
Abstract
Transmethylation in the context of psychiatry has historically referred to the enzymatic transfer of a methyl group from one biochemical to another, whose resulting function can change so dramatically that a biochemical like tryptamine, for example, is converted into the hallucinogen dimethyltryptamine. Central to endogenous methylation activity is the folate cycle, which generates the primary transferable methyl groups in mammalian biochemistry. The relevance of this cycle to mental health becomes clear when the cycle is dysregulated, often leading to a buildup of both homocysteine and S-adenosylhomocysteine (SAH), while accompanied by a transient reduction in the intended physiologic target, S-adenosylmethionine (SAM). This paper includes an in-depth review of the causes of folate cycle perturbations associated with psychotic symptoms, expounding on alternative downstream pathways which are activated and pointing toward potential etiologic agents of the associated psychosis, the methylated tertiary amines N-methyl-salsolinol, N-methyl-norsalsolinol, and adrenochrome, which appear in scientific reports concerning their association with hallucinogenic and/or neurotoxic outcomes. Electrotopological state (E-state) data has been generated for these compounds, illustrating a strong similarity with hallucinogens, particularly in terms of the E-state of the nitrogen in their tertiary amine moieties. In light of the role the folate cycle plays in transmethylation, neuroprotective strategies to prevent the transition to psychosis are suggested, including the advisory that folate supplementation can be harmful depending on the status of other relevant biochemicals.
Keywords
hallucinogens, psychotic disorders, folate cycle, homocysteine, vitamin B12, Folfirinox, MTHFR, electrotopologyIntroduction
Within the numerous research paths in psychosis research, important themes have emerged which deserve continued exploration but are often abandoned when novel discoveries result in a major shift in focus for the field as a whole. This can occur even in the absence of any refutation of the original research results, but is more likely when a clear path forward is missing. The biochemical cycling of methyl groups thought to relate to psychosis was one such area of endeavor. Termed the transmethylation hypothesis, the origins of this theory can be traced to the historical observation that many drugs capable of causing psychosis contained methylated amino groups, as reviewed by Grayson et al. [1]. The formation of tertiary amines is of particular interest, as will be made clear in a subsequent section. Alterations in pathways pertaining to DNA methylation in psychotic disorders [2] is also an important topic, but it is tangential to the work presented here. Much of the early emphasis on the transmethylation theory of psychosis sought validation through the tertiary amine dimethyltryptamine, which had been identified as a known hallucinogenic compound from its presence in certain hallucinogenic plants and has on occasion, been identified in human blood or urine samples. It can be measured easily after ayahuasca consumption [3]. However, positive findings for elevated DMT or its analogue 5-OH-DMT in individuals with schizophrenia have not been replicated well (reviewed by Dean [4]). Although the evidence for the presence of known endogenous methylated psychotogen may have been unconvincing in early work [5], the intention of this review is to revive interest in the transmethylation hypothesis of psychosis and to offer a potential path forward. The error in historical emphasis may have been the primary compound selected for research focus, dimethyltryptamine, diverting attention from the search for other relevant endogenous tertiary amines.
Central to the creation of methyl donating biochemicals is the folate (vitamin B9) cycle (Figure 1). How perturbations in this cycle may act to generate psychotic symptoms and what neuroprotective strategies might be adopted to overcome such dysregulation, will be covered in the pages that follow. The primary evidence for the relevance of methylation will be presented in detail. Perturbations of the folate cycle which increase homocysteine and its downstream metabolite, S-adenosylhomocysteine (SAH), are associated with psychotic symptoms and psychotic disorders and include the following: lower activity of the enzyme which degrades homocysteine, cystathionine-beta-synthase (CBS); deficiencies of vitamin B12, a vitamin crucial for folate cycle generation of the major methyl carrier compound, S-adenosylmethionine (SAM) which participates in many important methylation reactions (a deficiency in SAM is associated with an elevation in homocysteine and may be a marker for the diversion of methyl groups to undesirable alternative recipients); methionine, a methylated product of the folate cycle, when co-administered with a monoamine-oxidase inhibitor; administration of the drug Folfironox, which contains folinic acid, a methyl donor precursor, plus 5-fluorouracil (5-FU), an inhibitor of one destination for methyl groups, DNA synthesis; and some mutations in methylenetetrahydrofolate reductase (MTHFR), a key enzyme in the folic-acid cycle.
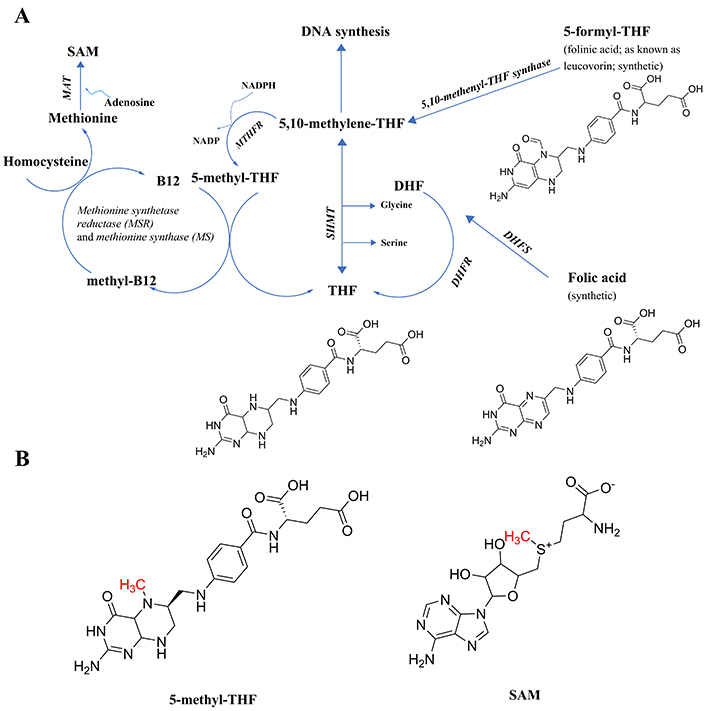
Components of the folate cycle. (A) Shown are the 2 points of entry to the cycle from synthetic B9 vitamins, and the single point of entry from the common form of B9 found in foods (THF, generally attached to polyglutamate residues, not shown), including their chemical structures to make clear that none have an available methyl group to transfer; thus, the reactions in the pathway are necessary to generate that methyl group. All enzymes are in italic font, and acronyms for enzymes are presented where available. References for the enzymatic steps are provided in the text. (B) The structure of 5-methyl-THF, the product of the enzyme MTHFR, with the methyl group available for transfer denoted in red font, and the structure of SAM, with the methyl group received from 5-methyl-THF via methylcobalamin and available for subsequent transfer, denoted in red font. Note that 5-methyl-THF is a tertiary amine, whereas SAM is not. The potential implications of the tertiary amine structure in 5-methyl-THF will be discussed in a later section on the E-state of tertiary amines. 5-methyl-THF: 5-methyltetrahydrofolate; DHF: dihydrofolate; DHFR: dihydrofolate reductase; DHFS: dihydrofolate synthase; E-state: electrotopological state; MAT: methionine adenosyltransferase; MTHFR: methylene tetrahydrofolate reductase; SAM: S-adenosylmethionine; SHMT: serine hydroxymethyl transferase; THF: tetrahydrofolate
The effect of these perturbations on either the concentration or flux of harmful tertiary amine metabolites of catecholamines will be explored, including N-methyl-salsolinol, N-methyl-norsalsolinol, and adrenochrome. The conclusion of this review will address how these endogenous tertiary amines compare to amine-containing drugs which possess opposing structure activity relationships dependent on substituent moieties, some with hallucinogenic and psychotomimetic effects. A potential basis for the difference in impact will be evaluated using data generated for atomic electrotopological state (E-state) predictors. Of relevance to the diversion of dopamine flux to undesirable products, convincing evidence for increased dopaminergic flux being involved in psychotic symptoms will be presented in the context of the literature on an effective inhibitor of dopamine synthesis, metyrosine. Finally, medical interventions intended to mitigate folate cycle dysregulation will be presented, including those that might be counterproductive as well as alternatives more likely to lead to neuroprotection.
Basic components of the folate cycle
The naturally occurring folate vitamin B9 present in foods, tetrahydrofolate (THF) polyglutamate, is not necessarily stable during storage [6] and requires hydrolysis of the polyglutamate residues to release THF [7] which led to the search for a more stable and rapid-acting form which turned out to be the chemically synthesized folic acid [6]. As can be seen in Figure 1A, folic acid differs in composition from THF (https://pubchem.ncbi.nlm.nih.gov/) and enters the cycle through the action of either the enzyme dihydrofolate synthase (DHFS; https://www.ncbi.nlm.nih.gov/gene/?term=DHFS; and [8]) and/or dihydrofolate reductase (DHFR [9]) to form dihydrofolate (DHF), followed by further reduction by DHFR [9] to form tetrahydrofolate (THF). Next, THF is acted upon by the enzyme serine hydroxymethyl transferase (SHMT; [10]), forming 5,10-methylene-THF. This is the intermediate in the pathway also generated by one enzymatic step from another synthetic form of folate, folinic acid (also known as leucovorin), in a reaction catalyzed by the enzyme 5,10-methenyl-THF synthetase [11].
At this point in the cycle, the methyl group to be generated can either be directed toward DNA synthesis or toward the generation of SAM, the most prevalent carrier for the methyl groups transferred in biochemical reactions. To get there [9], 5,10-methenyl-THF is a substrate for the enzyme methylene tetrahydrofolate reductase (MTHFR), creating the product 5-methyl-THF (Figure 1B), the first metabolite in the folate pathway to actually possess a readily transferable methyl group. Note that all of the enzymatic steps up until this point can be performed by intestinal enterocytes, which produce 5-methyl-THF for distribution in the blood [7].
The next step requires vitamin B12 (cobalamin) being methylated to form methylcobalamin, which in turn methylates homocysteine to form methionine in a two-step, two enzyme process [12], methionine synthase reductase (MSR), followed by the action of reduced methionine synthase (MS), to complete the methyl group transfer from methylcobalamin to homocysteine, forming methionine. Methionine is then acted upon by the enzyme methionine adenosyltransferase (MAT; [9]) to create SAM, utilizing adenosine as the second substrate and ATP as a cofactor.
Dysregulation of the folate cycle with psychosis: increases in SAH
One of the well-recognized indicators of perturbations in the folate cycle is an elevation in the concentration of homocysteine in the blood or urine because its methylation is somehow blocked. Any elevation in homocysteine would be expected to be associated with an increase in SAH, one of the products generated when methyl transfer occurs from SAM. With this on mind, Bao Ting Zhu authored a publication entitled “On the mechanism of homocysteine pathophysiology and pathogenesis: a unifying hypothesis” [13] which pointed towards SAH-mediated inhibition of catecholamine degradation as the mechanism underlying the association between high homocysteine and cardiovascular disorders, as well as Alzheimer’s disease and Parkinson’s. This review supports that premise and will expand upon it to explore possible downstream effectors of high SAH that could be relevant to psychosis.
As covered in ensuing sections, the most common causes of increases in homocysteine and SAH include: 1) a deficiency in the enzyme MTHFR, leading to a deficiency in the necessary methyl-group donor 5-methyl-THF; 2) a vitamin B12 deficiency such that the key methyl-transfer step to homocysteine is blocked; or 3) because the degradation of homocysteine is diminished by a deficiency in the enzyme CBS as reviewed by Bracken and Coll [14]. Another path to elevating homocysteine is less direct, i.e., the increase which occurs with L-DOPA treatment in Parkinson’s patients [15, 16] because increased methylation demands are placed on the system from the methylation that needs to occur while degrading the resulting excess of dopamine. The results of these perturbations can be seen in Figure 2.
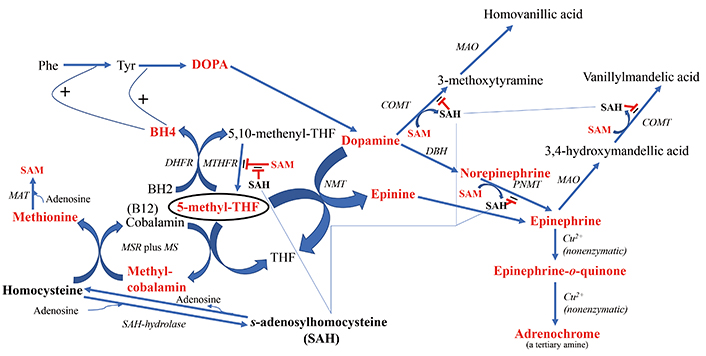
The downstream products of folate cycle activity. Shown in bold red font are the products expected to show higher flux with increased cycle activity. 5-methyl-THF is encircled to highlight its central role in the direction of downstream branch points. Shown in bold black font are biochemicals expected to potentially exert deleterious regulatory effects if in high concentration, i.e., homocysteine and SAH. The fine blue connectors illustrate the multitude of enzymes regulated by SAH. All enzymes are in italic font, and acronyms for enzymes are presented where available. References for the enzymatic steps can be found in the text. Note: the reaction between an NMT and 5-methyl-THF to form N-methyl-dopamine was reported by Laduron et al. in 1974 [297], but challenged by Meller et al. in 1975 [298] based on the poor chromatographic separation of N-methyl-dopamine from another product, tetrahydroisoquinoline. However, Meller et al. [298] did find that although the majority of product formed in their system was tetrahyudroisoquinoline, 12% of the product formed in their incubation was N-methyl-dopamine, thereby providing an alternative route to epinephrine when SAH inhibits the more commonly route via PNMT. BH4: tetrahydrobiopterin; BH2: dihydrobiopterin; COMT: catechol-O-methyltransferase; DBH: dopamine-beta-hydroxylase; DHFR: dihydrofolate reductase; DOPA: L-3,4-dihydroxyphenylalanine; MAO: monoamine oxidase; MAT: methionine adenosyltransferase; MS: methionine synthase; MSR: methionine synthase reductase; MTHFR: methylene tetrahydrofolate reductase; NMT: N-methyltransferase (nonspecific); Phe: phenylalanine; PNMT: phenylethanolamine NMT; SAH: S-adenosylhomocysteine; SAM: S-adenosylmethionine; THF: tetrahydrofolate; Tyr: tyrosine
There are numerous case reports of either homocysteinemia (high blood concentrations) or homocysteinuria (high urine concentrations) in psychosis. Kaeser et al. [17] admitted a patient with intermittent psychosis who suffered from homocystinuria as a result of a deficient activity of the CBS enzyme. After reviewing the literature on case reports at the time, and because pyridoxine is a cofactor for this enzyme, pyridoxine administration was initiated and resulted in a remarkable reduction in homocysteine concentration. Unfortunately, no follow-up was reported for the psychiatric symptoms. It has subsequently been observed that during remission from psychosis, homocysteine levels drop [18]. Golimbet et al. [19] identified a trend for a genetic association between the gene for CBS and schizophrenia in a study of 1,135 patients and 626 controls and Colafrancesco et al. [20] published a case report on a teen with sudden onset psychosis and bilateral subluxation of the lens, diagnostic of CBS deficiency, later confirmed by molecular testing.
The 22q deletion syndrome, which dramatically increases the risk of psychosis, can involve high levels of homocysteine [21]. Cannabis use is a well-established risk factor for psychosis [22], and in that regard, one study of first episode psychosis (FEP) patients found a significantly higher homocysteine concentration in the patients versus controls, and it was the subjects with a history of cannabis use who exhibited the highest homocysteine values [23]. This was a lingering effect of cannabis use, not an acute effect because the subjects had been screened to exclude those who had used cannabis in the past year. Patients taking vitamin B supplements were also excluded. Vitamin B12 was significantly lower in the patients with schizophrenia, the significance of which will be discussed in another section.
Onozato et al. [24] reported that both homocysteine and methionine were increased in FEP patients. Based on a series of 7 similar case reports, Chang et al. [25] recommended testing adolescents for homocysteine levels if they exhibit any gait disturbance and/or psychosis symptoms. Zhong et al. [26] compared Parkinson’s patients with hallucinations to those without and determined that homocysteine was significantly higher in those with hallucinations. In a case-control study of FEP patients versus healthy controls, Fan et al. [27] determined that FEP patients had significantly higher homocysteine. Neuroleptic treatment can reportedly diminish the levels of homocysteine [28], thus the homocysteine levels at first episode are most revealing.
Figure 2 also illustrates the downstream effects of upregulating homocysteine and consequently, SAH, when methyl transfer to homocysteine is impaired. The correlation between homocysteine and SAH levels was reported to be r = 0.413 [29]. SAH exerts feedback inhibition to decrease the activity of N-methyltransferases (NMTs) that utilize SAM, i.e., catechol-O-methyltransferase (COMT) [30, 31] and phenylethanolamine NMT (PNMT) [32], potentially leading to a buildup of dopamine, norepinephrine and epinephrine. In this regard, Silva et al. [33] found that a polymorphism in the gene SAH hydrolase (AHCY), which breaks down SAH, was significantly associated with, panic disorder, an endophenotype of psychosis, and Bönig et al. [34] presented data showing that elevation of SAH is one of the important factors in the psychosis associated with SAM depletion. Under the scenario of elevated SAH and depleted SAM, there are two neurotoxic types of downstream tertiary amine products that may either increase in concentration or experience an increase in flux: 1) adrenochrome from degradation of epinephrine and 2) increased production of the methyl-salsolinol (Figure 3). Another outcome associated with hyperhomocysteinemia is disruption of the integrity of the blood brain barrier (BBB) [35–37]. Yet, although evidence is emerging that BBB disruption may be a consequence of processes ongoing in psychotic disorders, there is as yet no consistent data showing how such disruption might be etiologic [38–40].
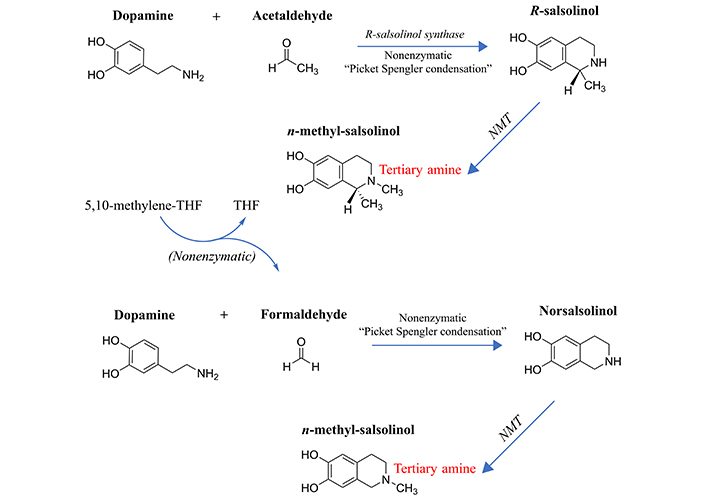
Formation of the methylated salsolinols. An enzyme has been characterized for the formation of salsolinol, R-salsolinol synthase, but the NMT catalyzing the methylation of salsolinol and norsalsolinol have not yet been well characterized. One endogenous source for the formaldehyde involved in forming norsalsolinol is 5,10-methenyl-THF, which can decompose nonenzymatically to form THF plus formaldehyde [78]. 1) The formation of salsolinol, norsalsolinol, N-methyl-salsolinol and N-methy-norsalsolinol from dopamine and the aldehydes, acetaldehyde and formaldehyde. 2) The formation of N-methyl-salsolinol and N-methyl-norsalsolinol from N-methyldopamine and acetaldehyde or formaldehyde, respectively. Note that and endogenous source of formaldehyde may occur in MTHFR enzyme deficiency when its substrate, 5,10-methenyl-THF builds up (Figure 2), leading to its spontaneous breakdown to THF and formaldehyde [78]. The numerous sources of acetaldehyde are discussed in the text, most prominently via metabolism of ethanol. All enzymes are in italic font, and acronyms for enzymes are presented where available. MTHFR: methylene tetrahydrofolate reductase; NMT: N-methyltransferase (nonspecific); THF: tetrahydrofolate
It is important to make clear at this juncture that much of the literature on homocysteine has focused on the association between homocysteine and oxidative stress, often with the implication that homocysteine itself is an agent of oxidative stress [24, 41–43]. However, the researchers failed to elucidate whether this association is an intrinsic characteristic of the compound, a result of its downstream its effects or whether it is merely a biomarker for oxidative stress, e.g., perhaps a compensatory mechanism when oxidative stress occurs. Indeed, the redox potential of homocysteine, which contains a sulfhydryl group, is far from being oxidizing, but rather has a reducing redox potential, according to the EPI-Suite algorithms of the website ChemSpider (https://www.chemspider.com/) which places the reactive hydroxyl-radical species reaction rate of homocysteine, 81.0376 E-12 cm3/molecule-second, above the reducing activity of the liver-sparing [44] antioxidant N-acetylcysteine, 54.9204 E-12cm3/molecule-second and only somewhat less than that of glutathione, 126.1477 E-12 cm3/molecule-second. This is consistent with the results of Ye et al. [45] who found homocysteine to have antioxidant properties in a study of reactive oxygen species in erythrocytes and leukocytes. In cardiovascular disease, rather than homocysteine mediating oxidative stress associated with atherosclerotic cardiovascular disease, Antoniades et al. [46] pointed towards 5-methyl-THF as the etiologic agent instead. Therefore, the effect of homocysteine to be associated with oxidative stress is more likely indirect via the corresponding increase in SAH, which inhibits COMT and PNMT methyltransferases, forcing the re-routing of catecholamine metabolism to the products reviewed in the ensuing two subsections.
It must be emphasized here that as with all known environmental and genetic perturbations that increase risk for psychosis, the effect of high homocysteine on a psychotic outcome is not absolute but rather depends on a host of other gene-environment interactions that are individual-specific. Many individuals with high homocysteine do not develop psychosis. In addition, the predilection of patients with psychosis to make poor dietary choices could theoretically result in high homocysteine levels unrelated to causation [47]. However, the cases presented in this section, and in subsequent sections, where normalization of high homocysteine results in normalization of psychosis, suggests a causal basis for those individuals.
Expected increase in the tertiary amine adrenochrome
Although SAH inhibits PNMT, the enzyme catalyzing the conversion of dopamine-derived norepinephrine to epinephrine, there is an alternative route to epinephrine from dopamine (Figure 2), involving the formation of N-methyldopamine (also known as epinine and deoxyepinephrine) via poorly characterized N-methyltransferase activity that does not involve PNMT [48]. Reports in the literature provide evidence that N-methyldopamine is then converted to epinephrine by dopamine-beta-hydroxylase [49, 50]. In point of fact, following the administration of ibopamine, the prodrug for N-methyldopamine, it is reported that copious amounts of the degradation product of epinephrine, adrenochrome (Figure 2), are subsequently formed in animal models [51]. The oxidation of epinephrine to form adrenochrome is nonenzymatic [52–55]. Evidence for the existence of endogenous N-methyldopamine was found in rat urine by Ziegler et al. [56] using the methodology available at the time, thin-layer chromatography. Elayan et al. [57] identified enzymatic production of N-methyldopamine from dopamine in the skin, and Kennedy et al. [58] detected N-methyldopamine formation in muscle tissue, although their analytical methodology did not involve direct ascertainment of the compound via chromatography, but more indirectly by measuring scintillation peaks from an elution method designed to capture N-methyldopamine. Of most relevance, Kajita et al. [59] identified N-methyldopamine in both normal and Parkinsonian postmortem human brains using liquid chromatography methods.
Nappi and Vass [60] pointed to the greater oxidizing free radical generation from Fenton reactions in the presence of N-methylated catecholamines such as N-methyldopamine versus the typical O-methylation route of degradation. This would include adrenochrome production from epinephrine. Because of its chemical reactivity profile, adrenochrome is regarded as a toxin for the cardiovascular system, where its effects have been somewhat more well-researched. As reviewed by Dhalla [61], the adverse impacts of stimulation of the sympathetic nervous system are not attributable to high concentrations of catecholamines per se but are exerted by the oxidation products of catecholamines, including adrenochrome.
Furthermore, adrenochrome has been reported to exert psychotomimetic effects [62–64], and a possible intersection for adrenochrome with upregulation of the kynurenine pathway has been identified [65] that would be expected to prolong the effects of adrenochrome. The kynurenine pathway has been shown to be overactive in the CNS of individuals with schizophrenia [66] and the balance of metabolites in the pathway during neurodevelopment is thought to be an important etiologic factor in predisposing individuals to schizophrenia [67]. Moreover, the production of adrenochrome from epinephrine administered to tissue extracts is regarded as a marker of oxidative stress [68, 69] and its concentration is utilized as an inverse measure of the activity of superoxide dismutase [55, 70, 71]. As pointed out by Smythies in 2002 [72], the reductant glutathione can lead to detoxification of adrenochrome and as another route to detoxification, Miller [73] demonstrated that 3 different classes of antipsychotic drugs remove adrenochrome from solution, effectively terminating its chemical reactivity.
Expected increase in the tertiary amines N-methylsalsolinol or N-methynorsalsolinol, of potential relevance to psychosis in Parkinson’s disease
The salsolinols have long been the focus of much research in Parkinson’s disease, as comprehensively reviewed by Naoi and Maruyama [74]. Among the key findings relevant to the methylsalsolinols is that the negative impact on dopaminergic neurons may depend on the enantiomer. The tertiary amine N-methyl [R] salsolinol has been found to exert selective neurotoxicity to dopaminergic neurons and to induce Parkinsonism in an animal model [75].
The propensity to form salsolinols should be greater as the dopamine concentration and/or flux goes up (Figure 3). The first step in their formation involves the reaction of an aldehyde with dopamine, which can either be accomplished in a stereo-selective manner by the enzyme R-salsolinol synthase, or nonenzymatically by what is known as Pictet-Spengler condensation [76], which usually generates a racemic mixture of the [R] and [S] enantiomers. The source of the aldehyde can be from alcohol metabolism to acetaldehyde [77], then forming salsolinol (Figure 3) or from the spontaneous dissociation of 5,10-methylene-THF (Figure 1) to form THF, releasing formaldehyde in the process [78]. Formaldehyde then condenses with dopamine to form norsalsolinol (Figure 3). This source of aldehyde would obviously be more likely to occur during dysregulation of the pathway when flux through the pathway is blocked at 5,10-methylene-THF. Subsequently, NMTs act to generate the methylated forms of salsolinol and norsalsolinol. These NMT enzymes are not yet fully characterized in terms of the substrate used as the source of the methyl group.
However, in another example of nonenzymatic reactions leading to alternative products, dopamine that has already been N-methylated can participate in the nonenzymatic Pictet-Spengler condensation [59, 79] with acetaldehyde or formaldehyde. N-methylsalsolinol is purported by some to be neurotoxic via generation of hydroxyl radicals [80], which would be of relevance to the finding of elevated CSF levels of N-methylsalsolinol in unmedicated Parkinson’s disease patients [81] and a significant difference in N-methylnorsalsolinol between cases and controls [82]. Indicative of a predominantly enzymatic process, the R-isomer has been found to predominate in the CNS [74]. It should be kept in mind, however, that although the non-enzymatic process would be expected to generate a racemic mixture, it would nevertheless contribute to the pool of R-enantiomers. The S-enantiomer of salsolinol has recently been demonstrated to show greater activity at the µ opioid receptor [83]; therefore, it is not possible to predict which enantiomer holds greater selectivity for a given receptor. It also has not been definitively established that total salsolinols are higher in Parkinsonian brain and there is no significant correlation between disease severity and salsolinol concentration [74]. Confounding by L-DOPA treatment is an issue [84], because as Naoi and Murayama have pointed out in their large review [74] salsolinol and related neurotoxins not only decline with age and disease progression in Parkinson’s, but they are increased by L-DOPA treatment. Some authors suggest that L-DOPA does not overpower the effect of the disease on salsolinols, though the studies have been small [85]. The metabolism of N-methylsalsolinol and N-methylnorsalsolinol would be expected to generate their respective quinones via the action of endogenous peroxidases [86] or even auto-oxidation in the presence of metal ions [87], similar to the auto-oxidation of epinephrine that leads to adrenochrome [52–55]. Although no relevant study has yet been carried out for the methylated salsolinol quinones in regard to psychotic symptoms or neurotoxic damage, the toxicity of salsolinol is thought to be exerted through formation of its quinone [88]. At the very least, agents that deplete the quinones of the salsolinols might therefore be expected to protect against their adverse effects, and draw down the concentration of the salsolinol and N-methylated forms via Le Chatelier’s principle.
Regardless of whether the salsolinols are an intrinsic part of the Parkinson’s disease process or not, the fact that most Parkinson’s patients are treated with L-DOPA means any resulting downstream effects of L-DOPA in regards to the salsolinols are relevant to mental symptoms. In point of fact, elevated homocysteine is associated with L-DOPA treatment [16, 89, 90], which would be expected to augment the increased flux and/or concentration of dopamine via SAH inhibition of the methyltransferase metabolism of dopamine and epinephrine, making the alternative routes of reaction to adrenochrome and salsolinols more likely (Figure 2 and Figure 3).
Salsolinol in particular, has been shown to be associated with impacts consistent with neuropathy [91] and unlike salsolinol itself, the R-stereoisomer of N-methylsalsolinol was reported to be neurotoxic to dopaminergic innervation [92], likely via its effect to trigger apoptosis [93], obviously of potential relevance to Parkinson’s and other diseases.
There are psychotic symptoms that can occur in Parkinson’s, most commonly visual hallucinations, though auditory hallucinations can also occur [94]. In this regard, it has been reported that salsolinols are higher in those patients with dementia [85]. Furthermore, levels of salsolinols in Parkinson’s disease have been found to predict the hallucinatory events [95]. The etiologic form of salsolinol has been assessed to be N-methylsalsolinol based on its effect in an animal model to upregulate expression of serotonin receptors thought to be involved in hallucinations [96]. At a minimum, the relationship between N-methylsalsolinol and psychosis is that it inhibits the degradation of catecholamines by monoamine oxidase (MAO)-type A oxidation [97].
The source of acetaldehyde required for condensation with dopamine range from food products like vinegar, fruits, fermented soy, dairy products like yoghurt, and desserts with fruity flavors to cigarettes, coffee and alcohol [98]. Of particular note with respect to alcohol, the long-recognized neuropathy caused by disulfiram (reviewed by Frisoni and Monda [99]) could very well be relevant to the increased production of acetaldehyde by alcohol metabolism in the presence of this drug, which inhibits acetaldehyde dehydrogenase-mediated degradation, including inhibition of the acetaldehyde dehydrogenase found in brain tissue [100, 101]. Although acetaldehyde concentrations in the brain following ethanol consumption are likely lower than in the periphery [102], any increase could either exert a direct neurotoxic effect or an indirect effect from the increased availability of acetaldehyde leading to more salsolinol production (Figure 3).
Xie et al. [103] demonstrated salsolinol activation of DA neurons and Melis et al. [104] have shown that salsolinol is able to excite ventral tegmental area (VTA) dopaminergic neurons in mice when dopamine is depleted, whereas ethanol and acetaldehyde cannot, thereby pointing towards a direct role for salsolinol in the reinforcing effects of alcohol intake. The neuropathy caused by disulfiram has been proposed to be the result of its inhibition of dopamine-beta-hydroxylase, but the Ki for that inhibition is higher than the Ki for inhibiting acetaldehyde dehydrogenase [105], thus the role of excess aldehyde in forming salsolinols becomes key.
Disulfiram is also capable of triggering psychosis (reviewed by Mohapatra and Rath [106]) and its effects can extend to periods of abstinence from alcohol, in which case the sources of acetaldehyde from food and other imbibed substances should be kept in mind. Karamanakos et al. [105] proposed that using a lower dose of disulfiram (25 mg/kg) might prevent the neuropathy, but such a level is known now to not be protective against neuropathy [107–110] or psychosis [110, 111].
To conclude this section on the consequences of increases in SAH, i.e., diverting catecholamines from the standard degradation routes towards production of the salsolinols, there are three ways in which salsolinols could potentially set up a positive feedback cycle with regard to the concentration of dopamine and salsolinol-associated products:
Salsolinols act as competitive inhibitors of COMT [112, 113], an enzyme involved in the processing of dopamine to metabolites that are eventually excreted (Figure 2).
Salsolinols act as competitive inhibitors of MAO-A [112–114] leading to higher steady state concentrations of dopamine. These in vitro experiments for salsolinols have been supported by in situ data [115]. Of note, the inhibition by salsolinol found by Yamanaka et al. [114] commenced with 13% inhibition at 0.25 mM, 23% the average concentration identified in 2 patients with dementia [85] and approached 50% inhibition at 2 mM, 1.8-fold the average concentration identified in 2 patients with dementia [85]. The 2 patients without dementia exhibited an average salsolinol concentration 33% lower than those with dementia [85]. Despite the small sample size, the study contributes to establishing the range of in situ concentrations.
Salsolinol has been found to excite VTA dopaminergic neurons studied in rodent brain slices and perpetuates their firing, with a peak effect at 0.1 μM, though with substantially diminishing effect at higher concentrations (reviewed by Xie et al. [103]).
Only one report was identified which illustrated a negative feedback loop of relevance to dopamine and its salsolinol products, though there could well be others: N-methylnorsalsolinol inhibits tyrosine hydroxylase [116].
Dysregulation of the folate cycle with psychosis: deficiency of vitamin B12
Possibly the most consistent finding concerning the folate cycle and psychosis is the connection between a vitamin B12 (cobalamin) deficiency and psychotic symptoms. Usually, the cause of a deficiency is dietary in nature, but as with all disorders affecting human health, there is an environment-gene interaction. Sobczyńska-Malefora et al. [117] reviewed the environmental and physiological factors contributing to vitamin B12 status and point to the many genes controlling the levels of B12 in the human body, the sheer number of which [59 single-nucleotide-polymorphisms (SNPs) from 19 genes] contribute to the complexity of this topic.
Carney and Sheffield observed in a 1978 study of 272 psychiatric patients [118] that low B12 was predominantly associated with what was termed ‘organic’ psychosis at the time, primarily indicating an external cause that could be reversed. Subsequently, Metzler et al. [119] published an overview of the negative impacts of a B12 deficiency on psychiatric status. Numerous case reports and a few case-control studies followed over the years. Payinda and Hansen [120] treated a patient with B12 deficiency whose psychosis manifested without B12 deficiency anemia. Goebels and Soyka [121] successfully treated two elderly patients with dementia and psychosis related to a B12 deficiency, evaluating the result in the context of a literature review. Similarly, Hanna et al. [122], Silva et al. [123] and Calle-Gonzalez et al. [124] were able to resolve psychosis in elderly patients with B12 treatment, while Carvalho et al. [125] estimated psychiatric symptoms attributable to B12 deficiency occur at a rate of 10% to 20% in the elderly.
At the other end of the age spectrum, Dogan et al. [126] reported on the case of a 16-year-old girl who had developed a B12 deficiency associated with psychosis and seizures, both of which resolved with B12 treatment. Raveendranathan et al. [127] investigated a case of psychosis that was clozapine resistant and was instead a case of B12 deficiency that responded to B12 treatment. Misiak et al. [23] found not only high homocysteine in series of 57 first episode schizophrenia patients, but low B12. Consistent with this theme, FEP patients often exhibit macrocytosis [128], a symptom of vitamin B12-deficiency anemia, consistent with the finding by Kapici and Almis [129] that low B12 distinguished FEP patients from controls rather than low folate.
Sahoo et al. [130] reviewed the literature on B12 and psychosis, and presented 3 case reports in which psychosis developed without the classic hematologic or neurologic signs, suggesting that the psychosis can often precede the other symptoms, consistent with a finding by Lindenbaum et al. [131]. Similarly, Zheng et al. [132] reported on a case of acute adult-onset psychosis attributable to a B12 deficiency, followed by successful treatment of the psychosis with B12. Relevant to a specific cause of acute B12 deficiency, Teodoro [133] treated a patient with celiac disease who had developed late onset psychosis which was responsive to B12 supplementation. Less study has been made of B12 levels in schizophrenia, one example being the research of Yazici et al. [134] who determined B12 levels were significantly lower in 189 schizophrenia cases than in 109 controls.
In B12 deficiency, the pathway is blocked at the level of 5-methyl-THF. Figure 2 illustrates the downstream pathways that would be affected, including increased shunting of homocysteine to SAH, and the upregulation of tetrahydrobiopterin (BH4) production by 5-methyl-THF [135, 136] with availability for increased catecholamine production as a consequence. In addition, there have been reports that 5-methyl-THF can act as a methyl donor for the methylation of dopamine by N-methyltransferase, forming N-methyldopamine, also known as epinine (covered in a previous section). This would facilitate an alternative route to forming epinephrine, and when the subsequent metabolism of epinephrine is compromised by inhibition of COMT and MAO, it can nonenzymatically break down to the putative psychotogen and endogenous tertiary amine, adrenochrome [52–55]. Alternatively, N-methyldopamine could condense with acetaldehyde to form an N-methylsalsolinol.
The added level of complexity for a B12 deficiency is made clear by what became known as The Methyl-Folate Trap (proposed by Scott and Weir [137]; cited by van der Westhuyzen et al. [138]), i.e., that folate supplementation during B12 deficiency leads to worsening of the related symptomatology, possibly attributable to the oversupply of 5-methyl-THF (Figure 2). This was first observed for the neuropathy of B12 deficiency in patients, during which folate supplementation made the neuropathy more severe, as observed by van der Westhuyzen [139] and reviewed by Reynolds [140]. A simple but very elegant series of experiments exploring this peculiar finding was carried out by van der Westhuyzen and colleagues. In their 1982 publication [138] they employed a fruit bat animal model which, unlike rodents, develops a neuropathy from B12 deficiency in the diet as well as from nitrous oxide (N2O) exposure. N2O inactivates vitamin B12 via irreversibly oxidizing the cobalt core from the monovalent to the trivalent state [141]. Van der Westhuyzen et al. convincingly demonstrated that either folic acid or folinic acid administration hastened onset of the neuropathy in both scenarios, whereas methionine was protective. In a medical hypothesis paper, they further proposed that the etiologic folate was 5-methyl-THF which would be expected to build up in a vitamin B12 deficiency [142], and be increased further by folate supplements. They proposed the 5-methyl-THF could be expected to act similar to the neurotoxin kainic acid, based on its structure. However, another scenario for triggering neuropathy is the buildup of SAH with deficiency of B12, the potential for increased levels of dopamine as a consequence, potentially leading to funneling of the methyl group from 5-methyl-THF into the formation of N-methyldopamine (Figure 2) and thence to either epinephrine and adrenochrome, or to the production of the salsolinols (Figure 3). Additional folate input to the pathway would also worsen the outcome in this scenario.
Nitrous oxide (N2O) abuse by individuals, disabling the function of B12, has been commonly associated with psychosis [141]. Sethi et al. [143] reported on a case of abuse of N2O, causing functional B12 deficiency and psychosis, wherein treatment with B12 was effective. Farmer et al. [144] reviewed the effects of N2O abuse on CNS function, and summarize treatment recommendations. Sood and Parent [145] admitted a young man suffering from N2O abuse, psychosis and ataxia, treated with B12, but the patient was lost to follow-up. Of likely relevance to B12 inactivation, Shay et al. [146] found that N2O administration for anesthesia can lead to high homocysteine, particularly in individuals with a deficiency in the enzyme MTHFR (covered in a subsequent section).
Van der Westhuyzen et al. [147] additionally revealed that although treatment of fruit bats with N2O decreases SAM in the liver, SAM is nevertheless not decreased in the brain, pointing towards a brain-sparing mechanism in that model. This likely involves the role that SAM levels play in regulating the activity of CBS in degrading homocysteine, stimulating CBS degradation of homocysteine when SAM is high [148], and when SAM is low, the buildup of homocysteine through lowered activity of CBS results in more favorable conditions for SAM formation from homocysteine according to Le Chatelier’s principle. Reynolds [140] maintained that although SAM can remain in the physiologic range, if homocysteine (and hence SAH) is elevated, the decreased SAM/SAH ratio nevertheless allows for feedback inhibition of methyltransferases (Figure 2).
Intriguingly consistent with the worsening of neurologic symptoms by administration of folate in the presence of a B12 deficiency, Smith and Oliver [149] reported on a case of B12 deficiency in which psychosis was precipitated with folic acid administration, followed by resolution of the psychosis though treatment with B12 and cessation of folate. This theme will be further developed in the section on neuroprotective strategies.
Dysregulation of the folate cycle with psychosis: worsening of symptoms with supplementation of methionine plus MAO inhibitors
Shortly after the inception of the transmethylation theory, numerous experiments were conducted involving individuals with psychosis to determine if, in fact, methyl-carrier molecules would worsen psychosis or be protective. The van der Westhuyzen et al. series of experiments, described in the previous section, demonstrated that methionine was protective for developing neuropathy during B12 deficiency in fruit bats, but the results have been quite the opposite for psychosis in humans if MAO inhibitors are co-administered. Pollin et al. [150] and Brune and Himwich [151] found that administration of methionine and the MAO inhibitor iproniazid to schizophrenia patients in partial remission caused a worsening of symptoms. The data are mixed for methionine alone, with Haydu et al. [152] finding no effect of methionine, yet Antun et al. [153] discerning a worsening effect, and when individuals without psychosis are folate deficient, Stahelin and Winchell [154] found a psychotic-symptom inducing effect of methionine when given to patients with a range of psychiatric diagnoses. This latter outcome could be interpreted as being the result of the expected buildup of homocysteine and SAH. When an individual is folate deficient (Figure 2), methionine should theoretically cause feedback inhibition of MS, further perturbing the processing of homocysteine. Indeed, Kaeser et al. [17] observed that administration of methionine to a psychotic patient with intrinsic homocystinuria worsened the homocysteine levels and the psychosis.
Of note, Fekkes et al. [155] and Regland et al. [156] reported methionine to be high in acutely psychotic patients, but van der Heijden et al. [157] found methionine to be lower in treated patients with schizophrenia. In contrast, Kriisa et al. [158] found that methionine was slightly higher in FEP, and with treatment, the concentration increased further. Thus, the homeostatic level of methionine in psychosis shows no consistent trend, whereas its administration, particularly when MAO is inhibited, appears to generally worsen symptoms in psychotic patients.
Dysregulation of the folate cycle with psychosis: treating cancer patients with Folfirinox leads to psychotic symptoms
There have been numerous case reports of one particular class chemotherapy drugs, Folfirinox and its predecessor Folfiri, eliciting psychotic features in patients [159–164], including mania with delusions [162] or, in some cases, triggering only visual or auditory hallucinations [165, 166]. Folfirinox is a combination of folinic acid, 5-fluorouracil (5-FU), irinotecan and oxaliplatin. 5-FU is considered the most powerful anticancer component of Folfirinox, acting to inhibit the rapid DNA synthesis which underlies cancer cell growth and metastasis. The mechanism of inhibition involves the binding of the active metabolite of 5-FU, 5-fluoro-2-deoxyuridine-5-monophospate, to 5,10-methylenetetrahydrofolate (Figure 2, top center portion) which, in that bound form, inhibits thymidylate synthase, an enzyme responsible for catalyzing the formation and transfer of a methyl group from 5,10-methylenetetrahydrofolate, as required for thymidine monophosphate synthesis [167]. The folinic acid component of the drug enhances the binding of 5-fluoro-2-deoxyuridine-5-monophospate to 5,10-methylenetetrahydrofolate, increasing the efficacy of treatment. It is likely the occurrences of psychosis with Folfirinox over the years may have been underreported because the drug is so often administered as a last-ditch attempt to save the life of a patient suffering from stage 4 cancers, including pancreatic cancer, and psychotic outcomes pale in comparison to the fate those patients are facing.
Prior to the addition of irinotecan and oxaliplatin to the treatment, 5-FU was found to be capable of eliciting or worsening psychotic symptoms [168–170] and although folinic acid was not specified in two of these reports, its use was considered standard protocol with 5-FU administration as far back as 1986–2003 [171, 172]. The year 1979 likely marked the turning point for its incorporation with “anti-metabolite” drugs, in order to overcome their associated toxicity for metabolism in noncancer cells [173], with the subsequent finding of the additional benefit conferred by folinic acid stabilizing the binding of the active metabolite of 5-FU to 5,10-methylene-THF and thymidylate synthase. Of note, no reports of the use of 5-FU prior to 1979 can be found that mention psychosis as a side effect.
In the case reports of Folfirinox causing psychotic symptoms, if there was attribution of the cause of the cases of psychosis, the authors have pointed towards 5-FU alone [160, 165], to steroid psychosis from co-administered steroids [163] or to the cancer mechanism itself [162] without evaluating what might happen if excess folate is administered when the normal destination of methyl groups towards DNA synthesis is blocked. Rustum [174] demonstrated that modest increases in what he termed “reduced folates” occurred with 5-FU infusion alone in an animal model (Table 2 of Rustum, [174]), likely because their consumption by the DNA synthetic pathway was blocked, but massive increases in 5-methyl-THF (which is indeed one form of “reduced folate”) specifically occurred with co-infusion of 5-FU and high-dose folinic acid (Table 3 of Rustum, [174]).
Lacking a head-to-head comparison of psychiatric symptoms in patients with 5-FU treatment alone versus 5-FU plus folinic acid, it is not certain that the addition of folinic acid might be precipitating psychotic outcomes when they occur. Yet, similar to the administration of folic acid when the pathway is substantially blocked by a B12 deficiency, leading to a projected buildup of 5-methyl-THF and worsening of B12-deficiency neuropathy [138] and the report of folic acid triggering psychosis in a B12-deficient patient [149], any excess methyl-group delivery via 5-methyl-THF distributed among the alternative downstream components of the pathway (Figure 2) might be problematic in some patients, depending on their methylation-cycle genes of relevance.
Dysregulation of the folate with psychosis: MTHFR mutations
Mutations in MTHFR are typically diagnosed when a decrease in MTHFR enzyme activity is detected, along with a corresponding hypomethionemia, hyperhomocysteinemia, homocystinuria [175] and a markedly low 5-methyl-THF concentration when the deficiency results from a homozygous SNPs in key MTHFR sequences [9]. However, publications on clinical results for MTHFR enzyme activity, are often plagued by incomplete data on the full spectrum of MTHFR polymorphisms in the affected individual and/or the specific impact on enzyme activity expected from each. MTHFR polymorphisms are unusually common, with the most prevalent minor frequency allele (the T allele in C667T) thought to be carried by as much as 10% to 16% of the population [146], and a meaningful number of those individuals carry a SNP variant at more than one location in the gene [176–182].
In regards to the corresponding enzyme activity reports, the field has also been somewhat compromised by the early existence of two methods, the first actually assaying the reverse direction because of the ease of obtaining and working with the natural folate product, 5-methyl-THF (as discussed above, the substrate for the forward reaction, 5,10-methylene-THF, can spontaneously decompose), and the ready availability of a reliable exogenous electron acceptor known as menadione [183]. The forward reaction, i.e., the physiologic reaction, is just slightly more problematic, utilizing 5,10-methylene-THF as the substrate and NADH as the electron donor [184]. But it affords the opportunity to incorporate in the assay what was once thought to be an essential co-factor, FAD, shown by Suormala et al. [184] to be most strikingly effective in the presence of adequate substrate only if there is a mutation in the binding site for FAD. The consensus is that the forward reaction is the best protocol [185]. Unfortunately, many publications on human MTHFR variants either do not address the putative effect of the polymorphism variant on enzyme activity, or if mentioned, do not specify the assay methodology. Why this poses a problem for understanding the impact of MTHFR mutations will become clear below.
Froese et al. [9] conducted an in-depth examination of the structure activity relationship of the enzyme in relation to its 5-methyl-THF substrate, FAD cofactor and allosteric inhibitor, SAM. Although Donaldson et al. [186] found the reaction catalyzed by MTHFR to be physiologically reversible in the pig, Froese et al. [9] characterized the reaction as physiologically irreversible. Trimmer [78] elucidates the kinetics and thermodynamics in more detail, describing that the half-reaction in which NADPH reduces the enzyme is kinetically irreversible, but the half-reaction wherein 5,10-methylene-THF oxidizes the reduced FAD-containing enzyme is reversible. Although the menadione used in clinical assays can act as an electron acceptor for the reduced form of the enzyme [183], NADP+ cannot because of thermodynamic constraints [78]. This raises the possibility that other more suitable endogenous electron acceptors might exist. What is known is that in the presence of high concentration of 5-methyl-THF, the production of BH4 (the reduced form), required for the first step leading to catecholamine synthesis (Figure 2), is increased [135, 136]. A few studies have indicated that DHFR can catalyze the BH2 to BH4 reduction in the salvage pathway [187] a reaction “stimulated” by folic acid [188] and even more so by 5-methyl-THF [189], but the possibility that 5-methyl-THF might act as an electron donor and BH2 its electron acceptor in that reaction has apparently not been investigated.
SAM feedback inhibition of MTHFR was shown by Froese et al. [9] to occur by its binding to the C-terminal domain, while phosphorylation of the N-terminal domain led to a further increase in feedback inhibition by SAM. Of note, Froese et al. identified 20 human mutations in the regulatory domain, the region where SAM would bind. The possibility should be kept in mind that individuals with compound mutations in MTHFR could theoretically have mutations in this region as well as the mutations that can lead to a decrease in maximal enzyme activity (Vmax).
Weile et al. [190] have created a cDNA library of a broad spectrum of possible mutations in human MTHFR, and modeled the expected change in enzyme activity using the algorithm OpenPyMol, as well as advancing to an in vitro enzyme assay of the expressed gene. They “considered only substitutions that can result from a single-nucleotide substitution (and are therefore more likely to be observed in human populations), finding 70 folinate-independent, 152 low-folinate, and 10 high-folinate suppressors”. In this regard, one should bear in mind that most clinical assays of MTHFR in study subjects are conducted at only one folate concentration. They found several variants that would affect FAD binding, and importantly, with regard to SAM feedback regulation, “identified 896 variants that provided growth rescue beyond that of WT MTHFR (FDR < 5%)”, a phenomenon previously described as ‘‘hyper-complementation”, meaning the enzyme might be constitutively active even when SAM is high because of loss of feedback inhibition. In such a scenario, it is theoretically possible that an enzyme showing lower activity in an in vitro clinical assay (typically in the absence of SAM), could continue to produce product after SAM reaches physiologic levels in situ, leading to an excess of 5-methyl-THF flux over time. However, the net effect of MTHFR mutations is generally to lower the concentration of total folates. As noted by Suormala et al. [184], in contrast to other perturbations that can lead to trapping folate and therefore higher concentrations of 5-methyl-THF, deficiencies in MTHFR activity generally do not cause a buildup in folates [185].
A smattering of case reports of MTHFR deficiency and psychosis can be found [191]. Iida et al. [192] presented a case of compound heterozygosity in MTHFR in a 15-year-old, with psychosis “triggered” by an infection, and normalized by betaine, a methyl source used by betaine-methyltransferases (e.g., methionine synthetase) in the periphery. Similarly, Bönig et al. [34] reported on a case of severe MTHFR deficiency with a psychosis that was successfully treated with betaine. Birnbaum et al. [193] treated a case of late-onset (30-year-old) psychosis, in a subject with compound heterozygosity for two loci in MTHFR, as well as homozygosity for the minor allele T in SNP C667T. Hyperhomocysteinemia was marked, but folate and B12 were near normal. Supplementation with betaine, vitamins B6 and B12, folic acid, riboflavin, and methionine was initiated and the psychosis went into remission, but the tetraparesis did not resolve completely. In a review of 24 case reports with MTHFR deficiency that became apparent only in adulthood, Gales et al. [194] found that 17 percent had psychotic symptoms. Furthermore, Regland et al. [195] demonstrated the homozygous form of the risk T allele in C667 to be present in 7 out of 11 FEP patients.
These limited case reports gave rise to genetic association studies. Some have shown a genetic association between the MTHFR SNP C667T and schizophrenia [175, 196–200], while other studies in a variety of ethnic groups failed to find such an association [201–203]. Gilbody et al. [204] concluded that the SNPs C667T and A1298C were only modestly associated with schizophrenia but were somewhat more strongly associated with depression and bipolar disorder. Wan et al. [205], reviewed many genetic association studies for schizophrenia and MTHFR, some of which paradoxically demonstrated a higher OR for the heterozygous than for the homozygous genotype, though those with the homozygous genotype of TT C667T, for example, exhibited the lowest global methylation values. Although the heterozygous state, CT, showed the strongest association with schizophrenia, the homozygotic state of TT was also associated but to a lesser extent, and the associated phenotype expanded to include bipolar disorder. TT was also more likely to increase negative symptoms and early onset of disease, while positive symptoms were generally lower than for CT heterozygotes. Compound heterozygosity for C667 and A1298C, as well as other added polymorphisms, tended to augment the severity of negative symptoms of schizophrenia. In a meta-analysis conducted by Rai et al. [206], the results showed that although the A1298C had less of an impact on enzyme function than C667T, it had a greater association with schizophrenia. Nevertheless, the OR for the C allele in A1298C remained modest, on the order of a 10% to 15% increase in risk. Of note, Kevere et al. [207] reported for cases of FEP that homocysteine is the highest and most problematic when the psychosis is episodic and recurrent. Again, it was in the patients heterozygous for C667T.
The basis for increased prevalence of affected cases exhibiting the heterozygous state of C667T could well lie in the fact that homozygosity in this SNP for the T allele appears to be incompatible for survival when it occurs in conjunction with the C allele in A1298C, as no such subjects were found in genetic association studies by Castro et al. [208] or Ulvik et al. [209]. Nevertheless, other potential causes for increased risk in heterozygotes should be pointed out. When the risk for disease is greater in heterozygotes it can indicate transacting SNPs within the gene or in closely linked genes, are at play. This phenomenon is part of the spectrum of gene-gene interactions that underlie what is known as the hybrid vigor of heterosis, or alternatively, in the case of worsening function in heterosis, “outbreeding depression” [210]. Intriguingly, Song et al. [211] found that in a eukaryotic model of heterosis, the expression of several 1-carbon metabolism genes was significantly involved, including some heterozygotes that showed a significantly decreased expression of the affected gene than seen in the parental lines. Yet, none of these differentially-expressed genes included MTHFR. At any rate, the first consideration for an enzyme that forms a dimer should be mutations that impact dimer formation, and in this regard, Ulvik et al. [209] identified human compound mutations in C677T and A1298C which led to instability in MTHFR, inferred from the high homocysteine concentration that was present in carriers. This occurred when C667T was heterozygous combined with AA or AC in the SNP A1298C. The prior report by Castro et al. [208] supports this finding for homocysteine in the heterozygous C667T condition combined with AC in A1298C. Also, of increased risk for instability in the dimer was the C667T homozygous state of TT when combined with AC for A12098C, a very rare combination in the Ulvik et al. study population and a result not supported by the work of Castro et al.
Interpretation of the impact of low MTHFR activity relevant to psychosis should first and foremost involve the predicted increase in SAH from elevated homocysteine levels (Figure 2). Interpretation of altered folate levels, however, must take into account the most relevant environmental variable, i.e., diet, and what type of sample is being assayed, blood or plasma. If plasma is the sample source for the folate assays, the predominant folate detected is 5,10-methylene-THF whereas if red blood cells are the sample source, the predominant folate detected is likely 5-methyl-THF [208]. The two compounds are generally not distinguished in clinical assay reports. Obviously, with low MTHFR activity, 5,10-methylene-THF would tend to increase but 5-methyl-THF should decrease. Although Kevere et al. [207] reported levels of folic acid were normal in all subjects with heterozygous C667T and elevated homocysteine, it is unclear if their report for “folic acid” involved cellular or blood plasma assays for folates. The measured biochemical that was significantly associated with a greater level of positive symptoms, including paranoia, was homocysteine and its level was also significantly associated with the CT heterozygous state in C667T. As discussed in the prior section on homocysteine, elevated levels of homocysteine typically result in elevated SAH, which would be expected to negatively impact the degradation pathway for the catecholamines dopamine and epinephrine (Figure 2).
In addition, there is the possibility that underactivity of MTHFR would lead to a buildup of its substrate, 5,10-methenyl-THF, which can spontaneously degrade to THF plus formaldehyde [78], a potential source of the formaldehyde required in the formation of the salsolinols (Figure 3).
To summarize in brief, MTHFR enzyme deficiencies most often result in elevated homocysteine which consequently raises SAH levels, potentially increasing the risk for the development of psychotic symptoms. Paradoxically, it is not the carriers of the polymorphism leading to the greatest decrease in enzyme activity who are most at risk of psychosis, pointing towards a role for compound mutations that may affect the modulation of enzyme activity by SAM or FAD.
Dysregulation of the folate cycle with psychosis: evidence for upper and lower limits on the mental health window for folate concentration
Low folate in FEP and established schizophrenia patients
FEP represents a time period when long term effects of antipsychotic drug treatment and the drug itself, have not yet obscured factors that may have been involved in triggering the psychotic break. Firth et al. [212] conducted a meta-analysis on vitamin and mineral levels in predominantly medication naïve FEP patients, including A, B12, C, D, E, and folate, as well as zinc, magnesium, sodium, potassium, calcium, copper, chromium, iron, manganese, and selenium. Only low folate and low vitamin D levels reached significance for association with FEP, and corresponded to negative symptoms (in the case of folate), and but not to positive symptoms. Dos Reis et al. [213] subsequently confirmed these results for low folate in a large FEP cohort study. Saidinejad et al. [214] observed a patient who experienced transient psychosis from Trimethoprim-Sulfamethoxazole, an inhibitor of DHFR which thereby results in a block of the metabolism of folic acid, preventing the generation of bioavailable methyl group carriers (Figure 1). The patient was treated with antipsychotic drugs to relieve the psychosis.
Numerous case reports can be found which identified low folate in FEP or in schizophrenia. Yazici et al. [134] found significantly lower folate levels in schizophrenia patients as compared to healthy controls. Frajerman et al. [215] reported on the folate, B12, and homocysteine levels in 188 subjects with either a diagnosis of FEP, schizophrenia or schizoaffective disorder, finding no significant differences by diagnosis. Unfortunately, no controls were included, but given the average folate levels reported for healthy individuals, the range found for folate values in the subjects encompassed very low values (< 6.8 nM) as well as subjects in the high normal range (> 30 nM). Not surprisingly, FEP patients were significantly more likely to be cannabis users, a result relevant to the etiology of psychosis onset [22]. Although Frajerman did not differentiate folate values in cannabis users from nonusers, Bemanian et al. [216] determined that use of cannabis was significantly associated with low folate status (< 6.8 nM) and yielded the largest effect estimate of all drug use examined, though the trajectory over time showed more improvement than for injectable drugs.
Numerous case reports also present results from treatment with folate (or its near equivalent, betaine) in either early-psychosis or FEP patients who are determined to have either low folate or high homocysteine. A precedent-setting example was reported by Freeman et al. [217] concerning a young patient with psychosis and homocysteinemia, coupled with homocystinuria, but normal folate levels. After pyridoxine (a cofactor for CBS, the enzyme which degrades homocysteine) was administered to mitigate the homocystinuria without much success, high dose folic acid treatment was commenced which alleviated both the homocystinuria and the psychotic symptoms. Michot et al. [218] reported on a case of a middle-aged female patient with compound mutations in MTHFR, impaired gait (paraplegia, commonly seen with severe MTHFR deficiency) and late onset psychosis. Her folate and methionine values were low and homocysteine was high. She was treated with betaine and high dose folinic acid (50 mg/day), along with hydroxocobalamin (a form of B12). Over a 2-month period, her psychosis resolved, although the paraplegia did not normalize. Regland et al. [195] found that 7 patients with schizophrenia-like psychosis and homozygous for the MTHFR T allele in C667T had high homocysteine concentrations that were responsive to folate supplementation, but unresponsive to vitamin B12 supplementation alone. In contrast, those without the T allele were responsive to vitamin B12 alone. Unfortunately, no clinical follow-up evaluation of psychotic symptoms was conducted. Stahelin and Winchell [154] applied methionine supplementation to 3 cases of severe folate deficiency (in 2, likely coupled with B12 deficiency as exhibited by macrocytic anemia). All 3 developed symptoms of psychosis. Kapici and Almis [129] measured folate and B12 in 67 FEP patients, 67 first episode mania (FEM) patients and 67 healthy controls, finding that low B12 was significantly associated with FEP, but not FEM. Folate trended lower in the FEP patients, but the difference did not reach significance (P = 0.10).
Treatment of schizophrenia patients with folate has been explored less often. Levine et al. [219] reported on a series of patients with elevated homocysteine who benefited from treatment with folic acid, B12 and pyridoxine, though it is impossible to ascertain which agent was the most beneficial. Hill et al. [220] administered folate to a series of chronic schizophrenia patients with residual negative symptoms who had been genotyped for the most common SNP in MTHFR (C667T), finding that folate treatment was beneficial in those with at least one copy of the T allele, i.e., the DNA sequence that reduces enzyme activity significantly, meaning less production of 5-methyl-THF and less transfer of methyl groups to homocysteine. Roffman et al. [221] conducted a multi-center trial of 2 mg folic acid plus 400 mg B12 daily in chronic schizophrenia patients that had been genotyped for the MTHFR SNP, C667T along with a SNP in folate hydrolase (C484T), MTR G1298A, and one in COMT, G675A. The subject’s levels of folate and B12 increased significantly over time, and a significant improvement in negative symptoms was noted for the patients with the folate hydrolase T allele in C484T. Godfrey et al. [222] treated a cohort of depressed patients and schizophrenia patients with 5-methyl-THF and found a significant improvement in clinical scores in both groups, but did not distinguish improvement in positive versus negative scores in the schizophrenia patients. Simamora et al. [223] studied B12 and folate administration to a group of male schizophrenia patients with predominantly negative symptoms of schizophrenia, all on risperidone, and found significant improvement in negative symptoms for the patients taking the supplements as compared to the patients not taking the supplements. However, following a large review and meta-analysis, Niu et al. [224] concluded that folate supplementation in schizophrenia carries more risk for unwanted side effects than does B12 supplementation.
Of note, Carney and Sheffield observed [118] that low folate tended to be associated with depression rather than the positive symptoms of psychosis, an observation supported by subsequent publications by that group [225], data from Wilkinson et al. [226], data from a meta-analysis by Roberts et al. [227] on the benefits of folate supplementation for depression, results from a review by Zheng et al. [228], the report of Muntjewerff and Blom [229] and from the effect of 5-methyl-THF to improve negative symptoms in depression [230] and negative symptoms in schizophrenia with no improvement in positive symptoms [231].
Folate administration or high folate levels linked to mania or psychotic symptoms
That folic acid excess can be problematic for the CNS was suggested decades ago by the observations that neurologic complications developed during its use as a sole agent to correct pernicious anemia (reviewed by Berk et al. [232]), as well as research on its pro-convulsant action in animal models not treated with anticonvulsants (e.g., Baxter et al. [233]) following the observation that anticonvulsant drugs possessed the common property of being anti-folate. The studies by Baxter et al., demonstrated that folic acid was more potent on a weight basis than Leptazol and much more potent than glutamate for inducing seizures in an animal model when injected ICV, but much less potent than Leptazol with IV injection. However, the oral administration of appropriate levels of folic acid to normalize folate levels in humans with seizure conditions being treated with anticonvulsants is beneficial for markers of their folate status and does not precipitate seizures (e.g., Semmler et al. [234]). Other studies indicated problems specifically for mental health status. Hunter et al. [235] investigated the effect of high dose folic acid (15 mg/day) in healthy volunteers, but the study was curtailed at 1 month instead of the planned 3 months because of adverse reactions in the majority of subjects, including mental disturbances. Eight out of the fourteen experienced irritability, all of whom also experienced sleep difficulties and five of whom also experienced excitability/overactivity, which could be characterized as mania or submanic behavior. The sleep difficulties encompassed “vivid anxiety dreams”. The authors covered in the discussion section how folic acid when given to those on anticonvulsants (which deplete folate) can result in loss of seizure control or even status epilepticus.
Iida et al. [192] reported on an individual with MTHFR deficiency who first presented at age 13 for progressive difficulty in walking. At that time, his homocysteine levels were high and his folates were below normal, while B12 was low-normal. High folic acid supplementation was initiated (15 mg/day) without supplemental B12, and his walking difficulties worsened. By age 15, he was exhibiting symptoms of psychosis, both positive and negative, concurrent with a fever-inducing gastroenterological infection. One month after the infection cleared, he was still bed ridden and still exhibiting signs of psychosis, and had developed convulsions which were at the time thought to be associated with high folate [233]. Indeed, by that time, his folate levels were high (20 ng/mL) and, suggestive of supplementation at some point, so was his B12 level (1,120 pg/mL) as well as homocysteine (230 µM), while his methionine was below normal (11 µM). Two compound missense mutations were identified in his MTHFR gene, and as a result, vitamins B1, B6, and B12 (750 μg/day) as well as folic acid were administered; however, oral betaine was eventually substituted for folic acid. Note that betaine is not thought to be a source of methyl groups for CNS methyltransferases, as covered in the upcoming section on neuroprotective strategies. After a few months, his psychosis resolved, though his walking difficulties were such that a walker was required. The authors conclude that reductions in his nutrition, exacerbated by intestinal malabsorption from gastroenteritis, as well as a fever rendering his thermolabile MTHFR gene less stable, might explain the rapid decline in his mental and physical status at age 15. The other possibility is that high amounts of supplemental folate administered without matching B12 levels for several months triggered the psychosis.
Prakash and Petrie [236] reported on two cases, one with schizoaffective disorder and the other with catatonic schizophrenia, in whom excited agitation, irritability and aggression was precipitated with folate administration (3 and 1 mg/day, respectively). Both were on antipsychotic medications at the time. At this juncture it is worth noting that alterations in symptoms with folate administration in established schizophrenia (e.g., Roffman et al. [231]) are often confounded by antipsychotic drug treatment, which could be expected to somewhat lessen the worsening of positive symptoms, and also lessen improvement in the negative symptoms because most neuroleptics act to increase not diminish negative symptoms at higher doses [237, 238].
Chaudhry et al. [239] administered methotrexate (a DHFR inhibitor) to a patient with NMDA-receptor autoimmune encephalitis and related psychosis, with the goal of reducing the heightened autoimmune status. By inhibiting DHFR, the generation of 5-methyl-THF would be markedly reduced. All trial participants were administered 5 mg/day of folic acid, except on the one day per week that methotrexate was given. The positive symptom scores were significantly improved in the methotrexate medication arm. However, Sur and Chauhan [240] describe the case of a young man prescribed methotrexate for rheumatoid arthritis (7.5 mg once per week), along with 5 mg of folic acid (once per week). He experienced an adverse reaction to the treatment, developing increased intracranial pressure (pseudotumor cerebri) and the methotrexate administration was stopped, though presumably the folic acid administration continued. One month later, he experienced auditory and visual hallucinations. It was noted that he had a positive 2nd-degree family history for schizophrenia. His psychotic symptoms were resolved by treatment with olanzapine. Here, the methotrexate administration may have resulted in upregulation of enzymes associated with 5-methyl-THF generation, and with any continuation of the folate administration in the acute absence of methotrexate, too much 5-methyl-THF may have been produced, resulting in diversion of methylation to produce more of the potentially harmful tertiary amine products shown in Figures 2 and 3.
Geddes et al. [241] found that folic acid when co-administered with the anticonvulsant lamotrigine for bipolar disorder, nullified the beneficial effect of lamotrigine on bipolar disorder symptoms.
Morris et al. [242] investigated the correspondence between red blood cell folate levels and postpartum mania and depression in a group of 233 women genotyped for the C667T MTHFR polymorphism and the majority of whom were on folate supplements. Those with the TT genotype paradoxically exhibited significantly higher red blood cell folate levels (P = 0.004) than the CT and CC genotypes, possibly because fewer were off folate supplements. The TT genotype also exhibited higher mania and depression scores that did not reach significance. The authors present the correlations between the red blood cell folate and psychiatric scores within in each genotype, which were of low r values and also did not reach significance (n.s.). More interesting, however, was the correlation between mean red blood cell folate levels and mania scores across genotypes, which show a large r value of +0.80 (though also n.s.). For the correlation between mean red blood cell folate and depression scores across genotypes, the r value was +0.97, and the result is trending towards significance (P = 0.07). Glaser et al. [243] found that maternal folate supplementation at 18 weeks trended toward an increase in the odds of psychosis-like behaviors evaluated in children at 12 years of age (OR 1.34; CI 1.00–1.76). However, Hunter et al. [244] found a beneficial impact of maternal folate supplementation on an electrophysiological measure of psychosis-prone behavior, the P50 ratio, in neonates. The discrepancy in the outcomes of the Glaser et al. and Hunter et al. studies may have to do with the timing of the folate administration [245] or with the timing and type of the measures of psychosis proneness in the children. It should be pointed out that neurodevelopment also represents a crucial time period for the potential interaction between folate dysregulation and dysregulation in the kynurenine pathway [67].
In a retrospective cohort analysis, Rainka et al. [246] observed adverse events of impaired sleep (5 patients) and increased anxiety (3 patients) from 139 teens being treated for anxiety disorders and mood disorders with 5-methyl-THF (7.5 mg to 15 mg per day), though their overall conclusion was the supplement was safe and well-tolerated. And in subjects with ADHD, a disorder with some features that can overlap with psychotic symptoms, Prades et al. [247] concluded from their large scoping review that folate levels were higher in children with ADHD and B12 was lower.
Dysregulation of the folate cycle with psychosis: how the importance of dopamine is evident in the effect of alpha-methyl-para-tyrosine
The dopamine theory of schizophrenia has suffered its ups and downs over the years as it has been difficult to document increased concentrations of dopamine or even increased flux through dopamine in individuals with schizophrenia. As put succinctly by Murray and coauthors [248] “Genome-Wide Association studies (GWAS) have not implicated genes directly involved in dopamine synthesis or release but instead point to upstream and downstream pathways linked to dopamine.” Furthermore, attempts to identify specific genotypes in the dopamine receptor DRD2, regarded by many as the primary mechanism of action for antipsychotic drugs, that might be associated with schizophrenia have not been compelling for a single locus result. D’Ambrosio et al. [249] emphasized the need for “indexing” specific interacting genes in regards to DRD2 function in GWAS studies, which essentially means going in with a priori theories about which interacting genes are central to the problem, certainly a wise approach that should help separate signal from noise.
Nothing makes more clear how upstream or downstream pathways of dopamine, described by Murray as being key to the question, must play a pivotal role in psychosis than does the use of alpha-methyl-para-tyrosine (AMPT or “metyrosine”). Metyrosine has been used effectively in psychotic patients with the 22q deletion syndrome who tend to be treatment resistant to antipsychotic drugs [250–252]. Metyrosine has also been administered to schizophrenia patients on occasion with some modest success in reducing biomarkers of excess dopaminergic function [253], and as an effective add-on therapy for positive symptoms [254–257], though more recent work has found metyrosine to only temporarily reduce a specific subset of positive symptoms, thought disorder and delusions [258]. There certainly can be adverse neurologic effects [256] and as one might expect, with lowering the amount of available dopamine, more prominent negative symptoms [259, 260]. It has also been studied recently in subjects at ultra-high risk for schizophrenia, with promising results [261]. Intriguingly, Imamura et al. [262] found that metyrosine could block the toxic effects of homocysteine in neuronal cultures.
Dysregulation of the folate cycle relates to hallucinogenic drugs: the E-state of amino moieties and psychoactive effect
Alexander Shulgin has made a remarkable career out of investigating how analogues of hallucinogenic drugs compare in potency, and which substituents in particular either enhance or diminish hallucinogenic effect [263]. However, it was Snyder and Merril [264] a few decades earlier who may have hit on an underlying mechanism involved in hallucinogenic efficacy. While referring to Shulgin’s very early work on hallucinogenic potency and following the approach of Karreman et al. [265] in their study of chlorpromazine, Snyder and Merril estimated the energy of the electrons in the highest occupied molecular orbital (HOMO) as distributed across the molecule for a variety of hallucinogens, finding the most potent hallucinogens had greater net values. High HOMO energies correspond to the ease of electron donation, and Snyder and Merril concluded “if an electron transfer mechanism is involved, the negative correlation with energy of the lowest empty orbital would indicate that the hallucinogens do not act via electron acceptance. Thus, despite the crudeness of the theoretical and experimental data discussed here, the close relationship between HOMO energy, an index of electron donation, and the hallucinogenic potency of drugs does favor an electron donation model of drug-receptor interaction”. However, the correspondence between potency and HOMO energy wasn’t perfect, the caveat being mescaline and psilocybin had higher HOMO energies than the much more potent hallucinogen, LSD.
One of the most thorough and detailed reviews of structure-activity relationships for hallucinogens was published by Nichols [266]. As expected, structures that pose problems for steric interference reduce activity of the compounds, while many of the structural alterations which increase overall electron availability across the aromatic rings enhance activity. As Nichols also concludes: “Although all of the hallucinogens possess a basic nitrogen atom, there is little apparent dependence on basicity. Whereas lysergide or substituted phenylcyclopropyl-amines have pKa values of ~8–8.3, the phenethylamines or substituted amphetamines have higher pKa values, at ~9.4–9.8.” Yet, what is clear is that none of the hallucinogens are highly basic or quaternary amines, i.e., protonated at physiological pH, whereas methylated amine compounds which do carry a positive charge on an amino group at neutral pH are not hallucinogenic: caffeine, nicotine, methyl-nicotinamide, histamine, acetylcholine [267–271].
Focusing on the weakly basic nitrogen atoms that Nichols referred to in particular, it is now possible to use programs to calculate the electrotopological state (E-state) of those nitrogen substituents taking into account the composition of the entire molecule. Two such programs provided free on the internet include VCC Labs (“p-client”; now offline) and ChemSar, which could be used to calculate the E-state originally defined by Kier and Hall [272]. As reviewed by Ivanciuc [273], the E-state index incorporates the availability of electrons intrinsic to a particular moiety of interest, while adjusting for the electrotopological impact of other atoms in the molecule. The lower the E-state, the lower the availability of electrons.
For the drugs with more than one nitrogen (e.g., LSD), the issue becomes which nitrogen moiety would be most likely to interact with a receptor. Wipf et al. [274] highlighted the importance of conformational flexibility in receptor binding as being best exemplified by a moiety attached to a rotatable bond (see also Montoro-García et al. [275] and von Helden [276]). Table 1 illustrates the E-state values for the most conformationally flexible amino-N in hallucinogens, either an amide (LSD), or a tertiary amine (PCP, psilocybin, dimethyltryptamine), a secondary amine (ketamine), or a primary amine (mescaline). The E-state values of the endogenous tertiary amine byproducts of transmethylation that have been discussed previously are presented with some of their precursors, along with 5-methyl-THF, homocysteine, aromatic amino acids as well as straight chain, nitrogen rich amino acids, in order to place the results in a broader biochemical context. Also shown is Scar, the total E-state indices for the carbon atoms in the molecule.
Hallucinogens, potential endogenous psychotogens, and related biochemicals sorted by E-state value of N1 and/or Scar2
| Compound | E-state of key N1 | Scar2 | Ratio* Scar to E-state of N | Evidence for effective dose (µg) |
|---|---|---|---|---|
| Adrenochrome | 1.765 | 4.25 | 2.41 | n.d. |
| 5-methyl-THF | 1.815 | 5.66 | 3.12 | mixed3 |
| LSD | 1.945 | 27.60 | 14.19 | 100 |
| Psilocybin | 2.043 | 14.04 | 6.87 | 4,000 |
| N-methyl-norsalsolinol | 2.193 | 10.40 | 4.74 | n.d. |
| N-acetylmescaline** | 2.208 | 5.67 | 2.57 | n.d. |
| Dimethyltryptamine | 2.211 | 21.00 | 9.50 | 14,000 |
| N-methyl-salsolinol | 2.235 | 11.99 | 5.36 | n.d. |
| Rows above sorted4 on E-state of key N1, rows below sorted on E-state of key N1 (+) Scar | ||||
| PCP | 2.811 | 27.11 | 9.64 | 1,000 |
| Ketamine | 3.197 | 14.29 | 4.47 | 70,000 |
| Tryptamine | 5.500 | 15.79 | 2.87 | none |
| Mescaline | 5.500 | 13.02 | 2.37 | 200,000 |
| Serotonin | 3.134 | 12.26 | 3.91 | none |
| Tryptophan | 3.084 | 11.09 | 3.60 | none |
| Salsolinol | 3.292 | 9.60 | 2.92 | n.d. |
| N-methyldopamine | 3.006 | 9.36 | 3.11 | n.d. |
| Phenylalanine | 5.349 | 8.92 | 1.67 | none |
| Norsalsolinol | 3.195 | 8.07 | 2.53 | n.d. |
| Dopamine | 5.305 | 6.73 | 1.27 | none |
| Epinephrine | 2.806 | 5.90 | 2.10 | none |
| Tyrosine | 5.325 | 5.64 | 1.06 | none |
| Norepinehrine | 5.198 | 3.43 | 0.66 | none |
| Arginine | 3.680 | –0.43 | –0.12 | n.d. |
| Homocysteine | 5.079 | –0.75 | –0.15 | none |
1 N represents the amide, tertiary, secondary or primary amine identified as the most conformationally flexible [266]. 2 Scar represents the total E-state value for carbons in the molecule. 3 In the presence of a B12 deficiency, administration of ≥ 1,000 µg/day of 5-methyl-THF or other folates could be problematic in otherwise normal individuals or those with psychosis [136, 221]. 4 The compounds were sorted from low to high values of the E-state of key N up to values < 2.5; from values of 2.5 to 3.2 they were sorted on E-state and then from high to low values of Scar; from values of E-state of 3.2 and above, they were sorted on Scar only (see text). * The ratio of Scar to E-state of N is of interest because of the potential interaction between the two variables. ** The E-state value of N for N-acetylmescaline predicted by ChemSAR (http://chemsar.scbdd.com/calcdes/index/) involved 3 possibilities: tertiary amine, double-stranded, and an aromatic component; the results were averaged to represent all 3 characteristics
The relevance of E-state values to hallucinogenic effect for the N in a primary versus a tertiary amine is best exhibited by tryptamine versus dimethyltryptamine, for which the pre-methylated E-state value of the N in question is 5.500, and the post methylated E-state value is 2.211. LSD, one of the most potent hallucinogens known, has a very low E-state value of 1.945 for its most flexible tertiary amine-N (Figure 4). Ketamine, which contains a secondary amine, is roughly 700 times less potent than LSD, and mescaline, containing a primary amine, is reported to be at least 30-times less potent than psilocybin and 1,000 to 3,000 times less potent than LSD (reviewed by Kovacic and Somanathan [277]), while the E-state value for its primary amine-N is 5.50.
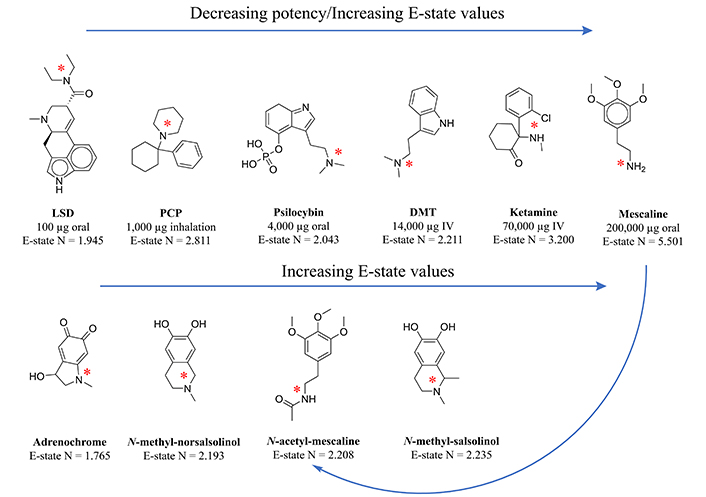
E-state values of amine-containing compounds of relevance to hallucinogenic activity. Top panel: The E-state of amine-N’s in hallucinogens placed in order of the dose required [268] for efficacy (increasing from left to right), calculated by both the online program VCC Labs “p-client” application (now offline) and the online program ChemSAR (http://chemsar.scbdd.com/calcdes/index/). The E-state value for most accessible amine-N was chosen based on steric constraints, i.e., selected if the N was not part of a ring structure, and if all were, then either the sole N or the N which was bound to a methyl group (itself not being part of a ring structure). The correlation between the E-state of the amine-N and dose required for efficacy was r = 0.97, P = 0.0013. Lower panel: The E-state of amine-N’s in potential endogenous and exogenous hallucinogens placed in order of increasing E-state value of the amine moiety. Note that the E-state value of N for the sole nitrogen in N-acetylmescaline predicted by ChemSAR involved 3 possible states: tertiary amine, double-stranded amino group, and an aromatic component; the results were averaged to represent all 3 characteristics. Ghafourian and Cronin [299] found E-state roughly on par with other atomic-level descriptors, depending on the application
Figure 4 illustrates the correspondence between the E-state of the most conformationally flexible N and dose required for hallucinogenic effect. Potency was derived from a 1996 publication by Abraham et al. [278]. A regression analysis of the dose required for hallucinogenic effect versus E-state values for the most conformationally flexible N in each compound revealed a strong and significant association (r = 0.97, P = 0.0013). Adrenochrome, the putative endogenous psychotogen described previously, exhibits a very low E-state value for its tertiary amine that is on par with that of the most conformationally flexible amino group (an amide) in LSD. The methylated salsolinols show equivalent values (Figure 4 and Table 1).
Antipsychotic drugs, which also have tertiary amines, and in most cases multiple tertiary amines, all contain either halogens and/or a sulfur moiety that would confer many other changes in the putative structure activity relationship at a receptor. For this reason, and because discernment of the E-state value of such multiple tertiary amines in the same molecule is not possible using current electrotopological programs, the antipsychotic drugs are not suitable for a direct comparison of amine E-state values to those of the hallucinogens. It also should be emphasized at this juncture that the mechanism of action of current antipsychotic drugs may not involve binding to the same receptor as the hallucinogenic drugs and endogenous hallucinogens, whatever that receptor or receptors may be, but may consist of another type of chemistry altogether [65, 73].
One framework to understand these data is to recognize that although total electron availability is high over the atoms in many hallucinogens as determined by Snyder and Merril [264], it is also high over the carbon atoms in antipsychotic drugs as represented by the molecular descriptor Scar (Table 1). Roughly similar to the results of Snyder and Merril [264], there is an inverse correlation between Scar (Table 1) and the dose of hallucinogen required (meaning potency would be positively correlated with electron density), but the correlation does not reach significance, r = –0.68, P = 0.14, likely in part because the N is small. However, the correlation between the descriptor for total E-state over the molecule, Shev, most similar to the approach of Snyder and Merril [264] shows a low r value, r = –0.227, P = 0.67.
Considering both factors, Scar and the E-state of the most conformationally flexible amino group, one possible scenario is that the “electron chain” interaction with the receptor originally proposed by Karreman et al. [265] and supported for hallucinogens by Snyder and Merril [264], Kang and Green [279], Nichols [266], and Kovacic and Pozos [280], actually pertains more to commonplace van der Waals electronic interaction, wherein carbon atoms with higher E-state values interact with low E-state areas on the receptor and high E-state areas of the receptor interact with the low E-state N in the hallucinogen. With this possibility in mind, Figure 5 illustrates the van der Waals conformation of PCP, the methylated salsolinols, adrenochrome, 5-methyl-THF, and N-acetylmescaline.
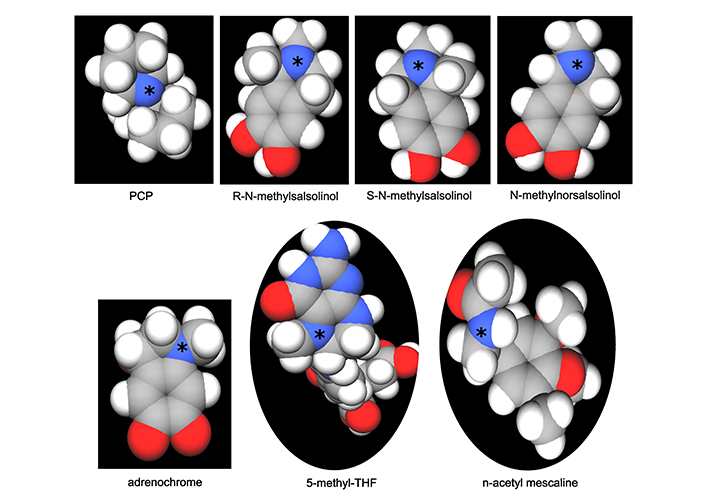
van der Waals models of PCP as compared to potential endogenous and exogenous psychotogens. The online program MolView (https://molview.org/) was utilized to generate the conformational figures. Hydrogen atoms are white, carbon atoms are grey, oxygen atoms are red, and nitrogen atoms (N) are blue. The amine-N’s for which E-state was calculated are designated with an asterisk. PCP, at the top left, is unusual in that all of its atoms are part of a ring structure, yet the van der Waals model shows good exposure of its nitrogen group, consistent with its potency as a hallucinogen. The methylated salsolinols and N-acetylmescaline also show good exposure for the amine-N, but those for adrenochrome and 5-methyl-THF are more structurally constrained in terms of the surface area exposed. N-acetylmescaline lies somewhere between 5-methyl-THF and the salsolinols for area of amine-N surface exposure. Note that the two enantiomers of N-methyl-salsolinol vary in conformation, with the S enantiomer exhibiting slightly greater accessibility to the amine-N
Representing a caveat to the above model of hallucinogenic action, is the primary amine-N of mescaline (Figure 4), with an E-state value higher than that for all other hallucinogens, suggesting that a metabolite of mescaline might instead be the etiologic factor for behavioral effects. In support of this premise, mescaline has a delayed onset of action ranging from 2 to 4 hours [51, 281, 282] as compared to faster onset of action for other hallucinogens, like LSD for example. This delay has been commonly attributed to a slower transit across the BBB, with mescaline concentrations in brain tissue peaking at 60 minutes post injection in an animal model [283] as compared to 15–20 minutes for LSD [284, 285], and in the same relative direction as the difference in time frame of peak behavioral effects for these two drugs [286]. It remains possible, however, that the delayed onset of peak effect for mescaline is not fully accounted for by the delay in BBB permeation and that production of an active metabolite is also involved. One potential candidate for a downstream metabolite of greater efficacy would be the secondary amine N-[2-(3,4,5-trimethoxyphenyl)ethyl]acetamide, also known as N-acetylmescaline, as illustrated in Figure 3 of Madrid-Gambin et al. [287], retaining the amino group yet involving the formation of an amide moiety predicted to lessen the electron E-state value over the nitrogen atom.
Table 1 also illustrates a preliminary and straightforward model for predicting hallucinogenic efficacy based on E-state values, wherein the E-state value of the most flexible amino moiety is more determinative of potency for values less than 2.5, above which (2.5–3.2) a Scar equivalent to that of ketamine (≥ 14.0) may compensate for the loss of efficacy associated with the higher E-state for N. This model is not intended to supplant a full scale QSAR analysis, but rather to suggest possible avenues to explore. Note that this approach omits mescaline from the hallucinogen category and relegates it to the position of being a prodrug. PCP would be included as a direct hallucinogen because it has a short time frame of action and even though it exhibits an E-state value for its sole N on par with that of the N in epinephrine, which is not a psychotogenic compound, the Scar value of PCP is much higher and on par with that of the other highly potent hallucinogens. The transmethylated by-products of an aberrant methylation cycle, adrenochrome, N-methylsalsolinol, and N-methylnorsalsolinol, all have low E-state values for their tertiary amine-N that place them in the range of the most potent hallucinogens, though based on the model presented here, their more modest Scar values might be predicted to mitigate their impact. Even a low-level hallucinatory effect could be of concern, however, for products whose flux endures over time.
Figure 5 exhibits the “van der Waals” conformation of PCP (generated by MolView) as having fairly accessible tertiary amine moiety, unusual in that its sole nitrogen is part of a sterically constrained ring structure. Thus, PCP is remarkably accessible in a steric sense. In addition, the “van der Waals” conformation for the putative endogenous psychotogens of interest is presented, as well as that for N-acetyl-mescaline, predicted to have some hallucinogenic potential because of low E-state value of its amide-N. PCP (top left) and the putative psychotogens to the right on the top row (the two enantiomers of methylsalsolinol plus N-methylnorsalsolinol), all show their tertiary amine is accessible, whereas the proportional exposure of tertiary amines in adrenochrome and 5-methyl-THF is noticeably smaller, which might serve to diminish the interaction with a receptor. N-acetylmescaline exhibits an exposure of its amide-N between the former two compounds and those on the top row. Of additional note are the two isomers of N-methylsalsolinol (modeling was not available for N-methylnorsalsolinol), the “R” and “S” enantiomers. To the eye, appears as though the “S” enantiomer is less hindered on one side than equivalent steric hindrance for the “R” enantiomer. This would have implications for any nonenzymatic formation of N-methylsalsolinol that can occur via Pictet-Spengler condensation of methyldopamine and acetaldehyde (discussed in a prior section), which generates a racemic mix, unlike the preference for the “R” enantiomer exhibited by the enzymatic process [74]. Based on the van der Waals conformations in Figure 5, the “S” enantiomer from the nonenzymatic process might provide more interaction of its tertiary amine with a receptor. It could be said that, as a general rule, the option of a nonenzymatic route to product formation (where it exists), is more likely to be utilized when well-regulated enzymatic pathways go awry.
To conclude this section, there is potential for endogenous psychotogens to be identified based on E-state analyses, but only experimental work will answer the question of whether or not any of these endogenous amines with low E-state N values are actually etiologic for psychotic symptoms.
Dysregulation of the folate cycle: neuroprotective strategies
Integrating the literature presented in the prior sections, there is clearly a strong argument for testing folate, B12 and homocysteine levels in FEP cases and evaluating neuroprotective options based on the results. Folate supplementation should not be initiated until B12 is well within normal range, and it should be accompanied by at least a modest level of B12 supplementation. Even in cases of low folate and high homocysteine, supplementation with folic acid, folinic acid, or 5-methyl-THF (a tertiary amine with a low E-state value for its N) should be done with care in an individual with psychosis or at ultra-high risk of psychosis when trying to mitigate positive symptoms. High doses of folic acid supplementation come with the mental health risks covered in previous sections, and as reviewed decades ago by Young and Ghadirian [288]. In a small clinical study, there is also new evidence to suggest that folic acid and folinic acid can either inhibit transport of 5-methyl-THF across the BBB [289], or diminish its synthesis. Consistent with this outcome, there is a more recent report that folinic acid acts as an inhibitor of MTHFR in lower organisms by changing the conformation of the active site [290], though no similar study of the mammalian enzyme has yet been conducted. Certainly, in a qualitative sense at least, there appears to be an inverted U-shaped curve for the benefit/risk ratio of folates, particularly if B12 is deficient or low-normal; thus, including B12 in any folate-treatment protocol would seem warranted.
For a more optimal and less risky route to lowering homocysteine, most clinicians seem to have gravitated towards the use of betaine (also known as trimethyl glycine) instead of folates to force methylation of homocysteine, which for betaine occurs via betaine-methyltransferase, an enzyme predominantly active in the periphery. Obeid and Herrmann [291] suggested that because betaine is effective in mitigating CNS consequences of high homocysteine levels, it is very likely that the transport mechanism for homocysteine identified by others in synaptosomes [292] contributes to its transport, while Büdy et al. [293] demonstrated that homocysteine is indeed transported by the large neutral amino-acid transport (LAT) proteins in endothelial membranes, and also present in the BBB, thereby explaining the efficacy of betaine for psychotic symptoms via its peripheral action. The advantage of betaine for an optimal outcome is that its methyl groups, unlike those generated by 5-methyl-THF, are not involved in the synthesis of epinephrine [32], N-methyldopamine (Figure 2) nor N-methyl-salsolinol [74].
In the unusual case of normal folate and high homocysteine, supplementation with pyridoxine should be considered, since a deficiency in the enzyme CBS could be the underlying etiologic factor, and pyridoxine is a cofactor which could assist in converting homocysteine to cystathionine.
With an MTHFR deficiency, it would be most informative to conduct “forward” physiologic assays [184, 185] to check for the impact of FAD supplementation. If FAD supplementation greatly increases the MTHFR activity, obviously administering riboflavin to the patient would be wise. Going one step further, investigating feedback inhibition of the enzyme by differing SAM and SAH levels would elucidate how the individual’s MTHFR responds to homeostatic levels of both. If feedback inhibition by SAM is missing, the potential for an untoward effect of excess folates would be greater.
Although methionine supplementation has been shown to be protective in B12 deficiency-induced neuropathy [138], its administration in cases of psychosis that are not necessarily associated with a B12-deficiency has proven to be risky [17, 153, 154]. Methionine supplementation certainly warrants consideration when methionine levels are subnormal and folate is normal, but care should be exercised to ensure the patients are not exposed to MAO inhibitors [150, 151] which include tricyclic antidepressants and the salsolinols as discussed previously. Levels of salsolinols in the CNS cannot be easily measured, but if worsening occurs with methionine supplementation, a lumbar puncture might be considered.
Disulfiram is a risky treatment for alcoholism because of the associated neuropathy and on occasion, psychosis. There is no easy neuroprotective approach to this outcome, other than immediately ceasing disulfiram should psychotic symptoms develop.
As an additional concern of note, BH4 has been reported to be lower in concentration in individuals with schizophrenia, and some may view this as a reason to explore supplementation with BH4. However, almost all of the prior research on the topic failed to adequately address the impact of neuroleptics on this biochemical. In point of fact, studies directly looking at that question have found lower concentrations of BH4 in human studies and in animal models of neuroleptic treatment [294, 295]. What can be seen in Figure 2 is that excess BH4 would be expected to increase catecholamine biosynthesis, which is not a metabolic shift expected to be helpful for the positive symptoms of psychosis. Although individuals with an inborn error of metabolism leading to severe BH4 deficiency can develop psychosis, as in the case report of Parikh and Panse [296], the patients are so quickly treated with L-DOPA that it is difficult to distinguish the effect of treatment from the effect of the genotype.
Metyrosine appears to work well for the psychosis associated with the 22q deletion syndrome, and has shown preliminary benefit in those in the clinically at high risk of psychosis group. But the potential for it to create or exacerbate negative symptoms is great, and possibly is best reserved for the subset of patients, similar to patients with a 22q deletion, who do not respond to other treatments.
Limitations:
Much of the research reviewed includes older publications on research conducted when the available biochemical and genetic methodology was somewhat limited. In addition, this review would have benefited from the existence of more animal model studies which followed up on the important results of van der Westhuyzen’s early publications on methylation cycle dysregulation [138, 142, 147]. The predilection of patients with psychosis to make poor dietary choices, though less likely to be a factor in FEP and clearly not a factor when treatment with a vitamin resolves the symptoms, may have facilitated a spurious association between measures of the folate-cycle and psychosis in some reports. Finally, comparing the ability of the electrotopological analyses to predict hallucinogenic potency to that of comprehensive structure activity relationship (SAR) analyses was beyond the scope of this review.
Conclusions
Transmethylation processes should be incorporated into any comprehensive, multi-component model of psychosis developed in the future. The implications of the methylation research covered here include the potential for mitigating the transition to psychosis in individuals with methylation cycle aberrations and the application of electrotopological analyses to predict psychotogenic effects of resulting endogenous amines. Although much can be done to rectify B-vitamin imbalances associated with dysregulation of methylation, there is also an opportunity to pharmacologically address potentially etiologic end-products of the dysregulation. Adrenochrome and the similar quinone metabolites of N-methylsalsolinol and N-methylnorsalsolinol represent easy targets for chemical polymerization reactions that would obscure problematic tertiary amine moieties and act as a natural sink for these potentially etiologic endogenous products [65]. The most promising strategy would be to select agents with minimal negative impact on CNS receptors or on enzymatic processes, thereby maximizing neuroprotection in FEP and potentially replacing pharmaceuticals with significant adverse side effects.
Abbreviations
| 5-FU: | 5-fluorouracil |
| BBB: | blood brain barrier |
| BH4: | tetrahydrobiopterin |
| CBS: | cystathionine-beta-synthase |
| COMT: | catechol-O-methyltransferase |
| DHFR: | dihydrofolate reductase |
| E-state: | electrotopological state |
| FEP: | first episode psychosis |
| HOMO: | highest occupied molecular orbital |
| MAO: | monoamine oxidase |
| MTHFR: | methylenetetrahydrofolate reductase |
| NMTs: | N-methyltransferases |
| PNMT: | phenylethanolamine N-methyltransferase |
| SAH: | S-adenosylhomocysteine |
| SAM: | S-adenosylmethionine |
| SNPs: | single-nucleotide-polymorphisms |
| THF: | tetrahydrofolate |
Declarations
Acknowledgments
I would like to extend my deep gratitude to Sebastian Colwell and to Spencer Pittman (Technologic Computer Services) for their assistance with computer program selection and performance.
Author contributions
CLM: Conceptualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The author has no conflict of interest to declare, has no commercial product related to this work, and holds no patents related to this work, nor are any under development at this time.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Where relevant, the site for availability of analytical tools is listed in the text.
Funding
Not applicable.
Copyright
© The Author(s) 2024.