
An Amplicon-Based Application for the Whole-Genome Sequencing of GI-19 Lineage Infectious Bronchitis Virus Directly from Clinical Samples
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Isolation and Propagation
2.2. RNA Extraction and IBV Real-Time RT-PCR Assay
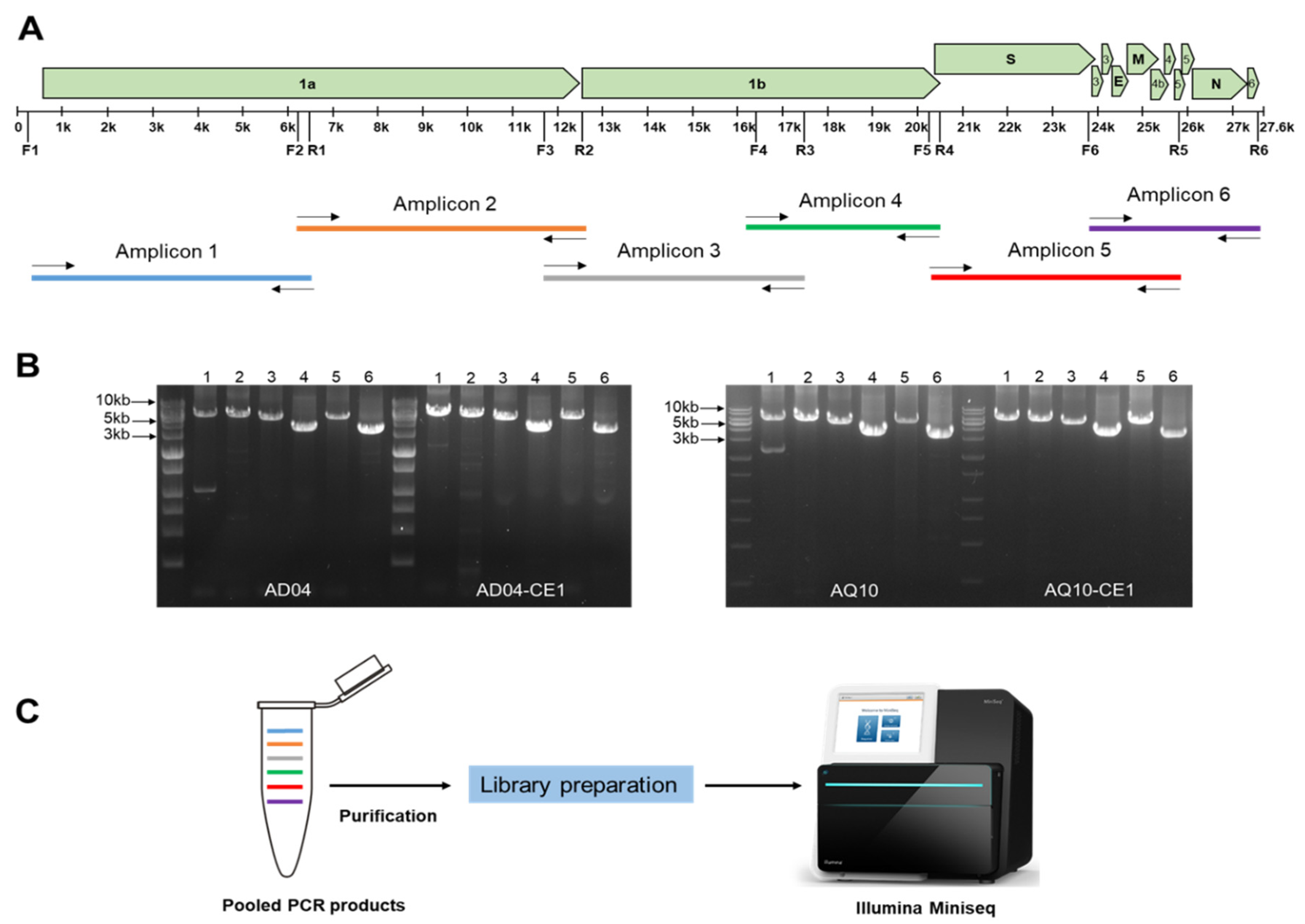
2.3. In Silico Design of the Primer Sets
2.4. cDNA Synthesis and Amplicon Analysis
2.5. SISPA
2.6. Amplicon and Metagenome Sequencing Libraries Preparation
2.7. Bioinformatic Analysis
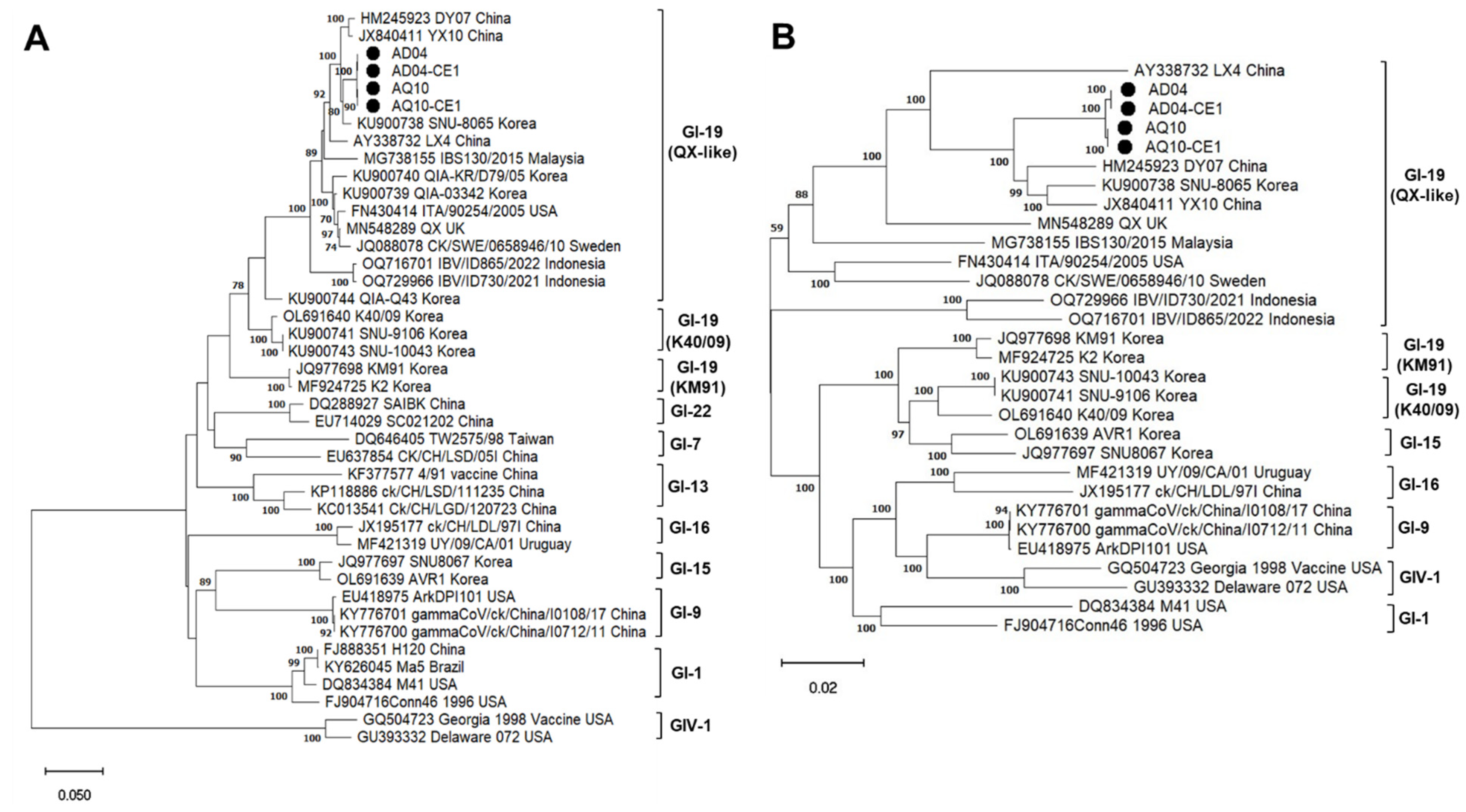
2.8. Phylogenetic Analysis and Sanger Sequencing
3. Results
3.1. RT-qPCR and Generation of Amplicons
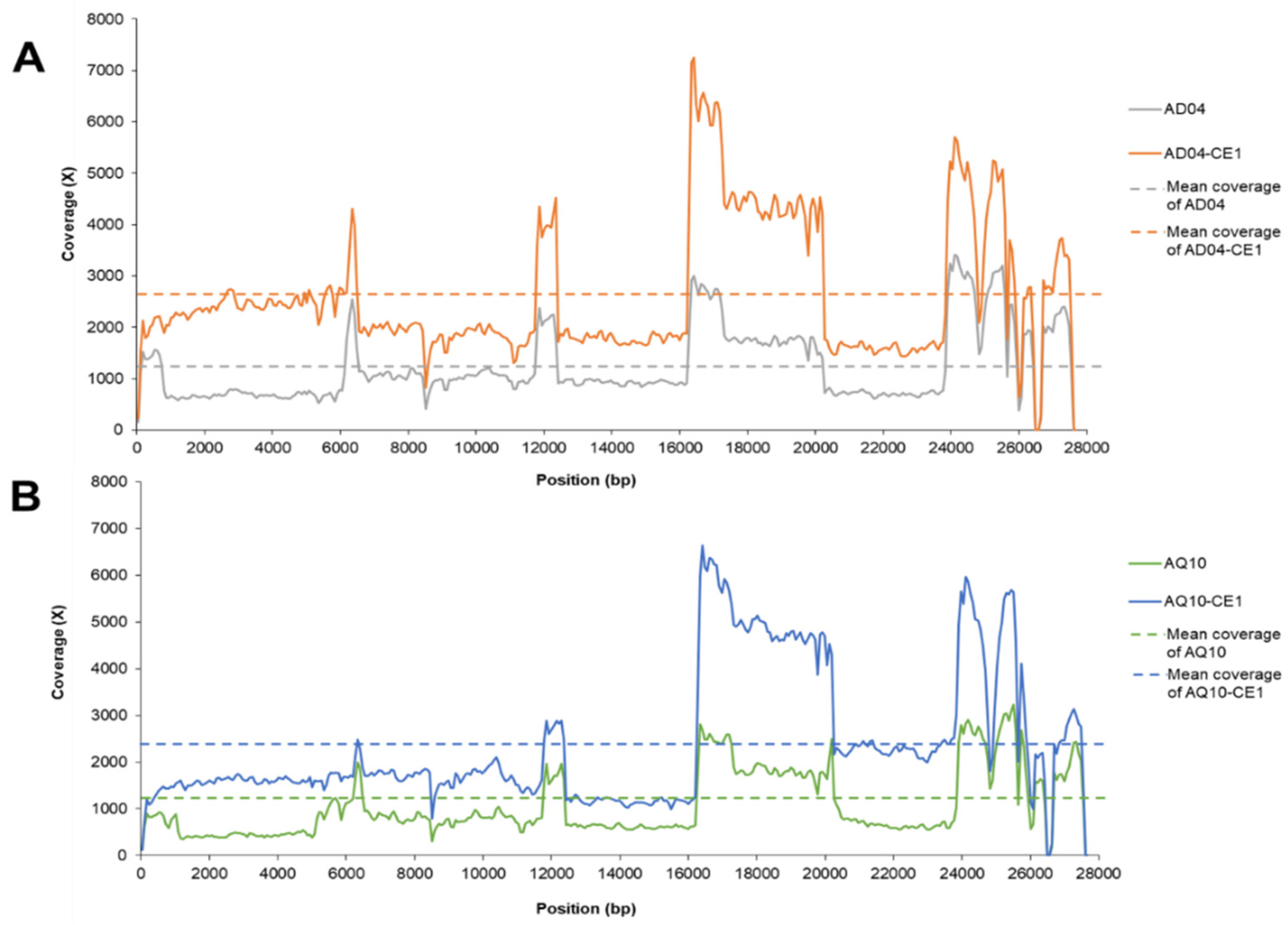
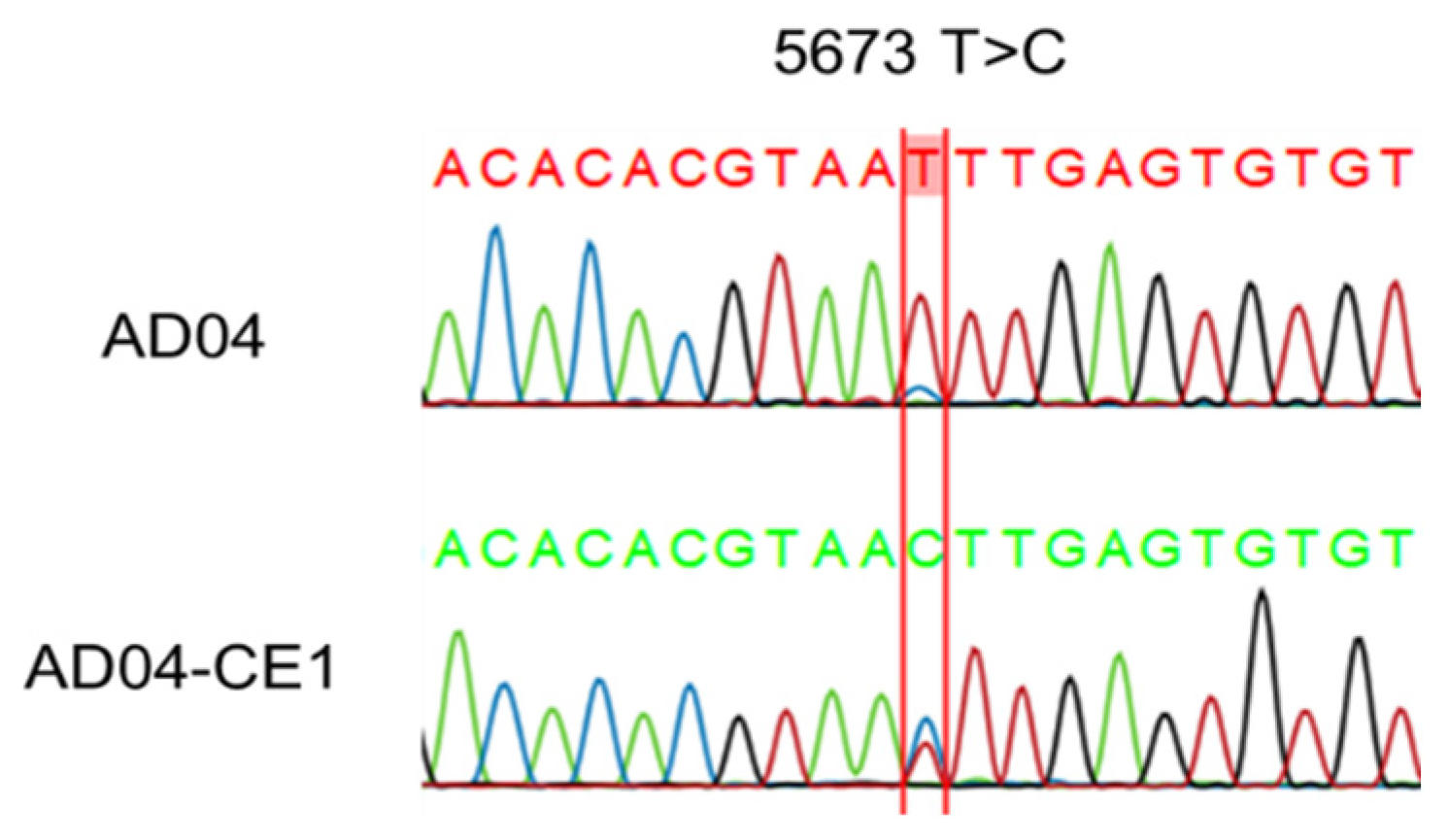
3.2. Complete Genome Characterization by Amplicon Sequencing
3.3. Comparison of Amplicon Sequencing and Metagenome Sequencing Approaches for IBV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; de Wit, S. Infectious Bronchitis. In Diseases of Poultry, 14th ed.; Swayne, D.E., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2020; pp. 167–188. [Google Scholar]
- Leghari, R.A.; Fan, B.; Wang, H.; Bai, J.; Zhang, L.; Abro, S.H.; Jiang, P. Full-length genome sequencing analysis of avian infectious bronchitis virus isolate associated with nephropathogenic infection. Poult. Sci. 2016, 95, 2921–2929. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, D.; Davis, P.J.; Cook, J.K.; Li, D.; Kant, A.; Koch, G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992, 21, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Hartog, L.; Kant, A.; van Roozelaar, D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: Correlation with biological functions. J. Gen. Virol. 1990, 71 Pt 9, 1929–1935. [Google Scholar] [CrossRef]
- Hong, S.M.; Kim, S.J.; An, S.H.; Kim, J.; Ha, E.J.; Kim, H.; Kwon, H.J.; Choi, K.S. Receptor binding motif surrounding sites in the spike 1 protein of infectious bronchitis virus have high susceptibility to mutation related to selective pressure. J. Vet. Sci. 2023, 24, e51. [Google Scholar] [CrossRef] [PubMed]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-González, L.; Marandino, A.; Panzera, Y.; Tomás, G.; Williman, J.; Techera, C.; Gayosso-Vázquez, A.; Ramírez-Andoney, V.; Alonso-Morales, R.; Realpe-Quintero, M.; et al. Research Note: High genetic diversity of infectious bronchitis virus from Mexico. Poult Sci. 2022, 101, 102076. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.; Thai, T.N.; Lee, J.I.; Kwon, Y.K.; Kim, H.R. Nationwide Surveillance for Infectious Bronchitis Virus in South Korea from 2020 to 2021. Avian Dis. 2022, 66, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Jeong, S.; Cho, A.Y.; Kim, K.J.; Kim, J.Y.; Park, D.H.; Kim, H.J.; Kwon, J.H.; Song, C.S. Genomic Analysis of Avian Infectious Bronchitis Viruses Recently Isolated in South Korea Reveals Multiple Introductions of GI-19 Lineage (QX Genotype). Viruses 2021, 13, 1045. [Google Scholar] [CrossRef]
- Lee, E.K.; Jeon, W.J.; Lee, Y.J.; Jeong, O.M.; Choi, J.G.; Kwon, J.H.; Choi, K.S. Genetic diversity of avian infectious bronchitis virus isolates in Korea between 2003 and 2006. Avian Dis. 2008, 52, 332–337. [Google Scholar] [CrossRef]
- Lee, S.K.; Sung, H.W.; Kwon, H.M. S1 glycoprotein gene analysis of infectious bronchitis viruses isolated in Korea. Arch. Virol. 2004, 149, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.H.; Lee, H.J.; Lee, D.H.; Lee, Y.N.; Park, J.K.; Youn, H.N.; Kim, M.S.; Lee, J.B.; Park, S.Y.; Choi, I.S.; et al. An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect. Genet. Evol. 2011, 11, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious Bronchitis Virus Evolution, Diagnosis and Control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Ni, R.; Qiu, R.; Wang, F.; Yan, W.; Wang, K.; Li, H.; Fu, X.; Chen, L.; Lei, C.; et al. Evaluation of a novel recombinant strain of infectious bronchitis virus emerged from three attenuated live vaccine strains. Microb. Pathog. 2022, 164, 105437. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, H.C.; Cho, A.Y.; Choi, Y.J.; Lee, H.; Lee, D.H.; Song, C.S. Novel recombinant avian infectious bronchitis viruses from chickens in Korea, 2019-2021. Front. Vet. Sci. 2023, 10, 1107059. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Qiu, R.; Wang, F.; Fu, X.; Li, H.; Cui, P.; Zhai, Y.; Li, C.; Zhang, L.; Gu, K.; et al. Genetic and pathogenic characterization of a novel recombinant avian infectious bronchitis virus derived from GI-1, GI-13, GI-28, and GI-19 strains in Southwestern China. Poult. Sci. 2021, 100, 101210. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.-Y.; Lee, J.-Y.; Bae, Y.-C.; Kwon, Y.-K.; Kim, H.-R. Genetic and Pathogenic Characterization of QX(GI-19)-Recombinant Infectious Bronchitis Viruses in South Korea. Viruses 2021, 13, 1163. [Google Scholar] [CrossRef] [PubMed]
- Armesto, M.; Cavanagh, D.; Britton, P. The replicase gene of avian coronavirus infectious bronchitis virus is a determinant of pathogenicity. PLoS ONE 2009, 4, e7384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, K.; Cheng, J.; Jia, W.; Zhao, Y.; Zhang, G. Replicase 1a gene plays a critical role in pathogenesis of avian coronavirus infectious bronchitis virus. Virology 2020, 550, 1–7. [Google Scholar] [CrossRef]
- Ramirez-Nieto, G.; Mir, D.; Almansa-Villa, D.; Cordoba-Argotti, G.; Beltran-Leon, M.; Rodriguez-Osorio, N.; Garai, J.; Zabaleta, J.; Gomez, A.P. New Insights into Avian Infectious Bronchitis Virus in Colombia from Whole-Genome Analysis. Viruses 2022, 14, 2562. [Google Scholar] [CrossRef]
- Butt, S.L.; Erwood, E.C.; Zhang, J.; Sellers, H.S.; Young, K.; Lahmers, K.K.; Stanton, J.B. Real-time, MinION-based, amplicon sequencing for lineage typing of infectious bronchitis virus from upper respiratory samples. J. Vet. Diagn. Investig. 2021, 33, 179–190. [Google Scholar] [CrossRef]
- Kariithi, H.M.; Volkening, J.D.; Leyson, C.M.; Afonso, C.L.; Christy, N.; Decanini, E.L.; Lemiere, S.; Suarez, D.L. Genome Sequence Variations of Infectious Bronchitis Virus Serotypes From Commercial Chickens in Mexico. Front. Vet. Sci. 2022, 9, 931272. [Google Scholar] [CrossRef]
- Brinkmann, A.; Uddin, S.; Krause, E.; Surtees, R.; Dinçer, E.; Kar, S.; Hacıoğlu, S.; Özkul, A.; Ergünay, K.; Nitsche, A. Utility of a Sequence-Independent, Single-Primer-Amplification (SISPA) and Nanopore Sequencing Approach for Detection and Characterization of Tick-Borne Viral Pathogens. Viruses 2021, 13, 203. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Mase, M.; Hiramatsu, K.; Watanabe, S.; Iseki, H. Complete Genome Sequences of Infectious Bronchitis Virus Genotype JP-II (GI-7) and JP-III (GI-19) Strains Isolated in Japan. Microbiol. Resour. Announc. 2023, 12, e0067022. [Google Scholar] [CrossRef]
- Bali, K.; Bálint, Á.; Farsang, A.; Marton, S.; Nagy, B.; Kaszab, E.; Belák, S.; Palya, V.; Bányai, K. Recombination Events Shape the Genomic Evolution of Infectious Bronchitis Virus in Europe. Viruses 2021, 13, 535. [Google Scholar] [CrossRef] [PubMed]
- Jude, R.; da Silva, A.P.; Rejmanek, D.; Crossley, B.; Jerry, C.; Stoute, S.; Gallardo, R. Whole-genome sequence of a genotype VIII infectious bronchitis virus isolated from California layer chickens in 2021. Microbiol. Resour. Announc. 2023, 12, e0095922. [Google Scholar] [CrossRef]
- Van Borm, S.; Steensels, M.; Mathijs, E.; Vandenbussche, F.; van den Berg, T.; Lambrecht, B. Metagenomic sequencing determines complete infectious bronchitis virus (avian Gammacoronavirus) vaccine strain genomes and associated viromes in chicken clinical samples. Virus Genes 2021, 57, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Callison, S.A.; Hilt, D.A.; Boynton, T.O.; Sample, B.F.; Robison, R.; Swayne, D.E.; Jackwood, M.W. Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J. Virol. Methods 2006, 138, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genomics 2006, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- D’Addiego, J.; Wand, N.; Afrough, B.; Fletcher, T.; Kurosaki, Y.; Leblebicioglu, H.; Hewson, R. Recovery of complete genome sequences of Crimean-Congo haemorrhagic fever virus (CCHFV) directly from clinical samples: A comparative study between targeted enrichment and metagenomic approaches. J. Virol. Methods 2024, 323, 114833. [Google Scholar] [CrossRef]
- Zakotnik, S.; Knap, N.; Bogovič, P.; Zorec, T.M.; Poljak, M.; Strle, F.; Avšič-Županc, T.; Korva, M. Complete Genome Sequencing of Tick-Borne Encephalitis Virus Directly from Clinical Samples: Comparison of Shotgun Metagenomic and Targeted Amplicon-Based Sequencing. Viruses 2022, 14, 1267. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.F.G.; Chaguza, C.; Gagne, L.; Doucette, M.; Smole, S.; Buzby, E.; Hall, J.; Ash, S.; Harrington, R.; Cofsky, S.; et al. Development of an amplicon-based sequencing approach in response to the global emergence of mpox. PLoS Biol. 2023, 21, e3002151. [Google Scholar] [CrossRef] [PubMed]
- Hourdel, V.; Kwasiborski, A.; Balière, C.; Matheus, S.; Batéjat, C.F.; Manuguerra, J.C.; Vanhomwegen, J.; Caro, V. Rapid Genomic Characterization of SARS-CoV-2 by Direct Amplicon-Based Sequencing Through Comparison of MinION and Illumina iSeq100(TM) System. Front. Microbiol. 2020, 11, 571328. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, K.W.; Park, D.; Hassan, Z.U.; Park, E.C.; Lee, C.S.; Rahman, M.T.; Yi, H.; Kim, S. Rapid and sensitive amplicon-based genome sequencing of SARS-CoV-2. Front. Microbiol. 2022, 13, 876085. [Google Scholar] [CrossRef] [PubMed]
- Oade, M.S.; Keep, S.; Freimanis, G.L.; Orton, R.J.; Britton, P.; Hammond, J.A.; Bickerton, E. Attenuation of Infectious Bronchitis Virus in Eggs Results in Different Patterns of Genomic Variation across Multiple Replicates. J. Virol. 2019, 93, e00492-19. [Google Scholar] [CrossRef]
- Tsai, C.T.; Wu, H.Y.; Wang, C.H. Genetic sequence changes related to the attenuation of avian infectious bronchitis virus strain TW2575/98. Virus Genes 2020, 56, 369–379. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Name | Sequence (5′ to 3′) | Position * | Product Size (bp) |
|---|---|---|---|---|
| Amplicon 1 | IBF1 | CTTAACAAAACGGACTTAAATACC | 57 | 6429 |
| IBR1 | GCAACYTCRGGAGACATAAATG | 6485 | ||
| Amplicon 2 | IBF2 | GCAGGDTTYTATTTCTGGC | 6226 | 6165 |
| IBR2 | GTATCAGCCGAGCCTCACTG | 12390 | ||
| Amplicon 3 | IBF3 | GAYCCACCATGTAARTTTGG | 11749 | 5589 |
| IBR3 | CRGGCTCRAAATTATTRCC | 17337 | ||
| Amplicon 4 | IBF4 | GTATGTTRACCAAYTAYGAATTG | 16264 | 3969 |
| IBR4 | GTAAATARTTACWATTCCKCC | 20232 | ||
| Amplicon 5 | IBF5 | GGTGGACAMTGTTYTGTACWG | 20083 | 5719 |
| IBR5 | CGMGCTTTTCKYGCTATTGC | 25801 | ||
| Amplicon 6 | IBF6 | GACCTAARAARTCTGTTTAATG | 23849 | 3721 |
| IBR6 | CCCTCGATCGTACTCCGCG | 27569 |
| Sample Name | Number of Read | Mapped Reads to Reference Sequence (%) a | Coverage (%) b | Mean of Depth Coverage c | SD | |
|---|---|---|---|---|---|---|
| Amplicon-based sequencing | AD04 | 271,693 | 265,341 (97.66%) | 100 | 1239 | 711 |
| AD04-CE1 | 577,061 | 551,186 (95.52%) | 100 | 2629 | 1313 | |
| AQ10 | 258,588 | 249,866 (96.63%) | 100 | 1069 | 714 | |
| AQ10-CE1 | 482,990 | 470,782 (97.47%) | 100 | 2385 | 1455 | |
| Metagenome sequencing | AD04 | 339,616 | 2,901 (0.85%) | 22.58 d | 14 | 98 |
| AD04-CE1 | 578,112 | 193,793 (33.52%) | 100 | 864 | 2170 | |
| AQ10 | 552,618 | 246,618 (44.63%) | 95.37 e | 1148 | 8406 | |
| AQ10-CE1 | 1,218,715 | 989,833 (81.22%) | 100 | 4525 | 24,717 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.D.; Thai, T.N.; Kim, J.-K.; Song, H.-S.; Her, M.; Tran, X.T.; Kim, J.-Y.; Kim, H.-R. An Amplicon-Based Application for the Whole-Genome Sequencing of GI-19 Lineage Infectious Bronchitis Virus Directly from Clinical Samples. Viruses 2024, 16, 515. https://doi.org/10.3390/v16040515
Le HD, Thai TN, Kim J-K, Song H-S, Her M, Tran XT, Kim J-Y, Kim H-R. An Amplicon-Based Application for the Whole-Genome Sequencing of GI-19 Lineage Infectious Bronchitis Virus Directly from Clinical Samples. Viruses. 2024; 16(4):515. https://doi.org/10.3390/v16040515
Chicago/Turabian StyleLe, Hoang Duc, Tuyet Ngan Thai, Jae-Kyeom Kim, Hye-Soon Song, Moon Her, Xuan Thach Tran, Ji-Ye Kim, and Hye-Ryoung Kim. 2024. "An Amplicon-Based Application for the Whole-Genome Sequencing of GI-19 Lineage Infectious Bronchitis Virus Directly from Clinical Samples" Viruses 16, no. 4: 515. https://doi.org/10.3390/v16040515