
A Customizable Suite of Methods to Sequence and Annotate Cattle Antibodies
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cattle Peripheral Mononuclear Cell (PBMC) Preparation
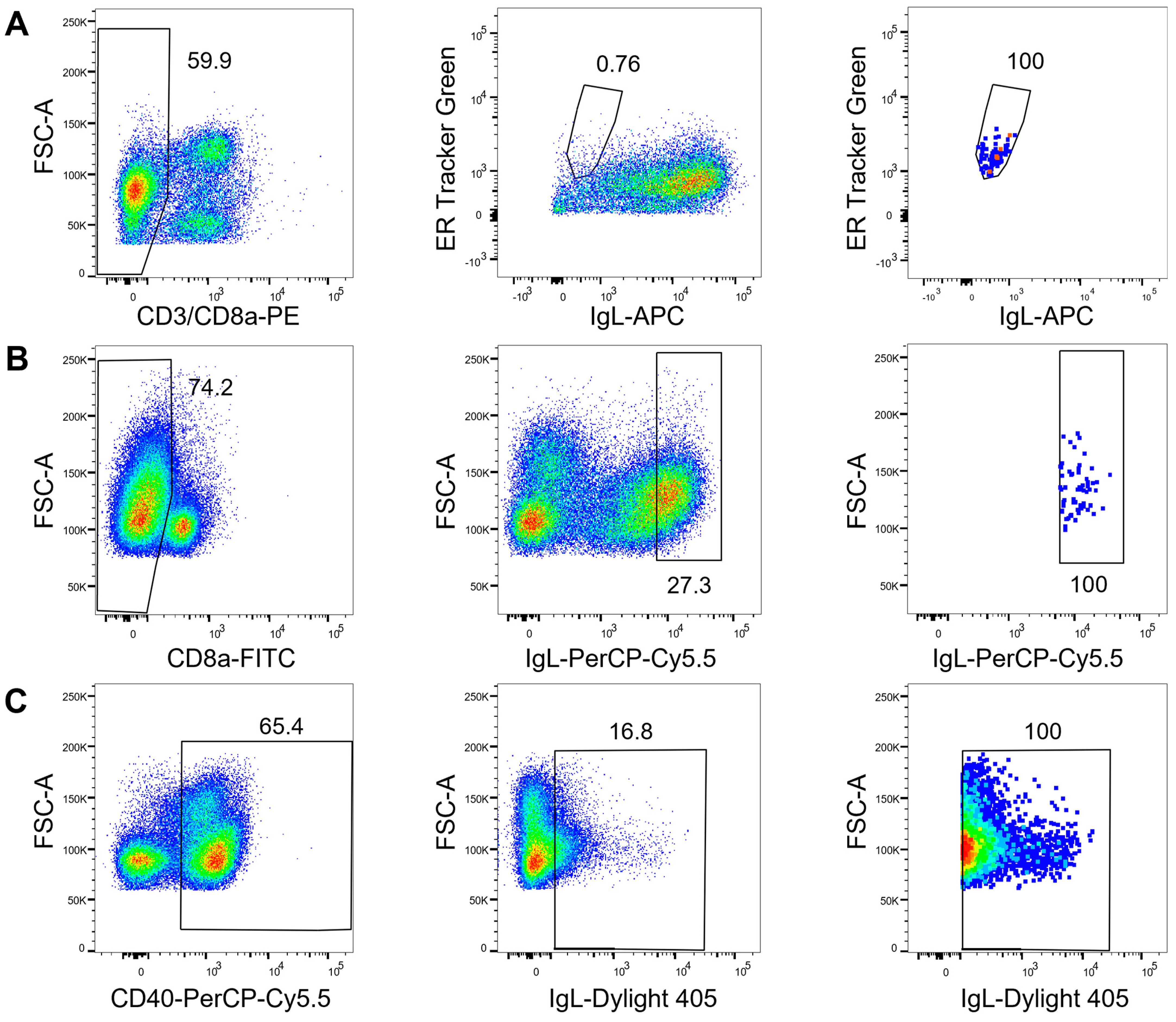
2.2. Cell Staining and Sorting
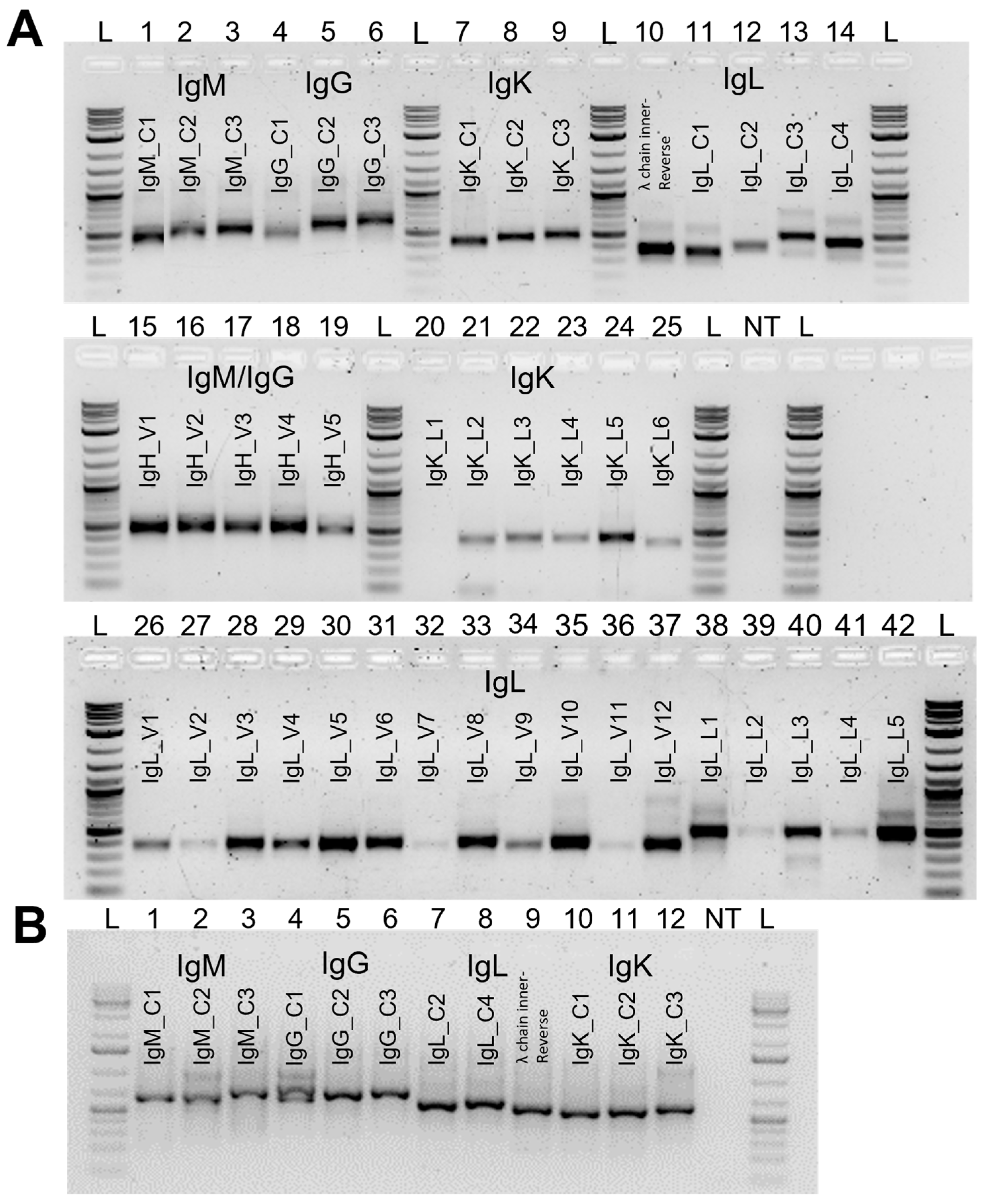
2.3. Primer Design
2.4. Immunoglobulin Gene Amplification and Library Preparation
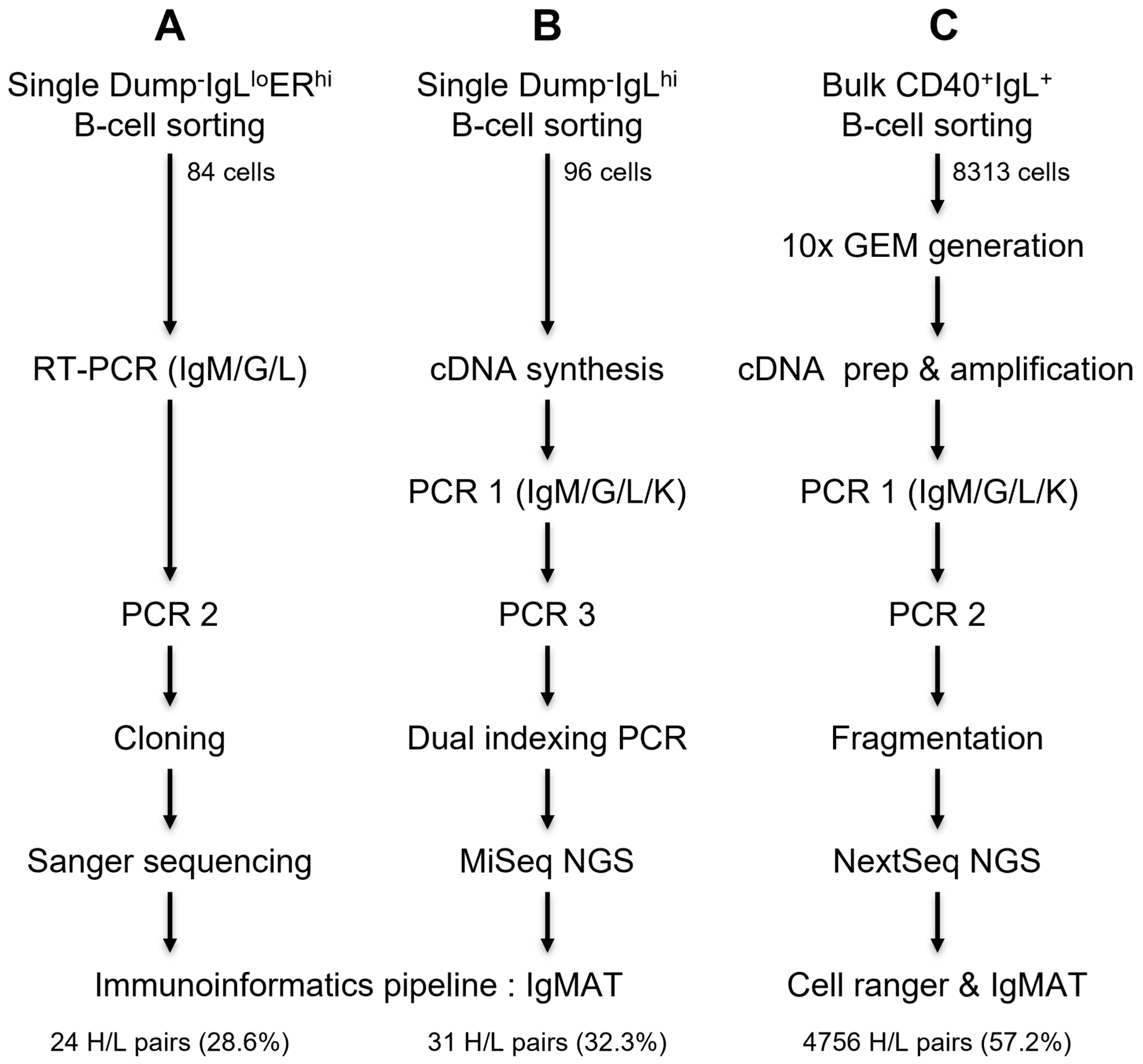
2.4.1. Strategy 1: PCR Amplification and Cloning Method Used in the Low-Throughput Workflow
2.4.2. Strategy 2: PCR Amplification and Indexing Method Used in the Medium-Throughput Workflow
2.4.3. Strategy 3: PCR Amplification Method Used in the High-Throughput Workflow
2.5. Sequencing
2.6. Bioinformatic Analysis
3. Results and Discussion
3.1. Flow Cytometry
3.2. Primer Design and Validation
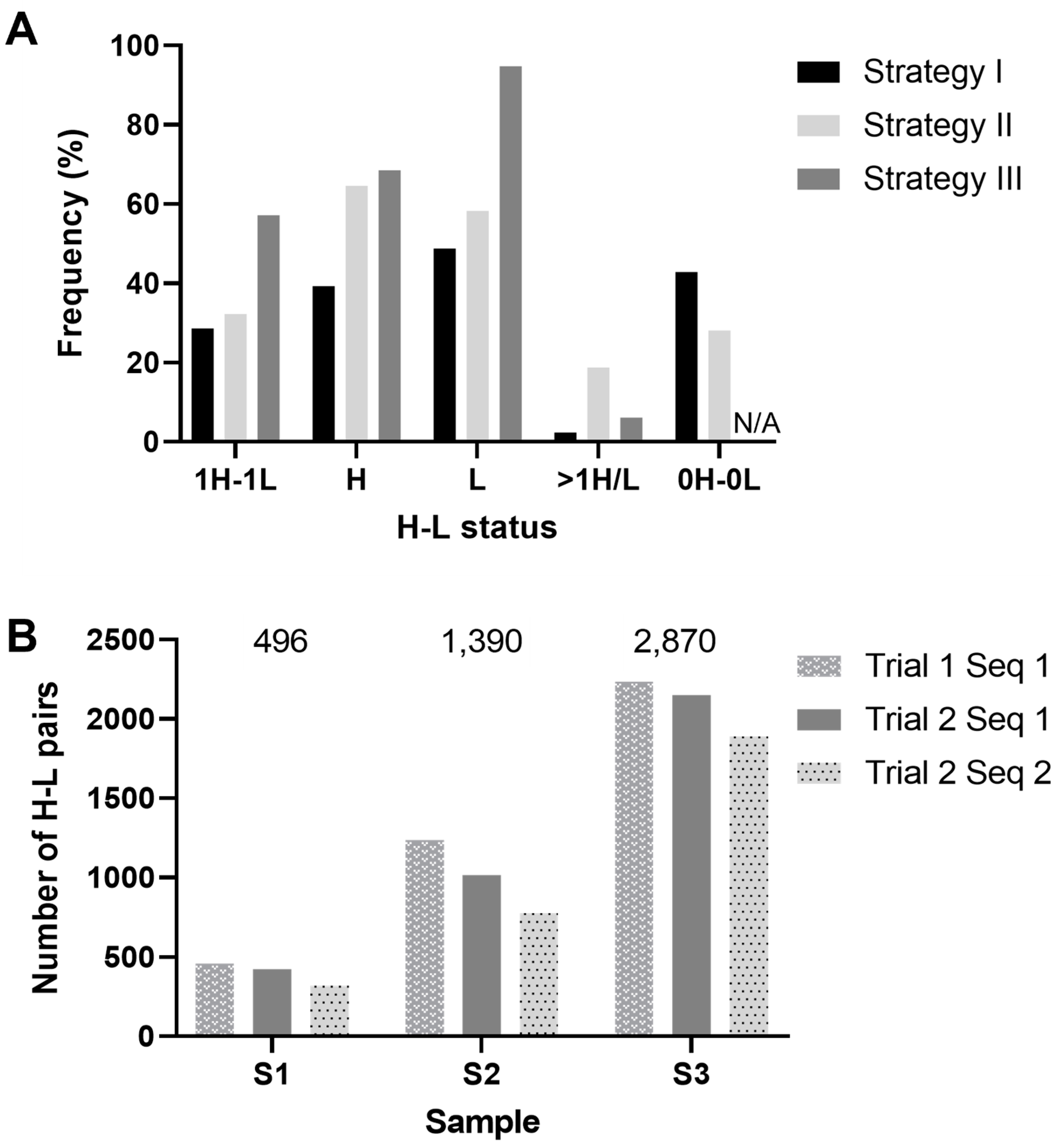
3.3. PCR Strategy and Sequence Analysis
3.3.1. Strategy 1: Antibody Amplification and Sequencing in a Low-Throughput Workflow
3.3.2. Strategy 2: Antibody Amplification and Sequencing in a Medium-Throughput Workflow
3.3.3. Strategy 3: Antibody Amplification and Sequencing in a High-Throughput Workflow
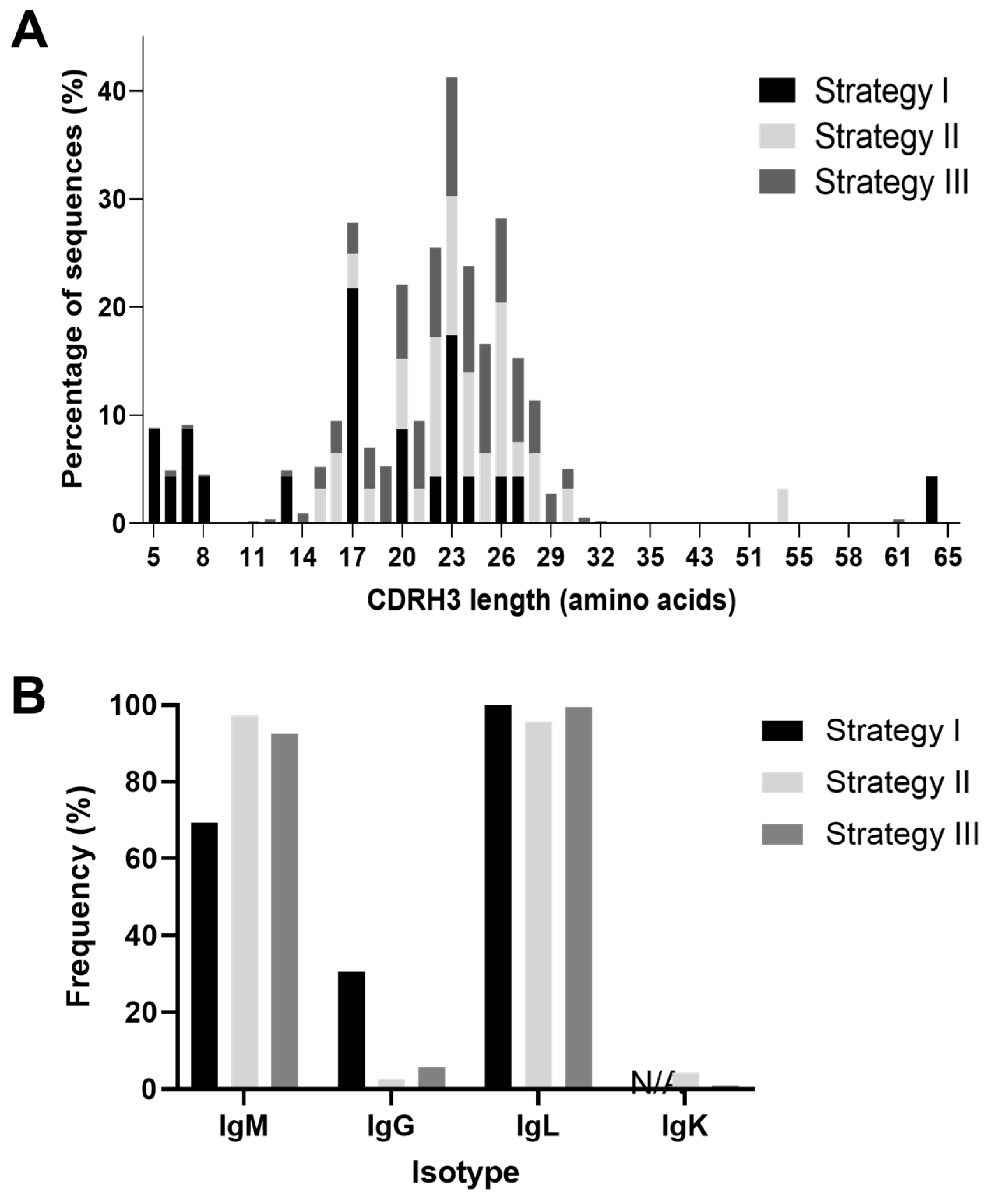
3.4. Cattle Antibody Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef] [Green Version]
- Bekker, L.-G.; Tatoud, R.; Dabis, F.; Feinberg, M.; Kaleebu, P.; Marovich, M.; Ndung’u, T.; Russell, N.; Johnson, J.; Luba, M.; et al. The complex challenges of HIV vaccine development require renewed and expanded global commitment. Lancet 2020, 395, 5. [Google Scholar] [CrossRef]
- Hatherill, M.; White, R.G.; Hawn, T.R. Clinical Development of New TB Vaccines: Recent Advances and Next Steps. Front. Microbiol. 2019, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Matz, K.M.; Marzi, A.; Feldmann, H. Ebola vaccine trials: Progress in vaccine safety and immunogenicity. Expert. Rev. Vaccines 2019, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Antos, A.; Miroslaw, P.; Rola, J.; Polak, M.P. Vaccination Failure in Eradication and Control Programs for Bovine Viral Diarrhea Infection. Front. Vet. Sci. 2021, 8, 11. [Google Scholar] [CrossRef]
- Belsham, G.J. Towards improvements in foot-and-mouth disease vaccine performance. Acta Vet. Scand. 2020, 62, 20. [Google Scholar] [CrossRef] [PubMed]
- Black, S.J.; Mansfield, J.M. Prospects for vaccination against pathogenic African trypanosomes. Parasite Immunol. 2016, 38, 9. [Google Scholar] [CrossRef]
- Guzman, E.; Taylor, G. Immunology of bovine respiratory syncytial virus in calves. Mol. Immunol. 2015, 66, 9. [Google Scholar] [CrossRef] [PubMed]
- Cancro, M.P.; Tomayko, M.M. Memory B cells and plasma cells: The differentiative continuum of humoral immunity. Immunol. Rev. 2021, 303, 11. [Google Scholar] [CrossRef]
- Stanfield, R.L.; Berndsen, Z.T.; Huang, R.; Sok, D.; Warner, G.; Torres, J.L.; Burton, D.R.; Ward, A.B.; Wilson, I.A.; Smider, V.V. Structural basis of broad HIV neutralization by a vaccine-induced cow antibody. Sci. Adv. 2020, 6, 12. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Mizuguchi, K. The diversity of H3 loops determines the antigen-binding tendencies of antibody CDR loops. Protein Sci. 2016, 25, 11. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.L.; Davis, M.M. Diversity in the CDR3 Region of VH Is Sufficient for Most Antibody Specificities. Immunity 2000, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Qin, T.; Chu, D.; Cheng, X.; Wang, J.; Wang, X.; Wang, P.; Han, H.; Ren, L.; Aitken, R.; et al. Internal Duplications of DH, JH, and C Region Genes Create an Unusual IgH Gene Locus in Cattle. J. Immunol. 2016, 196, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jackson, S.M.; Aitken, R. The bovine antibody repertoire. Dev. Comp. Immunol. 2006, 30, 12. [Google Scholar] [CrossRef] [PubMed]
- Oyola, S.O.; Henson, S.P.; Nzau, B.; Kibwana, E.; Nene, V. Access to ultra-long IgG CDRH3 bovine antibody sequences using short read sequencing technology. Mol. Immunol. 2021, 139, 9. [Google Scholar] [CrossRef] [PubMed]
- Deiss, T.C.; Vadnais, M.; Wang, F.; Chen, P.L.; Torkamani, A.; Mwangi, W.; Lefranc, M.-P.; Criscitiello, M.F.; Smider, V.V. Immunogenetic factors driving formation of ultralong VH CDR3 in Bos taurus antibodies. Cell. Mol. Immunol. 2019, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Smider, B.A.; Smider, V.V. Formation of ultralong DH regions through genomic rearrangement. BMC Immunol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Wang, F.; Ekiert, D.C.; Ahmad, I.; Yu, W.; Zhang, Y.; Bazirgan, O.; Torkamani, A.; Raudsepp, T.; Mwangi, W.; Criscitiello, M.F.; et al. Reshaping antibody diversity. Cell 2013, 153, 15. [Google Scholar] [CrossRef] [Green Version]
- Burke, M.J.; Stockley, P.G.; Boyes, J. Broadly Neutralizing Bovine Antibodies: Highly Effective New Tools against Evasive Pathogens? Viruses 2020, 12, 473. [Google Scholar] [CrossRef] [Green Version]
- Arun, S.S.; Breuer, W.; Hermanns, W. Immunohistochemical Examination of Light-chain Expression (Λ/k Ratio) in Canine, Feline, Equine, Bovine and Porcine Plasma Cells. J. Vet. Med. Ser. A 1996, 43, 4. [Google Scholar] [CrossRef]
- Barroso, R.; Morrison, W.I.; Morrison, L.J. Molecular Dissection of the Antibody Response: Opportunities and Needs for Application in Cattle. Front. Immunol. 2020, 11, 10. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 3. [Google Scholar] [CrossRef]
- Steinitz, M.; Klein, G.; Koskimies, S.; Makel, O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature 1977, 269, 3. [Google Scholar] [CrossRef]
- Smith, S.A.; Crowe, J.E., Jr. Use of Human Hybridoma Technology To Isolate Human Monoclonal Antibodies. Microbiol. Spectr. 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Crain, M.J.; Sanders, S.K.; Butler, J.L.; Cooper, M.D. Epstein-Barr virus preferentially induces proliferation of primed B cells. J. Immunol. 1989, 143, 6. [Google Scholar] [CrossRef]
- Borrebaeck, C.A.; Danielsson, L.; Möller, S.A. Human monoclonal antibodies produced by primary in vitro immunization of peripheral blood lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groves, D.J.; Tucker, E.M. The production and application of non-rodent monoclonal antibodies in veterinary science. Vet. Immunol. Immunopathol. 1989, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 5. [Google Scholar] [CrossRef] [PubMed]
- Tiller, T.; Meffre, E.; Yurasov, S.; Tsuiji, M.; Nussenzweig, M.C.; Wardemann, H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 2008, 329, 13. [Google Scholar] [CrossRef] [Green Version]
- Neu, K.E.; Guthmiller, J.J.; Huang, M.; La, J.; Vieira, M.C.; Kim, K.; Zheng, N.Y.; Cortese, M.; Tepora, M.E.; Hamel, N.J.; et al. Spec-seq unveils transcriptional subpopulations of antibody-secreting cells following influenza vaccination. J. Clin. Investig. 2019, 129, 13. [Google Scholar] [CrossRef]
- Li, K.; Wang, S.; Cao, Y.; Bao, H.; Li, P.; Sun, P.; Bai, X.; Fu, Y.; Ma, X.; Zhang, J.; et al. Development of Foot-and-Mouth Disease Virus-Neutralizing Monoclonal Antibodies Derived From Plasmablasts of Infected Cattle and Their Germline Gene Usage. Front. Immunol. 2020, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 16. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Wang, L.; Ding, Y.L.; Xie, Q.; Diao, H.Y. Current status and recent advances of next generation sequencing techniques in immunological repertoire. Genes Immun. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Chen, Y.J.; Wu, J.; Chaudhuri, S.; Hsiao, Y.C.; Schneider, K.; Hoi, K.H.; Lin, Z.; Guerrero, S.; Jaiswal, B.S.; et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun. Biol. 2019, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, J.R.; DeKosky, B.J.; Tanno, H.; Ellington, A.D.; Georgiou, G. Ultra-high-throughput sequencing of the immune receptor repertoire from millions of lymphocytes. Nat. Protoc. 2016, 11, 14. [Google Scholar] [CrossRef]
- DeKosky, B.J.; Kojima, T.; Rodin, A.; Charab, W.; Ippolito, G.C.; Ellington, A.D.; Georgiou, G. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat. Med. 2015, 21, 6. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, B.J.; Ippolito, G.C.; Deschner, R.P.; Lavinder, J.J.; Wine, Y.; Rawlings, B.M.; Varadarajan, N.; Giesecke, C.; Dörner, T.; Andrews, S.F.; et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 2013, 31, 4. [Google Scholar] [CrossRef] [Green Version]
- Saikia, M.; Burnham, P.; Keshavjee, S.H.; Wang, M.F.Z.; Heyang, M.; Moral-Lopez, P.; Hinchman, M.M.; Danko, C.G.; Parker, J.S.L.; De Vlaminck, I. Simultaneous multiplexed amplicon sequencing and transcriptome profiling in single cells. Nat. Methods 2019, 16, 4. [Google Scholar] [CrossRef]
- Wang, B.; DeKosky, B.J.; Timm, M.R.; Lee, J.; Normandin, E.; Misasi, J.; Kong, R.; McDaniel, J.R.; Delidakis, G.; Leigh, K.E.; et al. Functional interrogation and mining of natively paired human V(H):V(L) antibody repertoires. Nat. Biotechnol. 2018, 36, 4. [Google Scholar] [CrossRef]
- Rajan, S.; Kierny, M.R.; Mercer, A.; Wu, J.; Tovchigrechko, A.; Wu, H.; Dall Acqua, W.F.; Xiao, X.; Chowdhury, P.S. Recombinant human B cell repertoires enable screening for rare, specific, and natively paired antibodies. Commun. Biol. 2018, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- DeKosky, B.J.; Lungu, O.I.; Park, D.; Johnson, E.L.; Charab, W.; Chrysostomou, C.; Kuroda, D.; Ellington, A.D.; Ippolito, G.C.; Gray, J.J.; et al. Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires. Proc. Natl. Acad. Sci. USA 2016, 113, 10. [Google Scholar] [CrossRef] [Green Version]
- Asensio, M.A.; Lim, Y.W.; Wayham, N.; Stadtmiller, K.; Edgar, R.C.; Leong, J.; Leong, R.; Mizrahi, R.A.; Adams, M.S.; Simons, J.F.; et al. Antibody repertoire analysis of mouse immunization protocols using microfluidics and molecular genomics. MAbs 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; Mizrahi, R.A.; Spindler, M.J.; Adams, M.S.; Asensio, M.A.; Edgar, R.C.; Leong, J.; Leong, R.; Johnson, D.S. Rare, high-affinity mouse anti-PD-1 antibodies that function in checkpoint blockade, discovered using microfluidics and molecular genomics. MAbs 2017, 9, 12. [Google Scholar] [CrossRef]
- Adler, A.S.; Bedinger, D.; Adams, M.S.; Asensio, M.A.; Edgar, R.C.; Leong, R.; Leong, J.; Mizrahi, R.A.; Spindler, M.J.; Bandi, S.R.; et al. A natively paired antibody library yields drug leads with higher sensitivity and specificity than a randomly paired antibody library. MAbs 2018, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Jayaram, N.; Bhowmick, P.; Martin, A.C. Germline VH/VL pairing in antibodies. Protein Eng. Des. Sel. 2012, 25, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Nettleship, J.E.; Harris, G.; Mwangi, W.; Rhaman, N.; Grant, C.; Kotecha, A.; Fry, E.; Charleston, B.; Stuart, D.I.; et al. The role of the light chain in the structure and binding activity of two cattle antibodies that neutralize bovine respiratory syncytial virus. Mol. Immunol. 2019, 112, 8. [Google Scholar] [CrossRef] [PubMed]
- Curtis, N.C.; Lee, J. Beyond bulk single-chain sequencing: Getting at the whole receptor. Curr. Opin. Syst. Biol. 2020, 24, 7. [Google Scholar] [CrossRef]
- Smakaj, E.; Babrak, L.; Ohlin, M.; Shugay, M.; Briney, B.; Tosoni, D.; Galli, C.; Grobelsek, V.; D’Angelo, I.; Olson, B.; et al. Benchmarking immunoinformatic tools for the analysis of antibody repertoire sequences. Bioinformatics 2019, 36, 9. [Google Scholar] [CrossRef] [Green Version]
- Ehling, R.A.; Weber, C.R.; Mason, D.M.; Friedensohn, S.; Wagner, B.; Bieberich, F.; Kapetanovic, E.; Vazquez-Lombardi, R.; Di Roberto, R.l.B.; Hong, K.-L.; et al. SARS-CoV-2 reactive and neutralizing antibodies discovered by single-cell sequencing of plasma cells and mammalian display. Cell. Rep. 2022, 38, 21. [Google Scholar] [CrossRef]
- Qi, H.; Liu, B.; Wang, X.; Zhang, L. The humoral response and antibodies against SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 13. [Google Scholar] [CrossRef]
- Dorey-Robinson, D.; Maccari, G.; Borne, R.; Hammond, J.A. IgMAT: Immunoglobulin sequence multi-species annotation tool for any species including those with incomplete antibody annotation or unusual characteristics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mwangi, W.; Maccari, G.; Hope, J.C.; Entrican, G.; Hammond, J.A. The UK Veterinary Immunological Toolbox Website: Promoting vaccine research by facilitating communication and removing reagent barriers. Immunology 2020, 161, 3. [Google Scholar] [CrossRef]
- Roos, E.O.; Bonnet-Di Placido, M.; Mwangi, W.N.; Moffat, K.; Fry, L.M.; Waters, R.; Hammond, J.A. OMIP-085: Cattle B-cell phenotyping by an 8-color panel. Cytom. Part. A 2023, 103, 4. [Google Scholar] [CrossRef]
- Wang, L.; Madera, R.; Li, Y.; Gladue, D.P.; Borca, M.V.; McIntosh, M.T.; Shi, J. Development of Porcine Monoclonal Antibodies with In Vitro Neutralizing Activity against Classical Swine Fever Virus from C-Strain E2-Specific Single B Cells. Viruses 2023, 15, 863. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.F.; Lefevre, E.A.; Carr, B.V.; Prentice, H.; Gubbins, S.; Pollard, A.J.; Charreyre, C.; Charleston, B. Assessment of T-dependent and T-independent immune responses in cattle using a B cell ELISPOT assay. Vet. Res. 2012, 43, 68. [Google Scholar] [CrossRef] [Green Version]
- Holzer, B.; Rijal, P.; McNee, A.; Paudyal, B.; Martini, V.; Clark, B.; Manjegowda, T.; Salguero, F.J.; Bessell, E.; Schwartz, J.C.; et al. Protective porcine influenza virus-specific monoclonal antibodies recognize similar haemagglutinin epitopes as humans. PLoS Pathog. 2021, 17, 25. [Google Scholar] [CrossRef]
- 10x_Genomics. Chromium Next GEM Single Cell V(D)J Reagent Kits v1.1 User Guide. Available online: https://support.10xgenomics.com/single-cell-vdj/library-prep/doc/user-guide-chromium-single-cell-vdj-reagent-kits-v11-chemistry (accessed on 12 October 2020).
- Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; Guigó, R.; et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 7. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.C.; Philp, R.L.; Bickhart, D.M.; Smith, T.P.L.; Hammond, J.A. The antibody loci of the domestic goat (Capra hircus). Immunogenetics 2018, 70, 10. [Google Scholar] [CrossRef] [Green Version]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome Sequence Assembly Using Trace Signals and Additional Sequence Information. Comput. Sci. Biol. Proc. Ger. Conf. Bioinform. (GCB) 1999, 99, 12. [Google Scholar]
- Bonfield, J.K.; Whitwham, A. Gap5—Editing the billion fragment sequence assembly. Bioinformatics 2010, 26, 5. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.-A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 8. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, 11. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Illumina Adapter Sequences. Available online: https://support-docs.illumina.com/SHARE/AdapterSeq/illumina-adapter-sequences.pdf (accessed on 19 April 2021).
- Messing, J. New M13 vectors for cloning. Methods Enzym. 1983, 101, 59. [Google Scholar] [CrossRef]
- Illumina. 16S Metagenomic Sequencing Library Preparation. Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 9 March 2020).
- Potapov, V.; Ong, J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE 2017, 12, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefranc, M.-P.; Giudicelli, V.; Duroux, P.; Jabado-Michaloud, J.; Folch, G.; Aouinti, S.; Carillon, E.; Duvergey, H.; Houles, A.; Paysan-Lafosse, T.; et al. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res. 2014, 43, 10. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 7. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Caraux, A.; De Vos, J.; Fiol, G.; Larroque, M.; Cognot, C.; Bret, C.; Duperray, C.; Hose, D.; Klein, B. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood 2009, 114, 9. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosawa, N.; Yoshioka, M.; Fujimoto, R.; Yamagishi, F.; Isobe, M. Rapid production of antigen-specific monoclonal antibodies from a variety of animals. BMC Biol. 2012, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Diks, A.M.; Overduin, L.A.; van Leenen, L.D.; Slobbe, L.; Jolink, H.; Visser, L.G.; van Dongen, J.J.M.; Berkowska, M.A. B-Cell Immunophenotyping to Predict Vaccination Outcome in the Immunocompromised—A Systematic Review. Front. Immunol. 2021, 12, 21. [Google Scholar] [CrossRef]
- Lee, B.O.; Moyron-Quiroz, J.; Rangel-Moreno, J.; Kusser, K.L.; Hartson, L.; Sprague, F.; Lund, F.E.; Randall, T.D. CD40, but Not CD154, Expression on B Cells Is Necessary for Optimal Primary B Cell Responses. J. Immunol. 2003, 171, 5707–5717. [Google Scholar] [CrossRef] [Green Version]
- Frölich, D.; Giesecke, C.; Mei, H.E.; Reiter, K.; Daridon, C.; Lipsky, P.E.; Dörner, T. Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J. Immunol. 2010, 185, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galson, J.D.; Pollard, A.J.; Trück, J.; Kelly, D.F. Studying the antibody repertoire after vaccination: Practical applications. Trends Immunol. 2014, 35, 13. [Google Scholar] [CrossRef]
- Jiang, N.; He, J.; Weinstein, J.A.; Penland, L.; Sasaki, S.; He, X.-S.; Dekker, C.L.; Zheng, N.-Y.; Huang, M.; Sullivan, M.; et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci. Transl. Med. 2013, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laserson, U.; Vigneault, F.; Gadala-Maria, D.; Yaari, G.; Uduman, M.; Vander Heiden, J.A.; Kelton, W.; Taek Jung, S.; Liu, Y.; Laserson, J.; et al. High-resolution antibody dynamics of vaccine-induced immune responses. Proc. Natl. Acad. Sci. USA 2014, 111, 6. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.; Rice, J.S.; Wang, C.; Harris, S.L.; Diamond, B. Identification of an antigen-specific B cell population. J. Immunol. Methods 2003, 272, 11. [Google Scholar] [CrossRef]
- Illumina. How Short Inserts Affect Sequencing Performance. Available online: https://emea.support.illumina.com/bulletins/2020/12/how-short-inserts-affect-sequencing-performance.html (accessed on 12 October 2021).
- Shi, Z.; Zhang, Q.; Yan, H.; Yang, Y.; Wang, P.; Zhang, Y.; Deng, Z.; Yu, M.; Zhou, W.; Wang, Q.; et al. More than one antibody of individual B cells revealed by single-cell immune profiling. Cell. Discov. 2019, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Al-Eryani, G.; Carswell, S.; Ferguson, J.M.; Blackburn, J.; Barton, K.; Roden, D.; Luciani, F.; Giang Phan, T.; Junankar, S.; et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Setliff, I.; Shiakolas, A.R.; Pilewski, K.A.; Murji, A.A.; Mapengo, R.E.; Janowska, K.; Richardson, S.; Oosthuysen, C.; Raju, N.; Ronsard, L.; et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 2019, 179, 11. [Google Scholar] [CrossRef] [PubMed]
- Berens, S.J.; Wylie, D.E.; Lopez, O.J. Use of a single VH family and long CDR3s in the variable region of cattle Ig heavy chains. Int. Immunol. 1997, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Finn, J.A.; Larsen, P.A.; Smith, T.P.L.; Crowe, J.E., Jr. Structural Diversity of Ultralong CDRH3s in Seven Bovine Antibody Heavy Chains. Front. Immunol. 2019, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Saini, S.S.; Kaushik, A.K. Unusually long germline DH genes contribute to large sized CDR3H in bovine antibodies. Mol. Immunol. 2003, 40, 7. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, J.; Cico, A.; Poncin, R.; Le Bohec, M.; Morf, J.; Lipin, S.; Graindorge, A.; Eckert, H.; Saffarian, A.; Cathaly, L.; et al. RevGel-seq: Instrument-free single-cell RNA sequencing using a reversible hydrogel for cell-specific barcoding. Sci. Rep. 2023, 13, 11. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Orientation | Workflow 1 | Workflow 2 | Workflow 3 | |||
|---|---|---|---|---|---|---|---|---|
| RT-PCR | PCR 2 | PCR 1 | PCR 3 | PCR 1 | PCR 2 | |||
| IgG_C3 # | GGCACCCGAGTTCCAGGTCA | RV | ✓ | ✓ | ✓ | |||
| IgG_C2 | CACCGGCTCGGGCATGTAGCTGG | RV | ✓ | |||||
| IgG_C1 | GGGTAGACTTTCGGGGCTGTGGTGG | RV | ✓ | |||||
| IgG_C1_OH | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTAGACTTTCGGGGCTGTGGT | RV | ✓ | |||||
| IgM_C3 | AGGGCCACCGTGCTCTCATC | RV | ✓ | ✓ | ||||
| IgM_C2 | CGAGCTCACGCAGGACACCA | RV | ✓ | ✓ | ||||
| IgM_C1 | CTTTCGGGTGTGATTCACCT | RV | ✓ | |||||
| IgM_C2_OH | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTCTCRGGTGTGATTCAC | RV | ✓ | |||||
| IgM1_C1_OH | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTCTCGGGAGCGATTCAC | RV | ✓ | |||||
| IgH_V1 | ACTGTGGACCCTCCTCYTKGTGY | FW | ✓ | ✓ | ✓ | |||
| IgH_V2 | ACTGTGGACCCTSSTCTTTGTGC | FW | ✓ | ✓ | ✓ | |||
| IgH_V3 | ACTGTGGRCYCTCCTCTTTGTGC | FW | ✓ | ✓ | ✓ | |||
| IgH_V4 | VYYGTGGACCCTCCTCTTTGTGC | FW | ✓ | ✓ | ✓ | |||
| IgH_V5 | ACTGTGGACCCTCCTCTTTVTVC | FW | ✓ | ✓ | ✓ | |||
| IgH_V1_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACTGTGGACCCTCCTCYTKGTGY | FW | ✓ | |||||
| IgH_V2_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACTGTGGACCCTSSTCTTTGTGC | FW | ✓ | |||||
| IgH_V3_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACTGTGGRCYCTCCTCTTTGTGC | FW | ✓ | |||||
| IgH_V4_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGVYYGTGGACCCTCCTCTTTGTGC | FW | ✓ | |||||
| IgH_V5_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGACTGTGGACCCTCCTCTTTVTVC | FW | ✓ | |||||
| IgL_C4 | CGGGTAGAAGTCGCTGATGA | RV | ✓ | ✓ | ||||
| IgL_C2 | CCGTTGAGCTCCTCCGTGGAG | RV | ✓ | |||||
| IgL_C1 | CGAGGGTGSGGACTTGGGCTGAC | RV | ✓ | |||||
| Igλ chain Inner-Reverse # | GCGGGAACAGGGTGACCGAG | RV | ✓ | |||||
| IgL_C3_OH | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCGGGTAGAAGTCGCTGATGA | RV | ✓ | |||||
| IgL_V1 | CAGGCTSYACTGACTCAGCCR | FW | ✓ | ✓ | ||||
| IgL_V2 | CAGVCTGKSCTGACTCAGCCK | FW | ✓ | ✓ | ||||
| IgL_V3 | CAGGMTRTGCTGACKCAGCCG | FW | ✓ | ✓ | ||||
| IgL_V4 | CAGGMTVTRCTGACTCAGCCG | FW | ✓ | ✓ | ||||
| IgL_V5 | CAGGMTRTGCTGACKCAGCCG | FW | ✓ | ✓ | ||||
| IgL_V6 | CAGGCTGGYCTGACTCAGCCG | FW | ✓ | ✓ | ||||
| IgL_V7 | CAGGCTGTVCTRACBCAGCCG | FW | ✓ | ✓ | ||||
| IgL_V8 | CAGGCTGTGCTKRCTCARCCG | FW | ✓ | ✓ | ||||
| IgL_V9 | CAGGCTGTKYTGACTCAGCCR | FW | ✓ | ✓ | ||||
| IgL_V10 | CAGGGTGTGCTGACTCAGCCR | FW | ✓ | ✓ | ||||
| IgL_V11 | TCSTATGAACTGACMCAGYYG | FW | ✓ | ✓ | ||||
| IgL_V12 | TCTTCTCARCTGACTCAGCCG | FW | ✓ | ✓ | ||||
| IgL_L1 | ATGGCCYGGTCCCCTCTG | FW | ✓ | |||||
| IgL_L2 | ATGGCCTTGGCCCCTCTG | FW | ✓ | |||||
| IgL_L3 | ATGGCCTGGATGCTGCTT | FW | ✓ | |||||
| IgL_L4 | ATGGYCTGGGCTCTGCTY | FW | ✓ | |||||
| IgL_L5 | ATGGCCTGGACCCCTCTC | FW | ✓ | |||||
| IgL_L1_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGGCCYGGTCCCCTCTG | FW | ✓ | |||||
| IgL_L2_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGGCCTTGGCCCCTCTG | FW | ✓ | |||||
| IgL_L3_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGGCCTGGATGCTGCTT | FW | ✓ | |||||
| IgL_L4_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGGYCTGGGCTCTGCTY | FW | ✓ | |||||
| IgL_L5_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGGCCTGGACCCCTCTC | FW | ✓ | |||||
| IgK_C3 | TTCACCAAGCACACGACAGA | RV | ✓ | ✓ | ||||
| IgK_C2 | TTCAGCTGCTCATCAGATGGTT | RV | ✓ | |||||
| IgK_C1_OH | GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGAAGACGGATGGCTCAGCATCAGACC | RV | ✓ | |||||
| IgK_L1 | CCTTGGTCTCCTGCTGCTC | FW | ||||||
| IgK_L2 | CCCACTCAGCTCCTCAGTCT | FW | ||||||
| IgK_L3 | TGAGATTCYCTGCTCAGYTCC | FW | ✓ | |||||
| IgK_L4 | ATGAGGTTCCCTGTCAGCTC | FW | ✓ | |||||
| IgK_L5 | ARATTCCCTGCTCAGCTCCT | FW | ✓ | |||||
| IgK_L6 | CTGTTCTTCTGGCTCCCAGC | FW | ✓ | |||||
| IgK_L3_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGAGATTCYCTGCTCAGYTCC | FW | ✓ | |||||
| IgK_L4_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATGAGGTTCCCTGTCAGCTC | FW | ✓ | |||||
| IgK_L5_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGARATTCCCTGCTCAGCTCCT | FW | ✓ | |||||
| IgK_L6_OH | TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTGTTCTTCTGGCTCCCAGC | FW | ✓ | |||||
| Read1 primer # | TCTACACTCTTTCCCTACACGACG | FW | ✓ | |||||
| P5+ Read1 | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC | FW | ✓ | |||||
| Workflow 1 | Workflow 2 | Workflow 3 | ||
|---|---|---|---|---|
| Flow cytometry | Staining strategy | Stain PBMCs with fluorescently labeled B cell markers | ||
| Sorting | Single-cell in plates | Single-cell in plates | Bulk sorting in tubes | |
| Cost differences | Cost of staining and sorting is equivalent for the 3 strategies | |||
| Duration | 1 day | 1 day | 1 day | |
| Amplification | cDNA preparation | RT-PCR | cDNA preparation | 10x + cDNA preparation + amplification |
| PCR amplification | semi-nested PCR | semi-nested PCR | semi-nested PCR | |
| Purification | Gel purification | AMPure bead purification | AMPure bead purification | |
| Sample preparation for sequencing | pGEM-T cloning + test PCR | PCR-indexing | Fragmentation + indexing | |
| Duration | 10–12 days | 3–4 days | 2–3 days | |
| Cost of reagents * | 1750 USD/plate | 875 USD/plate | 1250 USD/sample | |
| Sequencing | Platform | Sanger | Illumina | Illumina |
| Analysis | V-Quest/IgMat | IgMat | Cell Ranger + IgMat | |
| Price/antibody * (excluding flow cytometry and FTE) | 80 USD | 80 USD | 2 USD | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramirez Valdez, K.; Nzau, B.; Dorey-Robinson, D.; Jarman, M.; Nyagwange, J.; Schwartz, J.C.; Freimanis, G.; Steyn, A.W.; Warimwe, G.M.; Morrison, L.J.; et al. A Customizable Suite of Methods to Sequence and Annotate Cattle Antibodies. Vaccines 2023, 11, 1099. https://doi.org/10.3390/vaccines11061099
Ramirez Valdez K, Nzau B, Dorey-Robinson D, Jarman M, Nyagwange J, Schwartz JC, Freimanis G, Steyn AW, Warimwe GM, Morrison LJ, et al. A Customizable Suite of Methods to Sequence and Annotate Cattle Antibodies. Vaccines. 2023; 11(6):1099. https://doi.org/10.3390/vaccines11061099
Chicago/Turabian StyleRamirez Valdez, Kristel, Benjamin Nzau, Daniel Dorey-Robinson, Michael Jarman, James Nyagwange, John C. Schwartz, Graham Freimanis, Angela W. Steyn, George M. Warimwe, Liam J. Morrison, and et al. 2023. "A Customizable Suite of Methods to Sequence and Annotate Cattle Antibodies" Vaccines 11, no. 6: 1099. https://doi.org/10.3390/vaccines11061099