of 2 and [IrCl(cod)]2 (5) unexpectedly promoted the ESR reaction of 6 to produce 6a in 99% yield with 93% ee (Entry 6). The pKa values of the bases in DMSO are as follows: CH3OH 29.0; CH3C(O)CH2C(O)CH3 (acac-H) 13.3; HCl 1.8 [29] [30] [31] . These values
![]()
Table 2. Catalytic performances of various metal catalyst precursors for the enantioselective reduction of 6 with (EtO)2MeSiH under the influence of 2a.
a2 (0.025 - 0.03 mmol), metal catalyst precursor (0.0125 - 0.03 mmol), and (EtO)2MeSiH (2.25 mmol) were added to THF (2 mL). After stirring the resulting mixture at room temperature for 20 h, ketone (0.5 mmol), K2CO3 (5 mg), and MeOH (2 mL) were added to it. Subsequently, the resulting mixture was reacted at room temperature for 2 h. bGC yield using internal standard method. cDetermined by GC using a chiral stationary phase.
indicate that the chloride ion in 5 could not act as an internal base. Indeed, as described above, no reaction was observed between 2 and 5. The use of the Ir catalyst precursors 28 and 5 will be discussed later.
Although good product yield and stereoselectivity were observed in the ESR reaction of 6 using 5 (Table 2, Entry 6), a lower yield was obtained when [IrCl(cyclooctene)2]2 (29) was used instead of 5; this reaction afforded 6a in 20% yield (Entry 7). Furthermore, cationic Ir(I) complexes, such as [Ir(cod)2]+{B [3,5-(CF3)2C6H3]4}⁻ (30), and Ir(III) complexes, such as [IrCp*Cl2]2 (31), were found to be inert (Entries 8 and 9). In addition, the execution of the ESR reaction was difficult when Rh catalyst precursors, such as [RhCl(cod)]2 (32) and [Rh(cod)2]+BF4⁻ (33), were used under typical reaction conditions (Entries 10 and 11). However, several well-defined chiral NHC/Rh complexes have been developed to date for the catalytic ESR reaction of ketones with Ph2SiH2 [32] [33] [34] .
Encouraged by our success with 5 as an Ir catalyst precursor, we continued to explore the use of Pd, Pt, Ru and Cu complexes in the ESR reaction. In the literature, the ESR reaction of ketone has been achieved using the well-defined NHC/Cu complex catalyst or an in situ-generated NHC/Ru catalyst derived from RuCl2(PPh3)2 and a chiral azolium salt [35] [36] . However, as shown in Table 2 (Entries 12-15), the metal catalyst precursors 34-37, which possess both anionic chloride and neutral cod ligands, could not be applied in the ESR reaction. These results showed that only Ir complexes promoted the catalytic ESR reaction using the pre-mixing reaction procedure.
Table 3 summarizes the results of the ESR reactions of several ketones catalyzed by the 2/28 and 2/5 catalytic system. These data show that 28 and 5 could be successfully used as Ir catalyst precursors. The use of the pre-mixing
![]()
Table 3. Enantioselective reduction of several ketones with (EtO)2MeSiH using the 2/28 or 2/5 catalytic systema.
a2 (0.025 mmol), Ir catalyst precursor (0.0125 or 0.025 mmol), and (EtO)2MeSiH (2.25 mmol) were added to THF (2 mL). After stirring the resulting mixture at room temperature for 20 h, the ketone (0.5 mmol), K2CO3 (5 mg), and MeOH (2 mL) were added. Then, the mixture was reacted at room temperature for 2 h. bIsolated yield. cDetermined by GC or LC using a chiral stationary phase. dKetone was reacted with (EtO)2MeSiH for 6 h.
reaction procedure was essential. Indeed, almost no reaction was observed when ketone, (EtO)2MeSiH, K2CO3 and MeOH were added at once to the reaction vessel after stirring a mixture of 2 and 28 (or 5) in THF for 20 h. Notably, the experiments could be performed under benchtop conditions at ambient temperature.
Finally, we investigated the independent reactions of 2 with 28 and 5. We assumed that the anionic acac ligand of 28 would serve as an internal base to deprotonate the C-H bond of 2. As expected, the reaction of 2 with 28 in THF at room temperature afforded 4 in 85% yield (Equation (2)). This result strongly suggests that the formation of the NHC species from 2 is promoted by the acac ligand of 28 in a manner similar to the reaction between 2 and 3 (Figure 1).
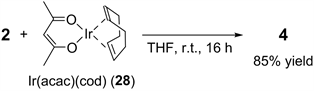 (2)
(2)
In the 2/5 catalytic system, we assumed that (EtO)2MeSiH would act as a base that deprotonates the C-H bond of 2 during the pre-mixing reaction process. Therefore, we next attempted to prepare an NHC/Ir complex by reacting 2 with 5 in the presence of (EtO)2MeSiH. However, we could not confirm the formation of the IrCl(NHC)(cod) complex in a pure form by analyzing the NMR spectra of the crude mixture, which was obtained after reacting 2, 5, and (EtO)2MeSiH. Meanwhile, we succeeded in synthesizing 4 by reacting 2 with 5 in the presence of tBuOK [37] . After stirring a mixture of 5 and tBuOK in THF for 10 min, 2 was added to the reaction vessel. The resulting mixture was then stirred at room temperature for 16 h, producing 4 in 89% yield (Equation (3)). Although the reaction mechanism of the 2/5 catalytic system is still unclear at this stage, we believe that 4 would be generated in situ during the catalytic cycle of the ESR reaction.
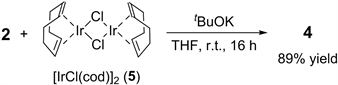 (3)
(3)
3. Conclusion
In summary, we investigated the use of a commercially available and air- and moisture stable Ir complex 3 in the ESR reaction of ketones. The complex 3 contains an OMe group, which acts as an internal base and deprotonates the C-H bond of the azolium ring of 2. The reaction of 2 with 3 furnished the well-defined IrCl(NHC)(cod) complex 4 in an almost quantitative yield. The ESR reaction of 6 with (EtO)2MeSiH was catalyzed by 4 and was performed using the pre-mixing reaction procedure. Additionally, we demonstrated that the in situ-generated NHC-Ir species derived from 2 and 3 promoted the catalytic ESR reaction of various ketones, which afforded the corresponding alcohols with moderate to excellent stereoselectivities. Throughout the course of this study, experiments were performed under benchtop conditions at room temperature. Hence, this study reports a useful and operationally simple method for the synthesis of optically active alcohols. Moreover, several transition metal complexes were evaluated as metal catalyst precursors; the evaluation revealed that 28 and 5 facilitated the catalytic ESR reaction using the pre-mixing reaction procedure. We believe that this protocol can be used for the practical and efficient synthesis of optically active alcohols from ketones.
4. Experimental
All other chemical reagents and solvents were obtained from commercial sources. Column chromatography was performed with silica gel 60 (63 - 210 μm) purchased from Kanto Chemical Co., Inc. 1H-NMR spectra were recorded on a JEOL ECA400 (400 MHz for 1H-NMR and 100 MHz for 13C-NMR) spectrometer. Chemical shifts were reported downfield from TMS (δ = 0 ppm) for 1H-NMR. For 13C-NMR, chemical shifts were reported on the scale relative to the solvent used as an internal reference. Elemental analyses were performed at Osaka University.
4.1. Procedure for Preparation of 4 from the Reaction of 2 with 3 or 28
2 (0.21 mmol, 95 mg) and 3 (0.1 mmol, 66 mg) were stirred in THF (2 mL) at room temperature for 16 h under Ar. After passing through a short silica gel column using THF as a solvent, the filtrate was dried in a rotary evaporator to afford 4 as a yellow solid (139 mg, 92% yield). 28 (0.2 mmol, 80 mg) could be used instead of 3 to obtain 4 (127 mg, 85% yield). 4 was very stable under air and could be stored as a solid for a minimum of one month at room temperature.
4: 1H-NMR (CDCl3, 400 MHz): Major isomer: δ 8.27 (d, J = 8.2 Hz, 1H, NH), 8.01 - 6.78 (m, 12H), 6.36 (d, J = 15.4 Hz, 1H, CH2CO), 6.29 (d, J = 15.4 Hz, 1H, NCH2Ar), 4.85 (d, J = 15.4 Hz, 1H, CH2CO), 4.76 (br, 1H, cod), 4.76 (br, 1H, NHCH), 4.10 (br, 1H, cod), 3.66 (br, 1H, cod), 3.53 - 3.48 (m, 1H, CH2OH), 3.08 - 3.01 (m, 1H, cod), 2.96 - 2.93 (m, 1H, CH2OH), 2.78 (t, J = 6.8 Hz, 1H, OH), 2.30 - 2.24 (m, 2H, cod), 1.99 - 1.97 (m, 1H, cod), 1.74 - 1.59 (m, 4H, cod), 1.41 (br, 1H, cod), 1.25-1.01 (m, 2H, CH2iBu), 0.93-0.86 (m, 1H, CHiBu), 0.61 (d, J = 6.8 Hz, 3H, CH3iBu), 0.56 (d, J = 6.8 Hz, 3H, CH3iBu). The following signals were attributed to minor isomer: δ 8.26 (d, J = 8.2 Hz, 1H, NH), 6.27 (d, J = 16.8 Hz, 1H, NCH2Ar), 6.21 (d, J = 16.8 Hz, 1H, NCH2Ar), 0.90 (d, J = 6.3 Hz, 3H, CH3iBu), 0.87 (d, J = 6.3 Hz, 3H, CH3iBu). 13C-NMR (CDCl3, 100 MHz): Major isomer: δ 192.4 (Ccarbene), 167.0 (C=O), 134.9, 134.3, 133.5, 130.8, 130.4, 129.0, 128.3, 126.8, 126.1, 125.2, 123.6, 123.5, 123.3, 122.3, 111.1, 110.4, 89.0 (CHcod), 88.6 (CHcod), 65.6 (CH2OH), 54.0 (CHcod), 53.6 (CHcod), 52.5 (NHCH), 50.4 (CH2CO), 49.6 (NCH2Ar), 39.2 (CH2iBu), 33.8 (CH2cod), 32.6 (CH2cod), 29.1 (CH2cod), 28.7 (CH2cod), 24.4 (CHiBu), 22.8 (CH3iBu), 21.4 (CH3iBu). Minor isomer: δ 192.3 (Ccarbene), 167.2 (C=O), 134.9, 134.4, 133.6, 130.9, 130.3, 128.9, 128.2, 126.7, 126.1, 125.2, 123.6, 123.4, 123.4, 122.4, 111.1, 110.6, 88.5 (CHcod), 85.0 (CHcod), 65.4 (CH2OH), 53.9 (CHcod), 53.7 (CHcod), 51.0 (NHCH), 50.5 (CH2CO), 49.5 (NCH2Ar), 39.2 (CH2iBu), 33.5 (CH2cod), 32.9 (CH2cod), 30.8 (CH2cod), 28.9 (CH2cod), 24.7 (CHiBu), 22.8 (CH3iBu), 22.1 (CH3iBu). Anal. Calc. for C34H41ClIrN3O2∙0.5H2O: C, 53.71; H, 5.57; N, 5.53. Found: C, 53.74; H, 5.60; N, 5.50%.
4-Et: 1H NMR (CDCl3, 400 MHz): Major isomer: δ 7.49 - 7.26 (m, 4H), 6.60 (d, J = 8.4 Hz, 1H, NH), 6.26 (d, J = 15.2 Hz, 1H, CH2CO), 4.76 (d, J = 15.2 Hz, 1H, CH2CO), 4.84 - 4.76 (br, 1H, cod), 4.84 - 4.76 (br, 1H, NHCH), 4.20 (s, 3H, NCH3), 3.88 - 3.83 (m, 1H, cod), 3.64 - 3.60 (m, 1H, cod), 3.49-3.46 (m, 1H, CH2OH) 3.15 - 3.10 (m, 1H, cod), 2.98 - 2.90 (m, 1H, CH2OH), 2.77 (t, J = 7.0 Hz, 1H, OH), 2.38 - 2.22 (m, 4H, cod), 1.94 - 1.83 (m, 2H, cod), 1.80 - 1.66 (m, 2H, cod), 1.38 - 1.25 (m, 1H, CH2CH3), 1.25 - 1.10 (m, 1H, CH2CH3), 0.53 (t, J = 7.2 Hz, 3H, CH2CH3). The following signals were attributed to minor isomer: δ 7.14 (d, J = 8.4 Hz, 1H, NH), 6.22 (d, J = 15.2 Hz, 1H, CH2CO), 4.80 (d, J = 15.2 Hz, 1H, CH2CO), 4.17 (s, 3H, NCH3), 1.54 - 1.47 (m, 2H, CH2CH3), 0.88 (t, J = 7.2 Hz, 3H, CH2CH3). 13C-NMR (CDCl3, 100 MHz): Major isomer: δ 191.5 (Ccarbene), 166.9 (C=O), 135.4, 134.0, 123.4, 123.3, 110.1, 109.8, 88.8 (CHcod), 88.2 (CHcod), 64.3 (CH2OH), 53.8 (CHcod), 53.4 (CHcod), 53.1 (CHNH), 52.3 (CH2CO), 34.3 (CH2cod), 33.4 (CH2cod), 33.2 (CH3N), 29.3 (CH2cod), 29.0 (CH2cod), 23.5 (CH2Et), 10.0 (CH3Et); Minor isomer: δ 167.4 (C=O), 135.3, 134.1, 123.4, 123.3, 110.9, 110.4, 88.6 (CHcod), 87.4 (CHcod), 64.7 (CH2OH), 54.5 (CHcod), 53.5 (CHcod), 53.0 (CHNH), 52.8 (CH2CO), 34.2 (CH2cod), 33.6 (CH2cod), 33.1 (CH3N), 29.6 (CH2cod), 28.9 (CH2cod), 23.4 (CH2Et), 10.5 (CH3Et); The carbene 13C-NMR resonance of the minor isomer was not observed. Anal. Calc. for C22H31ClIrN3O2: C, 44.25; H, 5.23; N, 7.04%. Found: C, 44.48; H, 5.56; N, 6.76%.
4-Me: 13C-NMR (CDCl3, 100 MHz): Major isomer: δ 191.5 (Ccarbene), 166.6 (C=O), 135.4, 134.0, 123.4, 123.4, 110.0, 109.8, 89.0 (CHcod), 88.2 (CHcod), 65.5 (CH2OH), 53.5 (CHcod), 53.1 (CHcod), 52.4 (CHNH), 48.4 (CH2CO), 34.3 (CH2cod), 33.4 (CH2cod), 33.2 (CH3N), 29.4 (CH2cod), 29.0 (CH2cod), 16.3 (CH3Me); Minor isomer: δ 191.3 (Ccarbene), 167.3 (C=O), 135.3, 134.0 123.4, 123.3, 110.4, 109.7, 88.5 (CHcod), 88.0 (CHcod), 66.7 (CH2OH), 53.3 (CHcod), 53.2 (CHcod), 52.7 (CHNH), 48.9 (CH2CO), 34.2 (CH2cod), 33.4 (CH2cod), 33.2 (CH3N), 29.4 (CH2cod), 29.2 (CH2cod), 16.2 (CH3Me). Anal. Calc. for C21H29ClIrN3O2: C, 43.25; H, 5.01; N, 7.21%. Found: C, 43.18; H, 5.31; N, 7.01%. Identity is known from 13C-NMR and elemental analysis, but our efforts to obtain the clean 1H-NMR spectra proved to be unsuccessful because of the broad signal.
4.2. Procedure for Preparation of 4 from the Reaction of 2 with 5 in the Presence of tBuOK
A mixture of 5 (0.07 mmol, 47 mg) and tBuOK (0.12 mmol, 14 mg) was stirred in THF (2 mL) at room temperature. After stirring for 10 min, 2 (0.12 mmol, 54 mg) was added to the reaction vessel. Then, the reaction mixture was stirred at room temperature for 16 h. After passing through a short silica gel using THF as a solvent, the filtrate was dried in a rotary evaporator to afford 4 as a yellow solid (80 mg, 89% yield).
4.3. General Procedure for ESR Reaction of Ketone with (EtO)2MeSiH
A mixture of 3 (0.015 mmol, 10 mg) and 2 (0.03 mmol, 14 mg) were stirred in THF (2 mL) at room temperature. After stirring for 3 h, (EtO)2MeSiH (2.25 mmol, 302 mg) was added to the reaction vessel. After further stirring at room temperature for 20 h, ketone (0.50 mmol), K2CO3 (5 mg) and MeOH (2 mL) were added. Then, the resulting mixture was stirred at room temperature for 2 h. After evaporation of the solvents, the corresponding alcohol product from the residue was purified by column chromatography on silica gel. The ee was measured by chiral GC or chiral LC according to our previously reported procedure.
1-Phenyl-1-propanol (6a): 1H-NMR (CDCl3, 400 MHz) δ 7.35 - 7.26 (m, 5H), 4.59 (t, J = 6.4 Hz, 1H), 3.53 (br, 1H), 1.84 - 1.71 (m, 2H), 0.92 (t, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 144.6, 128.4, 127.5, 125.9, 76.0, 31.9, 10.1.
1-Phenyl-1-butanol (7a): 1H-NMR (CDCl3, 400 MHz) δ 7.18 - 7.10 (m, 5H), 4.48 (t, J = 6.4 Hz, 1H), 1.64 - 1.58 (m, 1H), 1.55 - 1.46 (m, 1H), 1.32 - 1.21 (m, 1H), 1.19 - 1.08 (m, 2H), 0.77 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 144.9, 128.3, 127.3, 125.8, 74.3, 41.2, 18.9, 13.9.
1-Phenyl-1-pentanol (8a): 1H-NMR (CDCl3, 400 MHz) δ 7.33 - 7.24 (m, 5H), 4.64 - 4.60 (m, 1H), 1.83 - 1.64 (m, 3H), 1.43 - 1.34 (m, 2H), 1.28 - 1.19 (m, 2H), 0.88 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 144.9, 128.4, 127.4, 125.9, 74.6, 38.8, 27.9, 22.6, 14.0.
1,2-Diphenylethanol (9a): 1H-NMR (CDCl3, 400 MHz) δ 7.36 - 7.18 (m, 10H), 4.89 (dd, J = 8.2 and 5.2 Hz, 1H), 3.45 (br, 1H), 3.06 - 2.95 (m, 2H); 13C-NMR (CDCl3, 100 MHz) δ 143.8, 138.0, 129.5, 128.5, 128.4, 127.6, 126.6, 125.9, 75.3, 46.1.
1-Phenylethanol (10a): 1H-NMR (CDCl3, 400 MHz) δ 7.40 - 7.25 (m, 5H), 4.90 (q, J = 6.4 Hz, 1H), 3.53 (br, 1H), 1.50 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 145.8, 128.5, 127.4, 125.4, 70.4, 25.1.
1-Phenyl-2-methyl-1-propanol (11a): 1H-NMR (CDCl3, 400 MHz) δ 7.22 - 7.11 (m, 5H), 4.21 (d, J = 6.8 Hz, 1H), 3.67 (br, 1H), 1.86 - 1.78 (m, 1H), 0.86 (d, J = 6.8 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 143.6, 128.2, 127.4, 126.5, 80.0, 35.2, 19.0, 18.2.
Cyclohexyl(phenyl)methanol (12a): 1H-NMR (CDCl3, 400 MHz) δ 7.35 - 7.26 (m, 5H), 4.36 (dd, J = 7.2 and 3.2 Hz, 1H), 1.98 (d, J = 12.8 Hz, 1H), 1.83 - 1.75 (m, 2H), 1.65 - 1.57 (m, 4H), 1.40 - 1.35 (m, 1H), 1.24 - 0.91 (m, 4H); 13C-NMR (CDCl3, 100 MHz) δ 143.6, 128.2, 127.4, 126.6, 79.4, 44.9, 29.3, 28.8, 26.4, 26.1, 26.0.
1-(4-Methoxyphenyl)propanol (13a): 1H-NMR (CDCl3, 400 MHz) δ 7.28 - 7.24 (m, 2H), 6.90 - 6.86 (m, 2H), 4.53 (t, J = 6.4 Hz, 1H), 3.80 (br, 3H), 1.87 - 1.66 (m, 3H), 0.89 (t, J = 7.6 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 158.9, 136.7, 127.2, 113.8, 75.6, 55.2, 31.7, 10.2.
1-(4-Chlorophenyl)propanol (14a): 1H-NMR (CDCl3, 400 MHz) δ 7.32 - 7.25 (m, 4H), 4.57 (t, J = 6.4 Hz, 3H), 1.94 (br, 1H), 1.82 - 1.67 (m, 2H), 0.90 (t, J = 7.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 142.9, 133.0, 128.5, 127.3, 75.2, 31.9, 9.9.
1-(4-Butylphenyl)ethanol (15a): 1H-NMR (CDCl3, 400 MHz) δ 7.27 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 7.6 Hz, 2H), 4.86 (q, J = 6.4 Hz, 1H), 2.60 (t, J = 7.6 Hz, 2H), 1.91 (br, 1H), 1.63 - 1.55 (m, 2H), 1.48 (d, J = 6.4 Hz, 3H), 1.40 - 1.31 (m, 2H), 0.92 (t, J = 7.6 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 143.0, 142.2, 128.5, 125.3, 70.2, 35.2, 33.6, 25.0, 22.3, 13.9.
1-(4-Methoxyphenyl)ethanol (16a): 1H-NMR (CDCl3, 400 MHz) δ 7.31 - 7.26 (m, 2H), 6.89 - 6.86 (m, 2H), 4.84 (q, J = 6.4 Hz, 1H), 3.80 (s, 3H), 1.94 (br, 1H), 1.47 (d, J = 6.8 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 158.9, 138.0, 126.6, 113.8, 69.9, 55.2, 25.0.
1-(3-Methoxyphenyl)ethanol (17a): 1H-NMR (CDCl3, 400 MHz) δ 7.28 - 7.24 (m, 1H), 6.95 - 6.93 (m, 2H), 6.82 - 6.79 (m, 1H), 4.86 (q, J = 6.4 Hz, 1H), 3.81 (s, 3H), 2.00 (br, 1H), 1.48 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 159.7, 147.6, 129.5, 117.6, 112.8, 110.8, 70.3, 55.2, 25.1.
1-(4-Chlorophenyl)ethanol (18a): 1H-NMR (CDCl3, 400 MHz) δ 7.32 - 7.26 (m, 4H), 4.86 (q, J = 6.4 Hz, 1H), 2.14 (br, 1H), 1.46 (dd, J = 6.4 and 1.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 144.2, 133.0, 128.5, 126.8, 69.7, 25.2.
1-(2-Chlorophenyl)ethanol (19a): 1H-NMR (CDCl3, 400 MHz) δ 7.60 - 7.58 (m, 1H), 7.33 - 7.17 (m, 3H), 5.29 (q, J = 6.4 Hz, 1H), 2.15 (br, 1H), 1.48 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 143.0, 131.6, 129.4, 128.4, 127.2, 126.4, 66.9, 23.5.
1-(4-Dimethylaminophenyl)ethanol (20a): 1H-NMR (CDCl3, 400 MHz) δ 7.26 - 7.24 (m, 2H), 6.74 - 6.71 (m, 2H), 4.81 (q, J = 6.4 Hz, 1H), 2.94 (s, 6H), 1.74 (br, 1H), 1.48 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 150.2, 133.7, 126.4, 112.6, 70.1, 40.7, 24.6.
1-(2-Naphthyl)ethanol (21a): 1H-NMR (CDCl3, 400 MHz) δ 7.80 - 7.74 (m, 4H), 7.46 - 7.41 (m, 3H), 4.98 (q, J = 6.4 Hz, 1H), 1.87 (br, 1H), 1.53 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 143.2, 133.3, 132.8, 128.2, 127.9, 127.6, 126.1, 125.7, 123.8, 123.7, 70.4, 25.0.
1-(1-Naphthyl)ethanol (22a): 1H-NMR (CDCl3, 400 MHz) δ 7.32 - 7.26 (m, 4H), 4.86 (q, J = 6.4 Hz, 1H), 2.14 (br, 1H), 1.46 (dd, J = 6.4 and 1.2 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 141.3, 133.8, 130.2, 128.8, 127.9, 126.0, 125.5, 123.1, 122.0, 67.1, 24.3.
1-(2-Thiophenyl)ethanol (23a): 1H-NMR (CDCl3, 400 MHz) δ 7.26 - 7.23 (m, 1H), 6.98 - 6.95 (m, 2H), 5.12 (q, J = 6.4 Hz, 1H), 2.06 (br, 1H), 1.60 (d, J = 6.4 Hz, 3H); 13C-NMR (CDCl3, 100 MHz) δ 149.8, 126.6, 124.4, 123.2, 66.2, 25.2.
2-Chloro-1-phenylethanol (24a): 1H-NMR (CDCl3, 400 MHz) δ 7.38 - 7.25 (m, 4H), 4.89 (dd, J = 8.8 and 3.2 Hz, 1H), 3.73 (dd, J = 11.0 and 3.2 Hz, 1H), 3.64 (dd, J = 11.0 and 8.8 Hz, 1H), 2.77 (br, 1H); 13C-NMR (CDCl3, 100 MHz) δ 139.9, 128.6, 128.4, 126.0, 74.0, 50.8.
3-Chloro-1-phenylpropanol (25a): 1H-NMR (CDCl3, 400 MHz) δ 7.38 - 7.26 (m, 5H), 4.97 - 4.94 (m, 1H), 3.77 - 3.71 (m, 1H), 3.61 - 3.53 (m, 1H), 2.29 - 2.20 (m, 1H), 2.14 - 2.05 (m, 1H), 2.01 (br, 1H); 13C-NMR (CDCl3, 100 MHz) δ 143.7, 128.6, 127.9, 125.8, 71.3, 41.7, 41.4.
(2-Chlorophenyl)(4-chlorophenyl)methanol (26a): 1H-NMR (CDCl3, 400 MHz) δ 7.56 - 7.54 (m, 1H), 7.35 - 7.21 (m, 7H), 6.20 (d, J = 3.2 Hz, 1H), 2.42 (d, J = 3.6 Hz, 1H); 13C-NMR (CDCl3, 100 MHz) δ 140.7, 140.6, 133.5, 132.4, 129.6, 129.0, 128.6, 128.3, 127.9, 127.2, 72.0.
3,4-Dihydro-2H-chromen-4-ol (27a): 1H-NMR (CDCl3, 400 MHz) δ 7.30 - 7.26 (m, 1H), 7.22 - 7.17 (m, 1H), 6.93 - 6.82 (m, 2H), 4.75 (t, J = 4.0 Hz, 1H), 4.26 - 4.23 (m, 1H), 2.15 - 2.06 (m, 2H), 2.04 - 1.97 (m, 2H); 13C-NMR (CDCl3, 100 MHz) δ 154.6, 129.7, 129.6, 124.3, 120.6, 117.1, 63.2, 61.9, 30.8.