
JOURNAL OF PSYCHIATRY AND NEUROLOGY
Myasthenia Gravis with Idiopathic Inflammatory Myopathy and Sjögren Syndrome, A Triad of Immune-Mediated Diseases
Rafael Cobilt-Catana*1, Lily Rivera-Figueroa1, Jorge Ortiz-Quezada2, Francisca Fernández-Valverde3, María Lizeth Zertuche-Ortuño4, Irene Treviño-Frenk5 and Paul David Uribe-Jaimes5
1Department of Neurology, Star Medica Hospital, Naucalpan, State of Mexico
2Department of Neurology, National Autonomous University of Honduras, Tegucigalpa, Honduras
3Department of Experimental Pathology, Manuel Velasco Suárez National Institute of Neurology and Neurosurgery, Mexico City
4Department of Neurology, Medical Hospital, Tlalpan, Mexico City
5Department of Neurology, Division of Neuroimmunology, ABC Medical Center, Mexico City
*Corresponding Author: Cobilt-Catana R, MD, Department of Neurology, Star Medical Hospital, Avenida Lomas Verdes 2165, Los Alamos, Naucalpan de Juarez, State of Mexico.
| ReceivedMar 1, 2023 | RevisedMar 21, 2023 | AcceptedMar 22, 2023 | PublishedMar 28, 2023 |
Highlights
- The occurrence of myasthenia gravis (MG), idiopathic inflammatory myopathy (IIM), and Sjögren syndrome (SS) may overlap owing to their immune-mediated etiology.
- IIM patients with diplopia, ptosis, fluctuating weakness, or severe weakness due to increased steroid doses may exhibit atypical symptoms that overlap with those of MG.
- MG patients with muscle pain, elevated creatine kinase (CK) may exhibit atypical symptoms that overlap with those of IIM.
- In SS patients with symptoms of weakness aggravated by the effect of steroids, MG should be considered, and in the case of elevated CK, IIM should be considered.
Abstract
This report illustrates a 73-year-old woman with Sjögren's syndrome and idiopathic inflammatory myopathy with elevated creatine kinase that subsequently deteriorated with an increased dose of steroids, resulting in a myasthenic crisis requiring hospitalization in the intensive care unit; the patient responded successfully to treatment with intravenous immunoglobulin. The patient showed moderate elevation of creatine kinase at the initial diagnosis, with positive anti-Ro/SSA antibodies. Schirmer’s test, anti-Ro52 and anti-Ku antibodies, electromyography, and muscle biopsy were performed to confirm inflammatory myopathy. During the observation in the intensive care unit, creatine kinase increased to 1520 U/L, and a repetitive stimulation test and total anti-acetylcholine receptor antibodies supported the diagnosis of myasthenia gravis. The patient received intravenous immunoglobulin, evolving with clinical improvement. This case is being reported to illustrate the chronic clinical course of a patient with inflammatory myopathy who deteriorated following steroid adjustment, ultimately presenting with myasthenic crisis.
Keywords
Myasthenia gravis; Inflammatory myopathy; Sjögren's syndrome; Immune-mediated Disease; Antibody
Introduction
Myasthenia gravis (MG) is an autoimmune disease caused by antibodies directed against acetylcholine receptors or related molecules in the postsynaptic membrane at the neuromuscular junction, with the main characteristic symptoms of generalized or localized weakness of a fluctuating type that resolves with rest [1]. Idiopathic inflammatory myopathy (IIM) is a group of autoimmune diseases characterized by proximal weakness and elevation in creatine kinase (CK) and antibody levels that can be associated with different clinical phenotypes [2,3].
Polymyositis is characterized by the absence of typical skin lesions associated with dermatomyositis. Sjögren's syndrome (SS) is a chronic autoimmune disease accompanied by multiple lesions with main clinical manifestations, including dry mouth and eyes, together with systemic complications in various organs [4]. To date, there have been no reported instances of co-occurrence of the three diseases; however, a dual association between them has been reported [5], and some authors recommend that when such diseases coexist, more aggressive immunomodulatory treatments should be initiated. Herein, we present a case with three types of pathologies of immune-mediated origin: MG, IIM, and SS.
Case Report
A 73-year-old female patient had been experiencing pain for 5 years and decreased strength in the four extremities, predominantly proximal and symmetrical. The patient mainly felt pain when trying to lift heavy objects and raising the arms above the head, with fine tremors when picking up objects. The patient complained of tiredness during the day and experienced conjunctival foreign body sensation, tension-type headaches, and intermittent insomnia. The patient also lost 10kg in 5 years prior to diagnosis. The patient was evaluated by rheumatology, and diabetes mellitus, thyroid, liver, kidney disorders, and other infections were ruled out. Laboratory findings showed CK 550U/L, negative ToRCH, rheumatoid factor 40U/mL, erythrocyte sedimentation rate 49mm/h, C-reactive protein 1mg/dL, antinuclear antibody 1:80, positive anti-Ro/SSA, negative anti-Li/SSB, anti-dsDNA 5IU/mL, negative anti-Smith, anti-β2M blood 1.5mg/L, normal C3 and C4, negative lupus anticoagulant, negative anticardiolipin, negative pANCA and cANCA, normal electrocardiogram and chest X-ray, and positive Schirmer’s test with 4mm in the left eye and 5mm in the right eye. No salivary gland biopsy was performed. Antibodies to anti-Mi2 alpha, anti-M2 beta, anti-TIF1g, anti-MDA5, anti-NXP2, anti-SAE1, anti-Jo1, anti-PM-Scl100, anti-PM-Scl75, anti-SRP, anti-PL7, anti-PL12, anti-EJ, and anti-OJ were not detected; anti-Ro52 (++) and anti-Ku (+++) were positive. EMG and NCS showed fibrillations, positive sharp waves, and myopathic motor unit potentials with reduced amplitude, duration, and polyphasic potentials with early recruitment in the proximal muscles (deltoids and gluteals). A muscle biopsy was performed, and the tissues were analyzed using a cryoprecipitate technique; tissues were cut 9nm thick using a cryostat. Hematoxylin and eosin staining showed muscle fiber necrosis, abundant endomysial inflammatory cells around regeneration fibers, vesicular and internal nuclei, and characteristic extensive vacuolar degeneration (Figure 1A). Modified Gomori trichrome stain showed pale necrotic fibers around endomysial inflammatory cells and some angular shape atrophic fibers with regeneration fibers (Figure 1B).
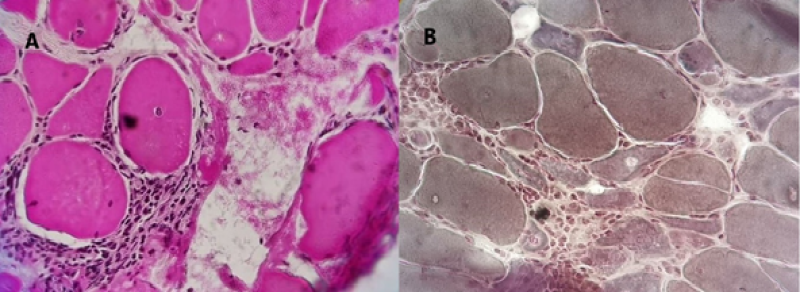
Figure 1: A: Hematoxylin and eosin staining of muscle biopsy-Muscle fiber necrosis and abundant endomysial inflammatory cells (black star) around two regeneration fibers (white star), one with vesicular and internal nuclei (black arrow) and one with vacuolar degeneration extensive (white arrow). B: Modified Gomori trichrome-Pale necrotic fibers (green arrow), endomysial inflammatory cells (black star), and some atrophic fibers of angular shape (blue star) with regeneration fibers (white star) and dark stained hypercontractile fibers (red arrow).
The patient was diagnosed with SS and IIM and treated with ocular lubricants and prednisone 10mg/day for a month. The patient’s condition did not improve; the patient experienced increased proximal pain in the extremities, fatigue that required three naps a day lasting for approximately 1 or 2h, with a slight recovery of muscle strength upon awakening, and decreased strength of the neck flexor muscles; hence, the dose of prednisone was increased to 1mg/kg/day. After a week, the patient presented greater deterioration in strength, dysphagia, respiratory distress, ptosis, and diplopia. The patient was evaluated in the emergency department, and endotracheal orointubation was performed. The patient was transferred to the intensive care unit and was approached by a neurologist owing to the clinical suspicion of a myasthenic crisis. Blood analysis revealed positive anti-AChR antibodies of 1.25 (normal<0.45) and CK of 1,250U/L. Jolly test at low frequency showed electrodecrement response (>10%) in the orbicularis oculi and nasal muscles. Simple and contrast-enhanced chest computed tomography was performed to rule out neoplastic or adnexal masses. The patient was treated with intravenous immune globulin (IVIG) at 0.4g/kg/day for 5 days, which resulted in the improvement of the airway in 1 week. The patient was discharged 2 weeks later with pyridostigmine, azathioprine, and prednisone.
Fifteen days post hospital discharge, the patient was clinically evaluated for cognitive function without alterations, preserved photomotor and consensual reflexes, fundus without edema or papillary atrophy, normal macula, eye movements without limitations, adequate facial sensitivity to thermoalgesic stimuli without facial palsy, preserved oropharyngeal muscle strength, proximal muscle strength of all four limbs, neck and knee flexion of 4/5, distal muscle strength of 5/5, muscle tone preserved, muscle stretch reflexes grade 2, normal sensation, gait, and cerebellar functions. The Quantitative Myasthenia Gravis Score was also measured, resulting in a score of 10 points. During the evaluation, an ice test was performed, which was positive.
Discussion
In this case, the evolution of a patient with SS and IIM who developed myasthenic crisis due to the effect of increased doses of steroids was analyzed. This triple association was not found in the literature; instead, it has been reported that each of these immune-mediated pathologies, such as SS and IIM, predominantly polymyositis, is difficult to diagnose [5,6]. The probability range of the inflammatory myopathy subtype using an online calculator was determined [7]; a score of 10.7 of the EULAR/ACR criteria was obtained [3]. Probability of 99% for the subclassification of polymyositis type, anti-Ro/SSA, anti-Ro52 (++), and anti-Ku (+++) antibodies were also obtained, which are associated with IIM and SS [1]. The treatment recommendations for both entities are high-dose steroids [6], mainly when they coexist. The patient received prednisone at 1mg/kg/day after a week of myasthenic crisis, requiring intensive care.
Conversely, the association between SS and MG, mainly in the early stage, has been reported. MG was reported before the diagnosis of SS, with initial bulbar symptoms of MG and predominantly positive anti-Ro/SSA antibodies, as in the case presented, and the presence of constitutional symptoms should be considered significant, as it may be associated with thymoma and mediastinal tumor masses [8-10]. In this case, there was no thymoma or mediastinal mass.
An association between IIM and MG with elevated CK levels have been reported. In cases where the initial diagnosis was MG, patients presented with typical MG symptoms, followed by myopathy, with a predominant association with polymyositis [11,12]. Thymoma has been associated with rhabdomyolysis-like features, respiratory failure, and dropped head; however, cardiac involvement and ocular symptoms are more commonly observed [13]. Nonetheless, the association between MG and IIM without thymic pathology occurs in 45% of cases [14,15]. In this case, the patient died owing to myasthenic crisis characterized by bulbar and ocular symptoms, such as dropped head syndrome, dysphagia, respiratory failure, ptosis, and diplopia. The patient received IVIG as recommended, considering the etiology to be immune-mediated [16].
Conclusions
Currently, MG is rarely associated with IIM and less frequently with SS because of the overlapping and diversity of clinical symptoms; hence, differentiating them during the clinical evolution of the patient is crucial, as diagnosis can be difficult and management can be erroneous.
Distinguishing atypical symptoms of these three diseases will enable detection of the possible superposition of these pathologies in a timely manner; for example, atypical symptoms of MG, IIM, and SS may overlap owing to their immune-mediated etiology. In IIM, symptoms such as diplopia, ptosis, fluctuating weakness, respiratory impairment, or severe weakness (due to increased doses of steroids), overlapping with MG should be considered. If there are atypical symptoms of MG with muscle pain and elevated CK, overlapping of MG with IIM should be considered. In SS, symptoms of weakness aggravated by the effect of steroids should raise suspicion for MG, while elevated levels of transaminases and CK should prompt consideration of IIM. In contrast, since there are constitutional symptoms in MG and MII without dysphagia or thymoma, we recommend ruling out other rheumatological and paraneoplastic diseases or completely ruling out infectious diseases. Therefore, close observation of the patient during their clinical evolution to detect unusual signs and symptoms, as well as the response to the drugs administered, may allow early detection of the coexistence of immune-mediated pathologies.
Funding statement
This study did not receive any specific grants from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of interests
None.
Acknowledgments
The author would like to thank “La Salle University” for providing permission to pursue the postgraduate course in Neuroimmunology.
References
1. Gilhus NE. N Engl J Med. 2016;375:2570-81. PubMed | CrossRef
2. Clark KE, Isenberg DA. A Review of Inflammatory Idiopathic Myopathy Focusing on Polymyositis. Eur J Neurol. 2018;25(1):13-23. PubMed | CrossRef
3. Bottai M, Tjärnlund A, Santoni G, Werth VP, Pilkington C, De Visser M, et al. EULAR/ACR Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and their Major Subgroups: A Methodology Report. RMD Open. 2017;3(2):e000507. PubMed | CrossRef
4. Zhan Q, Zhang J, Lin Y, Chen W, Fan X, Zhang D. Pathogenesis and Treatment of Sjogren’s Syndrome: Review and Update. Front Immunol. 2023;14. PubMed | CrossRef
5. Khosa S, Hovsepian DA, Khosa GS, Catherine Y, Trikamji B, Mishra SK. A Case of Sjögren's Syndrome Mimicking Inflammatory Myopathy. Cureus. 2018;10(10). PubMed | CrossRef
6. Koga T, Kouhisa Y, Nakamura H, Mizokami A, Motomura M, Kawakami A, et al. A Case of Primary Sjögren’s Syndrome Complicated with Inflammatory Myopathy and Interstitial Lung Disease. Rheumatol Int. 2012;32:3647-9. PubMed | CrossRef
7. EULAR/ACR Classification Criteria; Classification Criteria for Idiopathic Inflammatory Myopathies. 2023.
8. Hartert M, Melcher B, Huertgen M. Association of Early-Onset Myasthenia Gravis and Primary Sjögren’s Syndrome: A Case-Based Narrative Review. Clin Rheumatol. 2022;41(10):3237-43. PubMed | CrossRef
9. Li X, Zhao Y, Liao Q, Da Y. Myasthenia Gravis Coexisting with Primary Sjögren's Syndrome: Report of Three Cases and Literature Review. Front Neurol. 2020;11:939. PubMed | CrossRef
10. Fujiu K, Kanno R, Shio Y, Ohsugi J, Nozawa Y, Gotoh M. Triad of Thymoma, Myasthenia Gravis and Pure Red Cell Aplasia Combined with Sjögren’s Syndrome. JPN J Thorac Cardiovasc Surg. 2004;52:345-8. PubMed | CrossRef
11. Kanbayashi T, Tanaka S, Hatanaka Y, Uchio N, Shimizu J, Sonoo M. Myasthenia Gravis with Inflammatory Myopathy without Elevation of Creatine Kinase. Neuromuscul Disord. 2021;31(6):570-3. PubMed | CrossRef
12. Alessandro L, Kohler AA, Calandri IL, Taratuto A, Cammarota A, Barroso F. Overlap Syndrome between Myasthenia Gravis and Idiopathic Inflammatory Myopathies. Rev Neurol. 2016;62(11):526-7. PubMed | CrossRef
13. Uchio N, Taira K, Ikenaga C, Kadoya M, Unuma A, Yoshida K, et al. Inflammatory Myopathy with Myasthenia Gravis: Thymoma Association and Polymyositis Pathology. Neurol Neuroimmunol Neuroinflamm. 2019;6(2). PubMed | CrossRef
14. Huang K, Shojania K, Chapman K, Amiri N, Dehghan N, Mezei M. Concurrent Inflammatory Myopathy and Myasthenia Gravis with or without Thymic Pathology: A Case Series and Literature Review. Semin Arthritis Rheum. 2019;48(4):745-751. PubMed | CrossRef
15. Santos E, Coutinho E, Silva AM, Marinho A, Vasconcelos C, Taipa R, et al. Inflammatory Myopathy Associated with Myasthenia Gravis with and without Thymic Pathology: Report of Four Cases and Literature Review. Autoimmun Rev. 2017;16(6):644-9. PubMed | CrossRef
16. Illa I. IVIg in Myasthenia Gravis, Lambert Eaton Myasthenic Syndrome and Inflammatory Myopathies: Current Status. J Neurol. 2005;252:i14-8. PubMed | CrossRef
Rafael Cobilt-Catana*1, Lily Rivera-Figueroa1, Jorge Ortiz-Quezada2, Francisca Fernández-Valverde3, María Lizeth Zertuche-Ortuño4, Irene Treviño-Frenk5 and Paul David Uribe-Jaimes5
1Department of Neurology, Star Medica Hospital, Naucalpan, State of Mexico
2Department of Neurology, National Autonomous University of Honduras, Tegucigalpa, Honduras
3Department of Experimental Pathology, Manuel Velasco Suárez National Institute of Neurology and Neurosurgery, Mexico City
4Department of Neurology, Medical Hospital, Tlalpan, Mexico City
5Department of Neurology, Division of Neuroimmunology, ABC Medical Center, Mexico City
*Corresponding Author: Cobilt-Catana R, MD, Department of Neurology, Star Medical Hospital, Avenida Lomas Verdes 2165, Los Alamos, Naucalpan de Juarez, State of Mexico.
Copyright© 2023 by Cobilt-Catana R, et al. All rights reserved. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Cobilt-Catana R, Rivera-Figueroa L, Ortiz-Quezada J, Fernández-Valverde F, Zertuche-Ortuño ML, Trevino-Frenk I, et al. Myasthenia Gravis with Idiopathic Inflammatory Myopathy and Sjögren Syndrome, A Triad of Immune-Mediated Diseases. J Psy Neurol. 2023;1(1):21-26. DOI: https://doi.org/10.37191/Mapsci-JPN-1(1)-003