E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 58(3); 2024 > Article
-
Case Study
Primary epithelioid inflammatory myofibroblastic sarcoma of the brain with EML4::ALK fusion mimicking intra-axial glioma: a case report and brief literature review -
Eric Eunshik Kim1
 , Chul-Kee Park2, Koung Mi Kang3, Yoonjin Kwak1, Sung-Hye Park1, Jae-Kyung Won,1
, Chul-Kee Park2, Koung Mi Kang3, Yoonjin Kwak1, Sung-Hye Park1, Jae-Kyung Won,1 -
Journal of Pathology and Translational Medicine 2024;58(3):141-145.
DOI: https://doi.org/10.4132/jptm.2024.04.12
Published online: May 14, 2024
1Departments of Pathology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
2Departments of Neurosurgery, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
3Departments of Radiology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
- Corresponding Author: Jae-Kyung Won, MD, PhD, Department of Pathology, Seoul National University Hospital, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea Tel: +82-2-2072-4895, Fax: +82-2-743-5530, E-mail: 'jkwon@snuh.org'
© 2024 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 536 Views
- 49 Download
Abstract
- An aggressive subtype of inflammatory myofibroblastic tumor, epithelioid inflammatory myofibroblastic sarcoma occurs primarily inside the abdominal cavity, followed by a pulmonary localization. Most harbor anaplastic lymphoma kinase (ALK) gene rearrangements, with RANBP2 and RRBP1 among the well-documented fusion partners. We report the second case of primary epithelioid inflammatory myofibroblastic sarcoma of the brain, with a well-known EML4::ALK fusion. The case is notable for its intra-axial presentation that clinico-radiologically mimicked glioma.
- A 22-year-old male soldier was found lethargic, drooling, and breathing heavily in the middle of the night. His eyes showed upward deviation, and after a brief seizure episode, he regained consciousness in the ambulance. Emergency electroencephalogram was normal, but the emergency computed tomography revealed a mass in the right frontal lobe. A subsequent preoperative contrast-enhanced magnetic resonance imaging (MRI) glioma study (3.0T) revealed a 1.5× 1.4× 0.9 cm vividly enhancing mass with focal diffusion restriction and no intralesional hemorrhage or calcification (Fig. 1A, B). A craniotomy and tumor removal was done by the neurosurgical team at Seoul National University Hospital (SNUH), who noted intraoperatively that the tumor appeared grossly more yellow-orange than the surrounding parenchyma with no attachment to dura, and showed a 5-aminolevulinic acid (5-ALA) uptake (Fig. 1C, D).
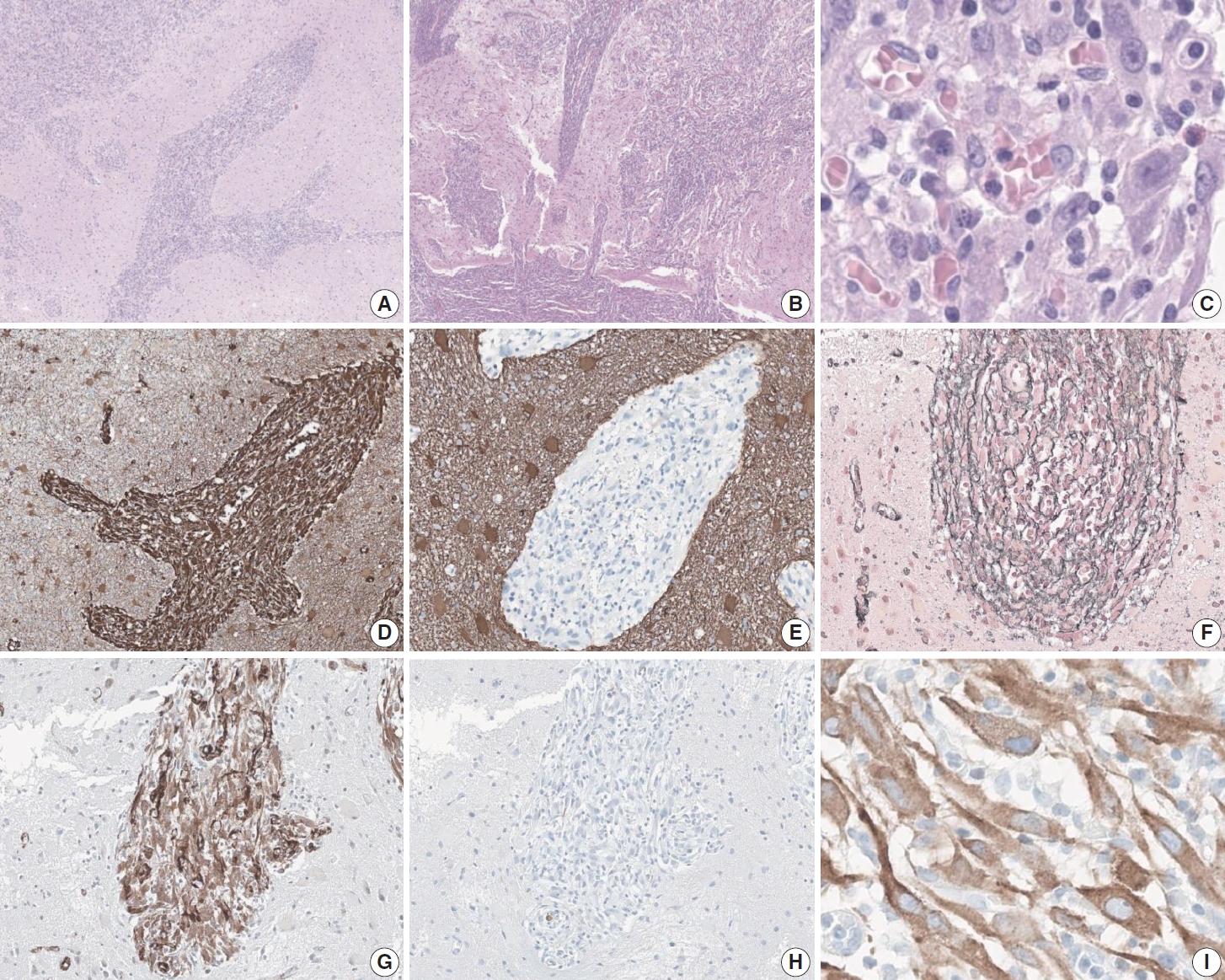
- Microscopic examination demonstrated an infiltrative epithelioid spindle neoplasm that seemed to spread in a perivascular pattern at low power (Fig. 2A). The low-power view also illustrated the appearance of the tumor to extend from the pia mater into the brain parenchyma (Fig. 2B). Even at high-power view, perivascular growth of the tumor can be appreciated, along with a lymphoplasmacytic infiltrate in the stroma and large nuclei with apparent nucleoli (Fig. 2C). The immunophenotype of the tumor is positive for vimentin, reticulin, and smooth muscle actin, while negative for glial fibrillary acidic protein and desmin (Fig. 2D–H). Anaplastic lymphoma kinase (ALK) showed a characteristic perinuclear staining for both the 5A4 clone (Fig. 2I) and the D5F3 clone (data not shown). In addition, the tumor cells were positive for alpha-thalassemia/intellectual disability syndrome X-linked, H3K27 trimethylation (H3K27me3), c-Myb, methylthioadenosine phosphorylase, while negative for isocitrate dehydrogenase 1, CD34, neuronal nuclear antigen, synaptophysin, microtubule associated protein-2, and Olig2. The tumor was negative for BRAF staining and also showed focal positivity to epithelial membrane antigen. The FiRST Brain Tumor Panel v3.3, which is a customized next-generation sequencing (NGS) panel developed at SNUH, was performed on the patient’s tumor to reveal an EML4::ALK fusion. Mitotic activity was 3 per 2 mm 2 as measured with phospho-histone H3 immunohistochemistry, and the Ki-67 proliferative index was 6.6%.
- The diagnosis of EIMS was rendered. An F-18 fluorodeoxyglucose positron emission tomography revealed no abnormal hypermetabolic lesions in the brain or the scanned body, helping to rule out the possibility of brain metastasis and confirming that the EIMS was indeed primary to the brain. Following the radiologic confirmation of the gross total resection of the tumor, the patient received adjuvant intensity-modulated radiation therapy at 60 Gy and 30 fractions for 2 months after the surgery. The patient is being followed up by the oncology team every 6 months, and by the neurology team for his anti-epileptic drug titration.
CASE REPORT
- EIMS is an aggressive subtype of IMT that has been recognized by the World Health Organization (WHO) Classification of Tumors since its fifth edition publication of various organ systems, with the notable exception of WHO Classification of Central Nervous System Tumors that does not feature either EIMS or IMT as a primary entity for their extreme rarity [5]. As such, whenever IMT is considered as a diagnosis, regardless of the primary site, EIMS is an important differential diagnosis. The clinicoradiological presentation of the primary EIMS of the CNS is distinct from the primary IMT of the CNS, as most of the intracranial IMT reported were extra-axial, requiring a differential diagnosis of meningioma, while the intracranial EIMS in our patient appeared intra-axial, mimicking a glioma [2,3]. EIMS also has a distinct histopathology in the CNS, with a perivascular growth pattern that seems to extend from the pia mater, which was not noted in the case report on the first primary EIMS of the CNS [4]. In our case, the region with less apparent perivascular growth pattern features the more epithelioid morphology with a heavier inflammatory infiltrate, while the perivascular region shows the more spindle and less inflamed features. While the first primary EIMS of the CNS was localized in the temporal horn of the left lateral ventricle of a 72-year-old patient [4], the EIMS of our 22-year-old patient was in the right frontal lobe. Notably, both primary EIMS of the CNS were supratentorial. The age of EIMS patients in any anatomic localization varies widely, from infants as young as 7 months [6] to a 72-year-old adult [4], with a median age of 39 years [1].
- The immunohistochemistry for ALK showed a characteristic perinuclear staining that was noted back when EIMS was first introduced as an aggressive subtype of IMT in 2011, as was the immunophenotype with the first primary EIMS of the CNS [1,4]. For IMT, it is known that the ALK staining pattern can vary from nuclear to perinuclear, depending on the driving fusion identified [1,7]. Of the EIMS that show ALK positivity on immunohistochemistry, the patients can benefit from ALK inhibitor therapy [8], although our patient opted for an adjuvant radiation therapy. Among the ALK-negative EIMS, ROS1 or even NTRK3 alterations can be expected [9], with TRK fusion-positive cases especially of note, since, while rare, their identification can give even pediatric patients the option for TRK inhibitor therapy that is tissue agnostic, crosses the blood-brain barrier, and shows mild adverse effects [10].
- In our patient’s EIMS, the NGS study revealed an EML4::ALK fusion from exon 2 and exon 20 of chromosome 2, respectively (Transcript ID–NM_019063/NM_004304). This fusion is well known in various malignancies, most famously in the non-small cell carcinomas of the lung, but also in intra-thoracic IMTs [11,12], and intra-abdominal EIMS [13].
- In the fourth edition of the WHO Classification of Tumors of Soft Tissue and Bone, published in 2013, the authors of the IMT chapter suggested that the IMT behaving aggressively could be labeled “inflammatory fibrosarcoma” instead of “atypical IMT” [14], but there are no known instances of primary inflammatory fibrosarcoma of the CNS in the literature. Therefore, not only is EIMS a relatively new entity with less than a decade of being diagnosed, but also this has led to some discrepancies in its descriptions by the WHO Classifications of Tumors, as summarized in Table 1.
- We need to include a unified sentence to the localization section for each of the WHO Classification of Tumors that lists IMT and EIMS as a chapter and try to recognize the clinical behavior of EIMS with the accumulation of more data. RANBP2::ALK and RRBP1::ALK fusions are noted specifically to EIMS in the abdominal cavity in the WHO Classification of Female Genital Tumors [1,7]. As fusions can be specific to the site of origin, this mention of reported fusions should be applied separately to other organ system publications. In the case of WHO Classification of Central Nervous System Tumors, not only should IMT/EIMS chapter be added in the next edition, it should also feature the VCL::ALK fusion [4] and EML4::ALK fusion, uniquely identified in the primary EIMS of the CNS. The WHO Classification of Breast Tumors lists IMT as a separate chapter, while noting that less than 25 cases have been reported [15]. In contrast, 55 cases of primary IMT in the intracranium has been reported [16]. The number of reported cases alone should therefore justify a separate chapter in the WHO Classification of Central Nervous System Tumors, while noting that, though rare, EIMS has been reported twice to arise from the CNS.
- In summary, we present the second case of EIMS primary to the CNS with EML4::ALK fusion to further elucidate the histomorphological characteristics and the immunophenotype of this rare entity. Given the atypical localization, the diagnosis of this tumor can be challenging to both neuropathologists and general pathologists alike. Recognizing the utility and limitations of ALK immunohistochemistry studies on a suspected EIMS lesion will guide pathologists to rely on NGS studies to identify fusions that will support their histomorphological diagnosis of EIMS.
DISCUSSION
-
Ethics Statement
The Institutional Review Board at Seoul National University Hospital approved the publication of this article under the number 2402-135-1514. Informed consent from the participant has been waived by Institutional Review Board, but a written consent form does exist.
-
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
-
Code Availability
Not applicable.
-
Author Contributions
Conceptualization: JKW. Data curation: EEK. Formal analysis: EEK, JKW. Investigation: EEK. Methodology: YK, JKW. Supervision: JKW. Validation: YK, CKP, KMK, SHP. Writing—original draft: EEK. Writing—review & editing: EEK, JKW. Approval of final manuscript: all authors.
-
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
-
Funding Statement
No funding to declare.
Notes


| WHO Classification of Tumors | 5th edition | IMT in 5th | IMT ICD-O | EIMS in 5th | EIMS ICD-O |
|---|---|---|---|---|---|
| Central nervous system | 2021 | – | – | – | – |
| Soft tissue and bone | 2020 | + | /1 | + | – |
| Pediatric | 2023 | + | /1 | + | /1a |
| Thoracic | 2021 | + | /1 | +b | – |
| Digestive system | 2019 | + | /1 | + | /3 |
| Breast | 2019 | + | /1 | + | – |
| Eye and orbit | 2022 | + | /1 | – | – |
| Urinary and male genital | 2022 | + | /1 | – | – |
| Female genital | 2020 | + | /1 | + | – |
| Head and neck | Online (beta) | + | /1 | – | – |
IMT, inflammatory myofibroblastic tumor; EIMS, epithelioid inflammatory myofibroblastic sarcoma; WHO, World Health Organization; ICD-O, International Classification of Diseases for Oncology; +, a dedicated chapter on IMT in the given organ system publication and the mention of EIMS as a subtype; –, no separate chapter for IMT or no mention of EIMS.
a It is uncertain whether the aggressive behavior of EIMS reported in adults is also seen in children;
b Recognized as epithelioid inflammatory myofibroblastic tumor.
- 1. Marino-Enriquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol 2011; 35: 135–44. PubMed
- 2. Coffin CM, Watterson J, Priest JR, Dehner LP. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor): a clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995; 19: 859–72. ArticlePubMed
- 3. Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol 2008; 61: 428–37. ArticlePubMed
- 4. Chopra S, Maloney N, Wang WL. Epithelioid inflammatory myofibroblastic sarcoma with VCL-ALK fusion of central nervous system: case report and brief review of the literature. Brain Tumor Pathol 2022; 39: 35–42. ArticlePubMedPDF
- 5. WHO Classification of Tumors Editorial Board. WHO classification of tumours: central nervous system tumours. 5th ed. Lyon: International Agency for Research on Cancer, 2021.
- 6. Yu L, Liu J, Lao IW, Luo Z, Wang J. Epithelioid inflammatory myofibroblastic sarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic analysis of five additional cases and review of the literature. Diagn Pathol 2016; 11: 67.ArticlePubMedPMC
- 7. Lee JC, Li CF, Huang HY, et al. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol 2017; 241: 316–23. ArticlePubMedPDF
- 8. Singh P, Nambirajan A, Gaur MK, et al. Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: a rare entity and a literature review. J Pathol Transl Med 2022; 56: 231–7. ArticlePubMedPMCPDF
- 9. Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 2016; 69: 72–83. ArticlePubMed
- 10. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018; 378: 731–9. PubMedPMC
- 11. Lovly CM, Gupta A, Lipson D, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 2014; 4: 889–95. ArticlePubMedPMCPDF
- 12. Sokai A, Enaka M, Sokai R, et al. Pulmonary inflammatory myofibroblastic tumor harboring EML4-ALK fusion gene. Jpn J Clin Oncol 2014; 44: 93–6. ArticlePubMed
- 13. Jiang Q, Tong HX, Hou YY, et al. Identification of EML4-ALK as an alternative fusion gene in epithelioid inflammatory myofibroblastic sarcoma. Orphanet J Rare Dis 2017; 12: 97.ArticlePubMedPMCPDF
- 14. Fletcher CD, Bridge JA, Hogendoorn PC, Mertens F. WHO classification of tumours of soft tissue and bone. 4th ed. Lyon: IARC Press, 2013.
- 15. WHO Classification of Tumors Editorial Board. WHO classification of tumours: breast tumours. 5th ed. Lyon: International Agency for Research on Cancer, 2019.
- 16. Zhou L, Pan W, Huang R, Lu Z, You Z, Li Y. Intracranial inflammatory myofibroblastic tumor: a literature review and a rare case misdiagnosed as acoustic neuroma. Diagnostics (Basel) 2023; 13: 2725.ArticlePubMedPMC
References
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-