


A Novel Tiled Amplicon Sequencing Assay Targeting the Tomato Brown Rugose Fruit Virus (ToBRFV) Genome Reveals Widespread Distribution in Municipal Wastewater Treatment Systems in the Province of Ontario, Canada
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
1.1. Tomato Brown Rugose Fruit Virus Global Incidence, Impact, and Phylogeny
1.2. Taxonomic Classification, Distinguishing between Species, Lineages, and Strains
1.3. ToBRFV Gene Content, Mode of Infection, and Host Immune-Escape
1.4. The ToBRFV NextStrain Database and Phylogeny
1.5. Use of PCR-Enrichment Sequencing Assays for Viral Detection
1.6. ToBRFV Occurrence and Transmission in Wastewater
1.7. ToBRFV Detection and Sequencing
2. Materials and Methods
2.1. Viral RNA Extraction from Wastewater Influent
2.2. Primer Design and Pooling
2.3. ToBRFV-Targeted Tiled Amplicon Sequencing Library Preparation
2.4. RNA Shotgun Sequencing Library Preparation
2.5. Library Pooling and Illumina NextSeq Sequencing
2.6. Read Processing, Taxonomic Classification, and ToBRFV Genome Alignment
2.7. Altob Implementation and Synthetic Read Simulation

3. Results
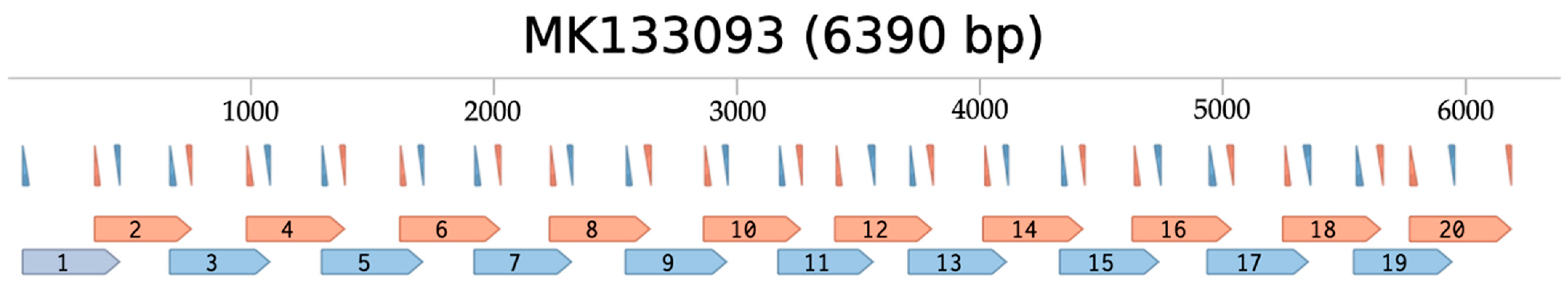
3.1. In Silico Primer Binding Analysis of ToBRFV Genome Sequences of 125 Strains
3.2. Read Quality Control and Adaptor Trimming
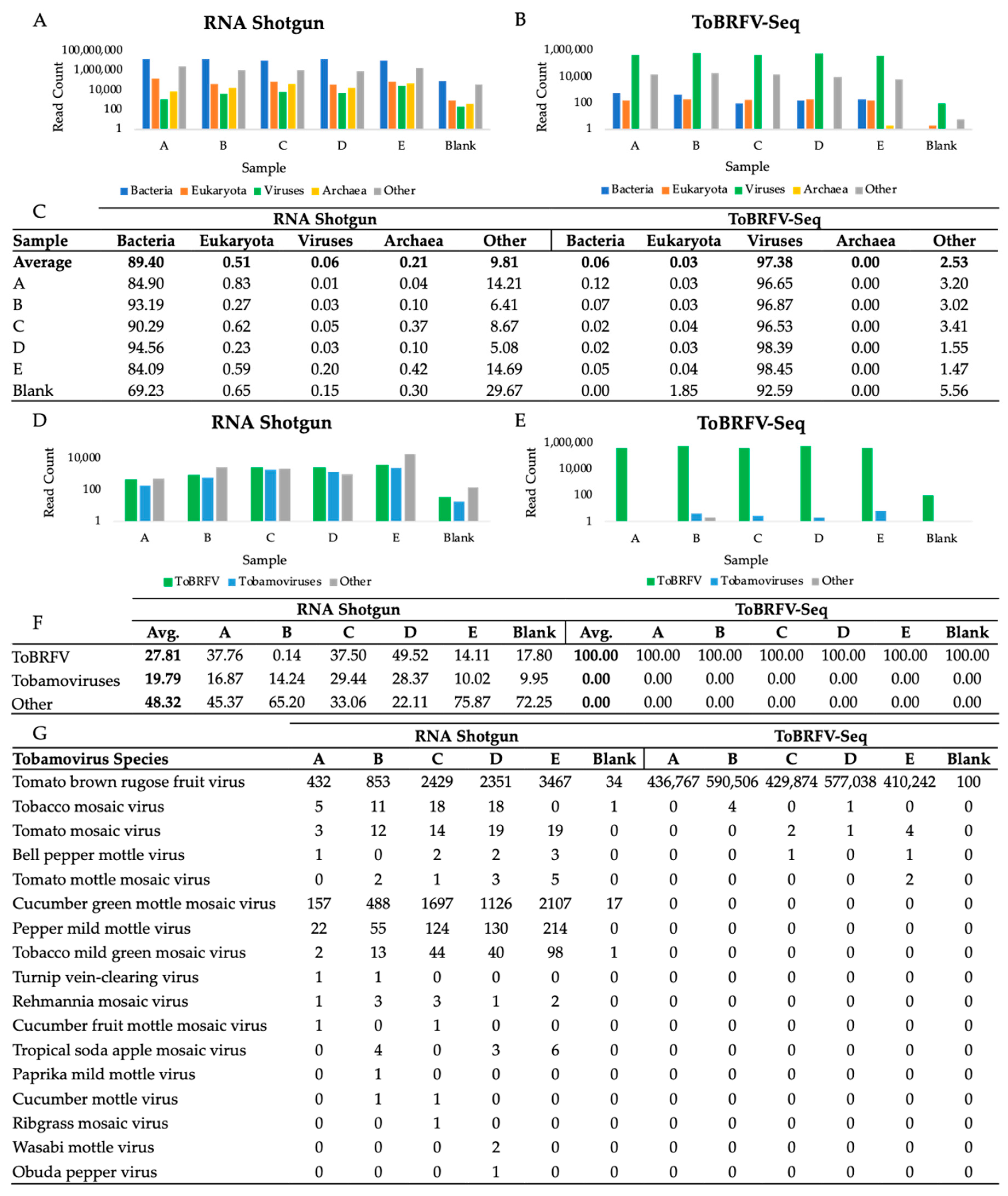
3.3. Taxonomic Profile of WWI Samples Prepared by RNA Shotgun and ToBRFV-Seq Methods
3.4. Mapping Reads to a ToBRFV Reference Genome
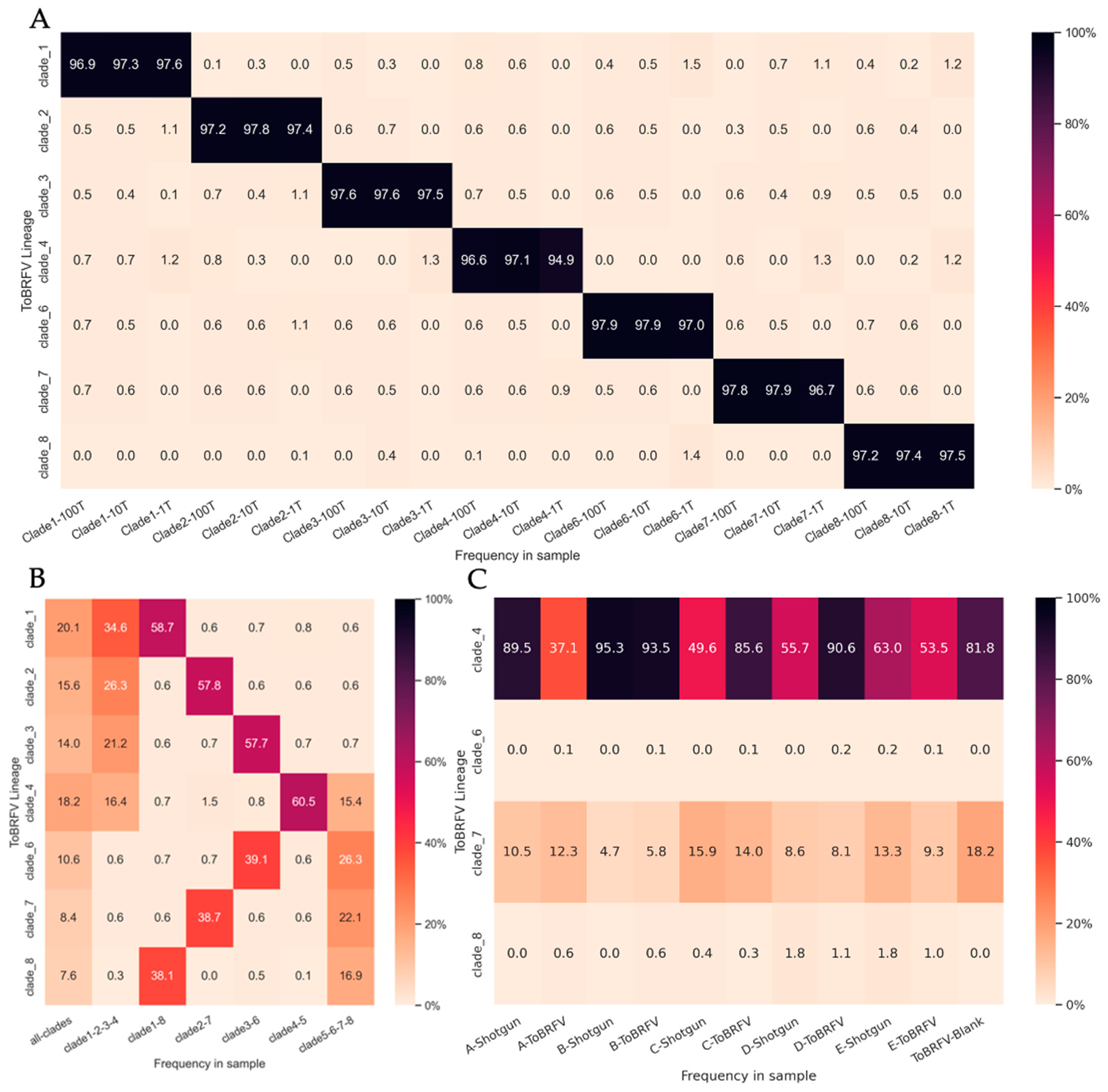
3.5. Estimating the Relative Abundance of ToBRFV Clades in Wastewater Influent
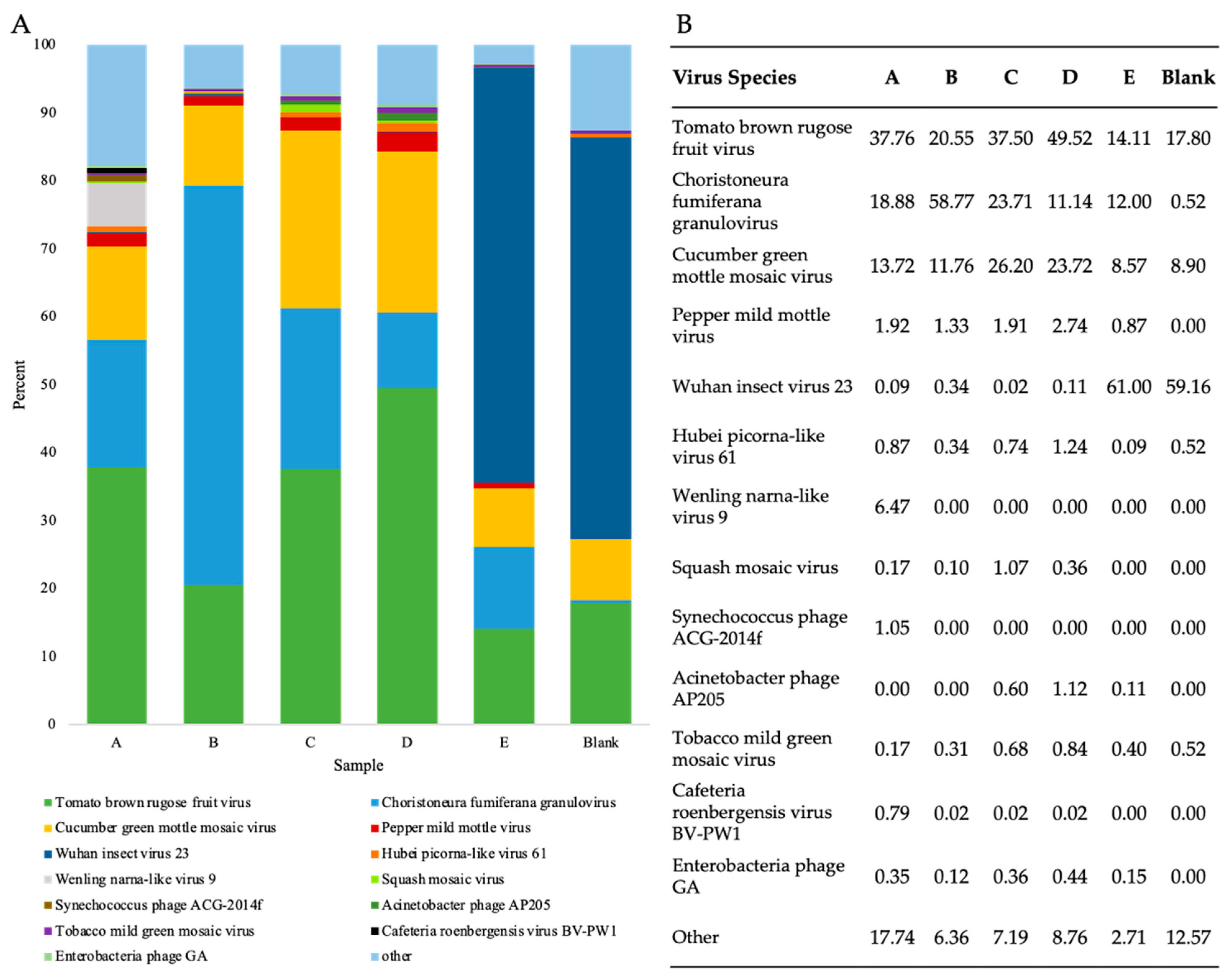
3.6. Assessment of Viral Shotgun Reads at a Species Level
4. Discussion
4.1. Possible Assay Applications to Determine ToBRFV Transmission and Prevalence
4.2. Effectiveness of and Possible Improvements to Viral Capture and ToBRFV-Seq Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Statistics Canada Table 32-10-0456-01 Production and Value of Greenhouse Fruits and Vegetables. Available online: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=3210045601 (accessed on 25 May 2023).
- Salem, N.; Mansour, A.; Ciuffo, M.; Falk, B.W.; Turina, M. A New Tobamovirus Infecting Tomato Crops in Jordan. Arch. Virol. 2016, 161, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.C.; Janssen, D. Global Plant Virus Disease Pandemics and Epidemics. Plants 2021, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Griffiths, J.S.; Marchand, G.; Bernards, M.A.; Wang, A. Tomato Brown Rugose Fruit Virus: An Emerging and Rapidly Spreading Plant RNA Virus that Threatens Tomato Production Worldwide. Mol. Plant Pathol. 2022, 23, 1262–1277. [Google Scholar] [CrossRef] [PubMed]
- Fresh Tomato Production and Top Producing Countries. Available online: https://www.tridge.com/intelligences/tomato/production (accessed on 21 February 2024).
- Fidan, H.; Sarikaya, P.; Yildiz, K.; Topkaya, B.; Erkis, G.; Calis, O. Robust Molecular Detection of the New Tomato Brown Rugose Fruit Virus in Infected Tomato and Pepper Plants from Turkey. J. Integr. Agric. 2021, 20, 2170–2179. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, H.; Ding, M.; Zhang, Z.; Yang, X.; Zhou, X. Molecular Characterization and Pathogenicity of an Infectious cDNA Clone of Tomato Brown Rugose Fruit Virus. Phytopathol. Res. 2021, 3, 14. [Google Scholar] [CrossRef]
- Botermans, M.; de Koning, P.P.M.; Oplaat, C.; Fowkes, A.R.; McGreig, S.; Skelton, A.; Adams, I.P.; Fox, A.; De Jonghe, K.; Demers, J.E.; et al. Tomato Brown Rugose Fruit Virus Nextstrain Build Version 3: Rise of a Novel Clade. PhytoFrontiersTM 2023, 3, 442–446. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Dawood, T.; Woźny, M.; Botermans, M. First Expansion of the Public Tomato Brown Rugose Fruit Virus (ToBRFV) Nextstrain Build; Inclusion of New Genomic and Epidemiological Data. PhytoFrontiersTM 2021, 1, 359–363. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Visser, M.; Bruinsma, M.; Koenraadt, H.M.S.; Westenberg, M.; Botermans, M. Real-Time Tracking of Tomato Brown Rugose Fruit Virus (ToBRFV) Outbreaks in the Netherlands Using Nextstrain. PLoS ONE 2020, 15, e0234671. [Google Scholar] [CrossRef]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.M.; Melcher, U.; Ratti, C.; Ryu, K.H. ICTV Virus Taxonomy Profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef]
- Gibbs, A. Evolution and Origins of Tobamoviruses. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1999, 354, 593–602. [Google Scholar] [CrossRef]
- Abrahamian, P.; Cai, W.; Nunziata, S.O.; Ling, K.-S.; Jaiswal, N.; Mavrodieva, V.A.; Rivera, Y.; Nakhla, M.K. Comparative Analysis of Tomato Brown Rugose Fruit Virus Isolates Shows Limited Genetic Diversity. Viruses 2022, 14, 2816. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Kreuze, J. Virgaviridae: A New Family of Rod-Shaped Plant Viruses. Arch. Virol. 2009, 154, 1967–1972. [Google Scholar] [CrossRef]
- Ochar, K.; Ko, H.C.; Woo, H.J.; Hahn, B.S.; Hur, O. Pepper Mild Mottle Virus: An Infectious Pathogen in Pepper Production and a Potential Indicator of Domestic Water Quality. Viruses 2023, 15, 282. [Google Scholar] [CrossRef]
- Hak, H.; Spiegelman, Z. The Tomato Brown Rugose Fruit Virus Movement Protein Overcomes Tm-22resistance in Tomato While Attenuating Viral Transport. Mol. Plant. Microbe Interact. 2021, 34, 1024–1032. [Google Scholar] [CrossRef]
- Bai, G.H.; Lin, S.C.; Hsu, Y.H.; Chen, S.Y. The Human Virome: Viral Metagenomics, Relations with Human Diseases, and Therapeutic Applications. Viruses 2022, 14, 278. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-Throughput Sequencing (HTS) for the Analysis of Viral Populations. Infect. Genet. Evol. 2020, 80, 104208. [Google Scholar] [CrossRef]
- Sandybayev, N.; Beloussov, V.; Strochkov, V.; Solomadin, M.; Granica, J.; Yegorov, S. Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome. Microorganisms 2022, 10, 2327. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Walls, S.D.; Gross, S.M.; Schroth, G.P.; Jarman, R.G.; Hang, J. Targeted Sequencing of Respiratory Viruses in Clinical Specimens for Pathogen Identification and Genome-Wide Analysis. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1838, pp. 125–140. [Google Scholar]
- Kumar, A.; Murthy, S.; Kapoor, A. Evolution of Selective-Sequencing Approaches for Virus Discovery and Virome Analysis. Virus Res. 2017, 239, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Glier, M.; Kuchinski, K.; Ross-Van Mierlo, T.; McVea, D.; Tyson, J.R.; Prystajecky, N.; Ziels, R.M. Assessing Multiplex Tiling PCR Sequencing Approaches for Detecting Genomic Variants of SARS-CoV-2 in Municipal Wastewater. mSystems 2021, 6, e0106821. [Google Scholar] [CrossRef] [PubMed]
- Artic Network. Available online: https://artic.network/ (accessed on 20 February 2024).
- Kareinen, L.; Ogola, J.; Kivistö, I.; Smura, T.; Aaltonen, K.; Jääskeläinen, A.J.; Kibiwot, S.; Masika, M.M.; Nyaga, P.; Mwaengo, D.; et al. Range Expansion of Bombali Virus in Mops Condylurus Bats, Kenya, 2019. Emerg. Infect. Dis. 2020, 26, 3007–3010. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.C.; de Vasconcelos, J.N.; Granja, B.G.; Thézé, J.; Jandondo, D.; Neto, Z.; Mirandela, M.; dos Santos Sebastiao, C.; Cândido, A.L.M.; Clemente, C.; et al. Early Genomic Detection of Cosmopolitan Genotype of Dengue Virus Serotype 2, Angola, 2018. Emerg. Infect. Dis. 2019, 25, 784–787. [Google Scholar] [CrossRef] [PubMed]
- ARTIC Multiplex PCR (Full Pathogens List)—Laboratory. Available online: https://community.artic.network/t/artic-multiplex-pcr-full-pathogens-list/494 (accessed on 20 February 2024).
- Bačnik, K.; Kutnjak, D.; Pecman, A.; Mehle, N.; Tušek Žnidarič, M.; Gutiérrez Aguirre, I.; Ravnikar, M. Viromics and Infectivity Analysis Reveal the Release of Infective Plant Viruses from Wastewater into the Environment. Water Res. 2020, 177, 115628. [Google Scholar] [CrossRef] [PubMed]
- Mehle, N.; Bačnik, K.; Bajde, I.; Brodarič, J.; Fox, A.; Gutiérrez-Aguirre, I.; Kitek, M.; Kutnjak, D.; Loh, Y.L.; Maksimović Carvalho Ferreira, O.; et al. Tomato Brown Rugose Fruit Virus in Aqueous Environments—Survival and Significance of Water-Mediated Transmission. Front. Plant Sci. 2023, 14, 1187920. [Google Scholar] [CrossRef] [PubMed]
- Rothman, J.A.; Whiteson, K.L. Sequencing and Variant Detection of Eight Abundant Plant-Infecting Tobamoviruses across Southern California Wastewater. Microbiol. Spectr. 2022, 10, e0305022. [Google Scholar] [CrossRef] [PubMed]
- Christou, A.; Agüera, A.; Bayona, J.M.; Cytryn, E.; Fotopoulos, V.; Lambropoulou, D.; Manaia, C.M.; Michael, C.; Revitt, M.; Schröder, P.; et al. The Potential Implications of Reclaimed Wastewater Reuse for Irrigation on the Agricultural Environment: The Knowns and Unknowns of the Fate of Antibiotics and Antibiotic Resistant Bacteria and Resistance Genes—A Review. Water Res. 2017, 123, 448–467. [Google Scholar] [CrossRef] [PubMed]
- Goh, P.S.; Ahmad, N.A.; Lim, J.W.; Liang, Y.Y.; Kang, H.S.; Ismail, A.F.; Arthanareeswaran, G. Microalgae-Enabled Wastewater Remediation and Nutrient Recovery through Membrane Photobioreactors: Recent Achievements and Future Perspective. Membranes 2022, 12, 1094. [Google Scholar] [CrossRef]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.L.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA Viral Community in Human Feces: Prevalence of Plant Pathogenic Viruses. PLoS Biol. 2006, 4, 0108–0118. [Google Scholar] [CrossRef]
- Natarajan, A.; Han, A.; Zlitni, S.; Brooks, E.F.; Vance, S.E.; Wolfe, M.; Singh, U.; Jagannathan, P.; Pinsky, B.A.; Boehm, A.; et al. Standardized Preservation, Extraction and Quantification Techniques for Detection of Fecal SARS-CoV-2 RNA. Nat. Commun. 2021, 12, 5753. [Google Scholar] [CrossRef]
- Rizzo, D.; Da Lio, D.; Panattoni, A.; Salemi, C.; Cappellini, G.; Bartolini, L.; Parrella, G. Rapid and Sensitive Detection of Tomato Brown Rugose Fruit Virus in Tomato and Pepper Seeds by Reverse Transcription Loop-Mediated Isothermal Amplification Assays (Real Time and Visual) and Comparison with RT-PCR End-Point and RT-qPCR Methods. Front. Microbiol. 2021, 12, 640932. [Google Scholar] [CrossRef]
- Ellmen, I.; Lynch, M.D.J.; Nash, D.; Cheng, J.; Nissimov, J.I.; Charles, T.C. Alcov: Estimating Variant of Concern Abundance from SARS-CoV-2 Wastewater Sequencing Data. medRxiv 2021. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 February 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C.; et al. Community-Led, Integrated, Reproducible Multi-Omics with Anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shawand, P.D.; Marshall, D. Using Tablet for Visual Exploration of Second-Generation Sequencing Data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Tannier, E.; Comte, N.; Parsons, D.P. Seaview Version 5: A Multiplatform Software for Multiple Sequence Alignment, Molecular Phylogenetic Analyses, and Tree Reconciliation. Methods Mol. Biol. 2021, 2231, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Rencher, A.C.; Christensen, W.F. Methods of Multivariate Analysis|Wiley Series in Probability and Statistics. Available online: https://onlinelibrary.wiley.com/doi/book/10.1002/9781118391686 (accessed on 21 February 2024).
- Nh13/DWGSIM: Whole Genome Simulator for Next-Generation Sequencing. Available online: https://github.com/nh13/DWGSIM/tree/main (accessed on 21 February 2024).
- Cloud-Based Platform for Biotech R&D|Benchling. Available online: https://www.benchling.com/ (accessed on 21 February 2024).
- Pérez-Cataluña, A.; Cuevas-Ferrando, E.; Randazzo, W.; Sánchez, G. Bias of Library Preparation for Virome Characterization in Untreated and Treated Wastewaters. Sci. Total Environ. 2021, 767, 144589. [Google Scholar] [CrossRef] [PubMed]
- Elbehery, A.H.A.; Deng, L. Insights into the Global Freshwater Virome. Front. Microbiol. 2022, 13, 953500. [Google Scholar] [CrossRef] [PubMed]
- Guajardo-Leiva, S.; Chnaiderman, J.; Gaggero, A.; Díe, B. Metagenomic Insights into the Sewage RNA Virosphere of a Large City. Viruses 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed]
- Mackie, J.; Kinoti, W.M.; Chahal, S.I.; Lovelock, D.A.; Campbell, P.R.; Tran-Nguyen, L.T.T.; Rodoni, B.C.; Constable, F.E. Targeted Whole Genome Sequencing (TWG-Seq) of Cucumber Green Mottle Mosaic Virus Using Tiled Amplicon Multiplex PCR and Nanopore Sequencing. Plants 2022, 11, 2716. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.W.; Avery, A.; Schandl, C.A. Managing a PCR Contamination Event in a Molecular Pathology Laboratory. Methods Mol. Biol. 2023, 2621, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Rashidan, K.; Guertin, C.; Cabana, J. Granulovirus. In Encyclopedia of Entomology; Capinera, J.L., Ed.; Springer: Dordrecht, The Netherlands, 2008; pp. 1653–1660. ISBN 978-1-4020-6359-6. [Google Scholar]
- Karthikeyan, S.; Levy, J.I.; De Hoff, P.; Humphrey, G.; Birmingham, A.; Jepsen, K.; Farmer, S.; Tubb, H.M.; Valles, T.; Tribelhorn, C.E.; et al. Wastewater Sequencing Reveals Early Cryptic SARS-CoV-2 Variant Transmission. Nature 2022, 609, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Ronquillo, N.; Belda-Ferre, P.; Alvarado, D.; Javidi, T.; Longhurst, C.A.; Knight, R. High-Throughput Wastewater SARS-CoV-2 Detection Enables Forecasting of Community Infection Dynamics in San Diego County. mSystems 2021, 6, e00045-21. [Google Scholar] [CrossRef] [PubMed]
- Symonds, E.M.; Sinigalliano, C.; Gidley, M.; Ahmed, W.; McQuaig-Ulrich, S.M.; Breitbart, M. Faecal Pollution along the Southeastern Coast of Florida and Insight into the Use of Pepper Mild Mottle Virus as an Indicator. J. Appl. Microbiol. 2016, 121, 1469–1481. [Google Scholar] [CrossRef]
- Torii, S.; Oishi, W.; Zhu, Y.; Thakali, O.; Malla, B.; Yu, Z.; Zhao, B.; Arakawa, C.; Kitajima, M.; Hata, A.; et al. Comparison of Five Polyethylene Glycol Precipitation Procedures for the RT-qPCR Based Recovery of Murine Hepatitis Virus, Bacteriophage Phi6, and Pepper Mild Mottle Virus as a Surrogate for SARS-CoV-2 from Wastewater. Sci. Total Environ. 2022, 807, 150722. [Google Scholar] [CrossRef]
- Maal-Bared, R.; Qiu, Y.; Li, Q.; Gao, T.; Hrudey, S.E.; Bhavanam, S.; Ruecker, N.J.; Ellehoj, E.; Lee, B.E.; Pang, X. Does Normalization of SARS-CoV-2 Concentrations by Pepper Mild Mottle Virus Improve Correlations and Lead Time between Wastewater Surveillance and Clinical Data in Alberta (Canada): Comparing Twelve SARS-CoV-2 Normalization Approaches. Sci. Total Environ. 2023, 856, 158964. [Google Scholar] [CrossRef]
- Ciannella, S.; González-Fernández, C.; Gomez-Pastora, J. Recent Progress on Wastewater-Based Epidemiology for COVID-19 Surveillance: A Systematic Review of Analytical Procedures and Epidemiological Modeling. Sci. Total Environ. 2023, 878, 162953. [Google Scholar] [CrossRef]
- Oloye, F.F.; Xie, Y.; Challis, J.K.; Femi-Oloye, O.P.; Brinkmann, M.; McPhedran, K.N.; Jones, P.D.; Servos, M.R.; Giesy, J.P. Understanding Common Population Markers for SARS-CoV-2 RNA Normalization in Wastewater—A Review. Chemosphere 2023, 333, 138682. [Google Scholar] [CrossRef]
- Farkas, K.; Walker, D.I.; Adriaenssens, E.M.; McDonald, J.E.; Hillary, L.S.; Malham, S.K.; Jones, D.L. Viral Indicators for Tracking Domestic Wastewater Contamination in the Aquatic Environment. Water Res. 2020, 181, 115926. [Google Scholar] [CrossRef]
- Hsu, S.Y.; Bayati, M.; Li, C.; Hsieh, H.Y.; Belenchia, A.; Klutts, J.; Zemmer, S.A.; Reynolds, M.; Semkiw, E.; Johnson, H.Y.; et al. Biomarkers Selection for Population Normalization in SARS-CoV-2 Wastewater-Based Epidemiology. Water Res. 2022, 223, 118985. [Google Scholar] [CrossRef]
- Kitajima, M.; Sassi, H.P.; Torrey, J.R. Pepper Mild Mottle Virus as a Water Quality Indicator. NPJ Clean Water 2018, 1, 19. [Google Scholar] [CrossRef]
- Rainey, A.L.; Liang, S.; Bisesi, J.H.; Sabo-Attwood, T.; Maurelli, A.T. A Multistate Assessment of Population Normalization Factors for Wastewater-Based Epidemiology of COVID-19. PLoS ONE 2023, 18, e0284370. [Google Scholar] [CrossRef]
- Aguado-García, Y.; Taboada, B.; Morán, P.; Rivera-Gutiérrez, X.; Serrano-Vázquez, A.; Iša, P.; Rojas-Velázquez, L.; Pérez-Juárez, H.; López, S.; Torres, J.; et al. Tobamoviruses Can Be Frequently Present in the Oropharynx and Gut of Infants during Their First Year of Life. Sci. Rep. 2020, 10, 13595. [Google Scholar] [CrossRef]
- Rivera-Gutiérrez, X.; Morán, P.; Taboada, B.; Serrano-Vázquez, A.; Isa, P.; Rojas-Velázquez, L.; Pérez-Juárez, H.; López, S.; Torres, J.; Ximénez, C.; et al. The Fecal and Oropharyngeal Eukaryotic Viromes of Healthy Infants during the First Year of Life Are Personal. Sci. Rep. 2023, 13, 938. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Fremin, B.J.; Schmidtke, D.T.; Wolfe, M.K.; Zlitni, S.; Graham, K.E.; Brooks, E.F.; Severyn, C.J.; Sakamoto, K.M.; Lacayo, N.J.; et al. The Tomato Brown Rugose Fruit Virus Movement Protein Gene Is a Novel Microbial Source Tracking Marker. Appl. Environ. Microbiol. 2023, 89, e0058323. [Google Scholar] [CrossRef] [PubMed]
- Luria, N.; Smith, E.; Reingold, V.; Bekelman, I.; Lapidot, M.; Levin, I.; Elad, N.; Tam, Y.; Sela, N.; Abu-Ras, A.; et al. A New Israeli Tobamovirus Isolate Infects Tomato Plants Harboring Tm-22 Resistance Genes. PLoS ONE 2017, 12, e0170429. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Bivins, A.; Korajkic, A.; Metcalfe, S.; Smith, W.J.M.; Simpson, S.L. Comparative Analysis of Adsorption-Extraction (AE) and Nanotrap® Magnetic Virus Particles (NMVP) Workflows for the Recovery of Endogenous Enveloped and Non-Enveloped Viruses in Wastewater. Sci. Total Environ. 2023, 859, 160072. [Google Scholar] [CrossRef] [PubMed]
- Nanotrap® Microbiome Particles|Ceres Nanosciences, Inc.|United States. Available online: https://www.ceresnano.com/microbiome (accessed on 21 February 2024).
- Fernandez-Cassi, X.; Timoneda, N.; Martínez-Puchol, S.; Rusiñol, M.; Rodriguez-Manzano, J.; Figuerola, N.; Bofill-Mas, S.; Abril, J.F.; Girones, R. Metagenomics for the Study of Viruses in Urban Sewage as a Tool for Public Health Surveillance. Sci. Total Environ. 2018, 618, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Hjelmsø, M.H.; Hellmér, M.; Fernandez-Cassi, X.; Timoneda, N.; Lukjancenko, O.; Seidel, M.; Elsässer, D.; Aarestrup, F.M.; Löfström, C.; Bofill-Mas, S.; et al. Evaluation of Methods for the Concentration and Extraction of Viruses from Sewage in the Context of Metagenomic Sequencing. PLoS ONE 2017, 12, e0170199. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tobamovirus Species | Accession | Primer Count |
|---|---|---|
| All 125 unique ToBRFV genomes | Supplementary Table S2 | 40 |
| Tobacco mosaic virus (TMV) | NC_001367.1 | 15 |
| Tomato mottle mosaic virus (ToMMV) | NC_022230.1 | 9 |
| Tomato mosaic virus (ToMV) | NC_002692.1 | 14 |
| Pepper mild mottle virus (PMMoV) | NC_003630.1 | 3 |
| Bell pepper mottle tobamovirus (BPeMV) | NC_009642.1 | 6 |
| Obuda pepper virus (ObPV) | NC_003852.1 | 1 |
| Paprika mild mottle virus (PaMMV) | NC_004106.1 | 3 |
| Tobacco middle green mosaic virus (TMGMV) | NC_001556.1 | 2 |
| Cucumber green mottle mosaic virus (CGMMV) | NC_001801.1 | 0 |
| Cucumber fruit mottle mosaic virus (CFMMV) | NC_002633.1 | 0 |
| Brugmansia mild mottle virus (BrMMV) | NC_010944.1 | 6 |
| Zucchini green mottle mosaic virus (ZGMMV) | NC_003878.1 | 0 |
| RNA Shotgun | ToBRFV-Seq | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample | Total Gb | Pairs | QF Pairs | % PF | Total Gb | Pairs | QF Pairs | % PF |
| Average | 4.640000 | 15,627,313 | 14,631,264 | 94.07 | 0.1798200 | 597,554 | 502,007 | 83.72 |
| A | 5.5000000 | 18,528,523 | 17,138,798 | 92.50 | 0.1688000 | 560,908 | 451,928 | 80.57 |
| B | 4.9000000 | 16,434,198 | 15,693,587 | 95.49 | 0.2103000 | 698,848 | 609,572 | 87.23 |
| C | 3.6000000 | 12,035,815 | 11,788,803 | 97.95 | 0.1589000 | 527,913 | 445,326 | 84.36 |
| D | 5.4000000 | 18,266,390 | 16,248,968 | 88.96 | 0.2054000 | 682,673 | 586,488 | 85.91 |
| E | 3.8000000 | 12,871,637 | 12,286,164 | 95.45 | 0.1557000 | 517,430 | 416,720 | 80.54 |
| Blank | 0.0396000 | 131,614 | 123,528 | 93.86 | 0.0000571 | 190 | 108 | 56.84 |
| RNA Shotgun | ToBRFV-Seq | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | ToBRFV Reads | % Aligned | % Total Cov. | % Target Cov. | Avg. Depth | Max. Depth | ToBRFV Reads | % Aligned | % Total Cov. | % Target Cov. | Avg. Depth | Max. Depth |
| Average | 3840 | 0.02 | 99.06 | 99.71 | 155.31 | 472 | 977,491 | 99.86 | 95.69 | 99.92 | 35,694.79 | 206,304 |
| A | 867 | 0.00 | 97.45 | 98.53 | 29.94 | 110 | 873,270 | 99.78 | 95.67 | 99.89 | 32,084.65 | 227,756 |
| B | 1732 | 0.01 | 99.34 | 100.00 | 67.63 | 184 | 1,180,695 | 99.85 | 95.70 | 99.85 | 44,893.64 | 312,224 |
| C | 4891 | 0.02 | 99.5 | 100.00 | 211.20 | 623 | 859,568 | 99.90 | 95.67 | 99.95 | 31,268.41 | 154,096 |
| D | 4742 | 0.02 | 99.44 | 100.00 | 177.90 | 556 | 1,153,735 | 99.90 | 95.74 | 99.95 | 40,645.85 | 227,468 |
| E | 6968 | 0.03 | 99.58 | 100.00 | 289.89 | 886 | 820,185 | 99.85 | 95.67 | 99.95 | 29,581.39 | 109,976 |
| Blank | 70 | 0.04 | 72.96 | 73.36 | 2.93 | 16 | 199 | 96.60 | 53.00 | 56.53 | 6.33 | 36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nash, D.; Ellmen, I.; Knapp, J.J.; Menon, R.; Overton, A.K.; Cheng, J.; Lynch, M.D.J.; Nissimov, J.I.; Charles, T.C. A Novel Tiled Amplicon Sequencing Assay Targeting the Tomato Brown Rugose Fruit Virus (ToBRFV) Genome Reveals Widespread Distribution in Municipal Wastewater Treatment Systems in the Province of Ontario, Canada. Viruses 2024, 16, 460. https://doi.org/10.3390/v16030460
Nash D, Ellmen I, Knapp JJ, Menon R, Overton AK, Cheng J, Lynch MDJ, Nissimov JI, Charles TC. A Novel Tiled Amplicon Sequencing Assay Targeting the Tomato Brown Rugose Fruit Virus (ToBRFV) Genome Reveals Widespread Distribution in Municipal Wastewater Treatment Systems in the Province of Ontario, Canada. Viruses. 2024; 16(3):460. https://doi.org/10.3390/v16030460
Chicago/Turabian StyleNash, Delaney, Isaac Ellmen, Jennifer J. Knapp, Ria Menon, Alyssa K. Overton, Jiujun Cheng, Michael D. J. Lynch, Jozef I. Nissimov, and Trevor C. Charles. 2024. "A Novel Tiled Amplicon Sequencing Assay Targeting the Tomato Brown Rugose Fruit Virus (ToBRFV) Genome Reveals Widespread Distribution in Municipal Wastewater Treatment Systems in the Province of Ontario, Canada" Viruses 16, no. 3: 460. https://doi.org/10.3390/v16030460