
Cross-Species Transmission Potential of H4 Avian Influenza Viruses in China: Epidemiological and Evolutionary Study
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Virus Isolation and Identification
2.3. RNA Extraction and Genome Sequencing
2.4. Sequence Collection and Collation
2.5. Information Collation and Epidemiological Analysis
2.6. Sequence Alignment and Phylogenetic Analysis
2.7. Genotypic Analysis
3. Results
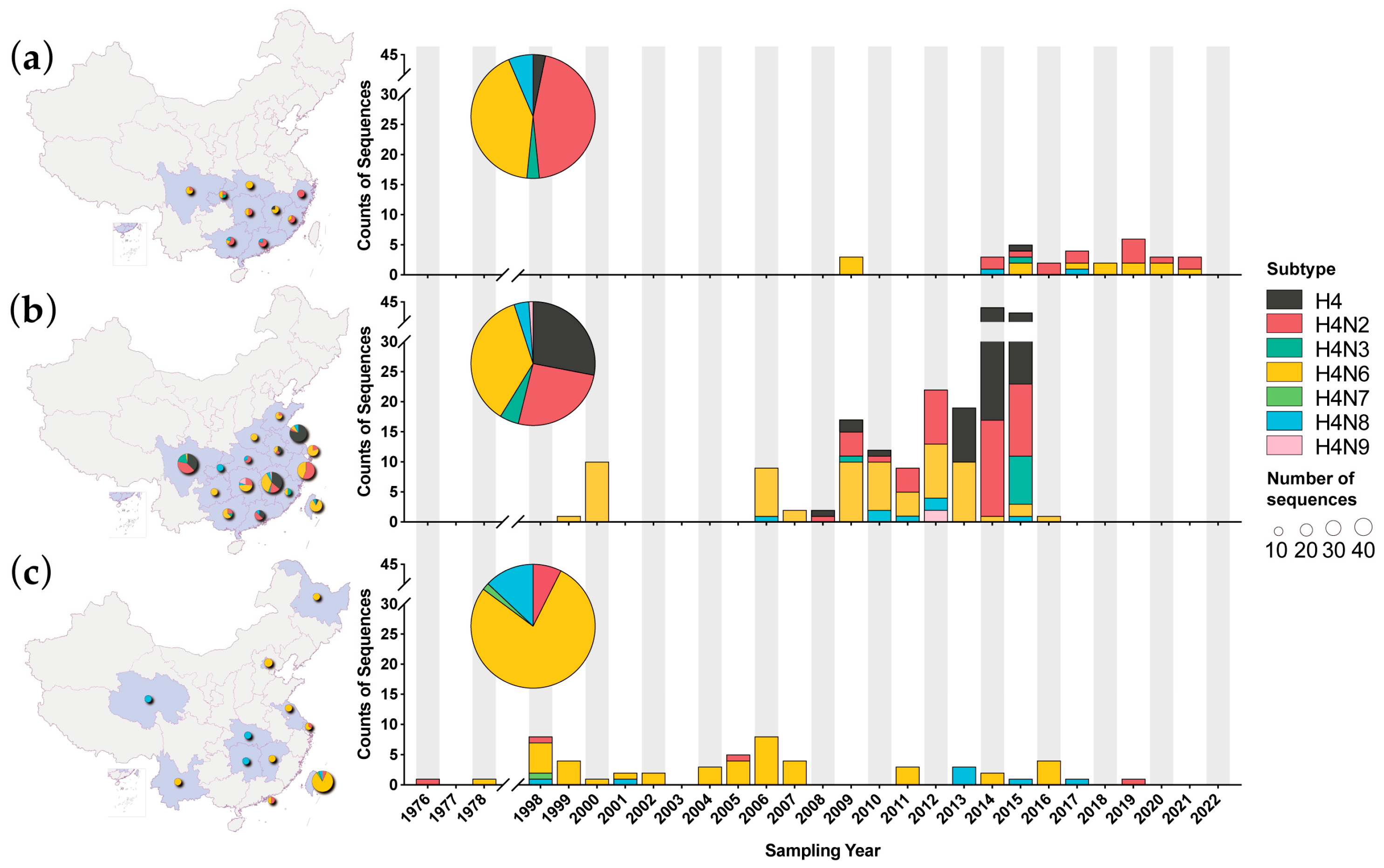
3.1. H4 AIVs in China
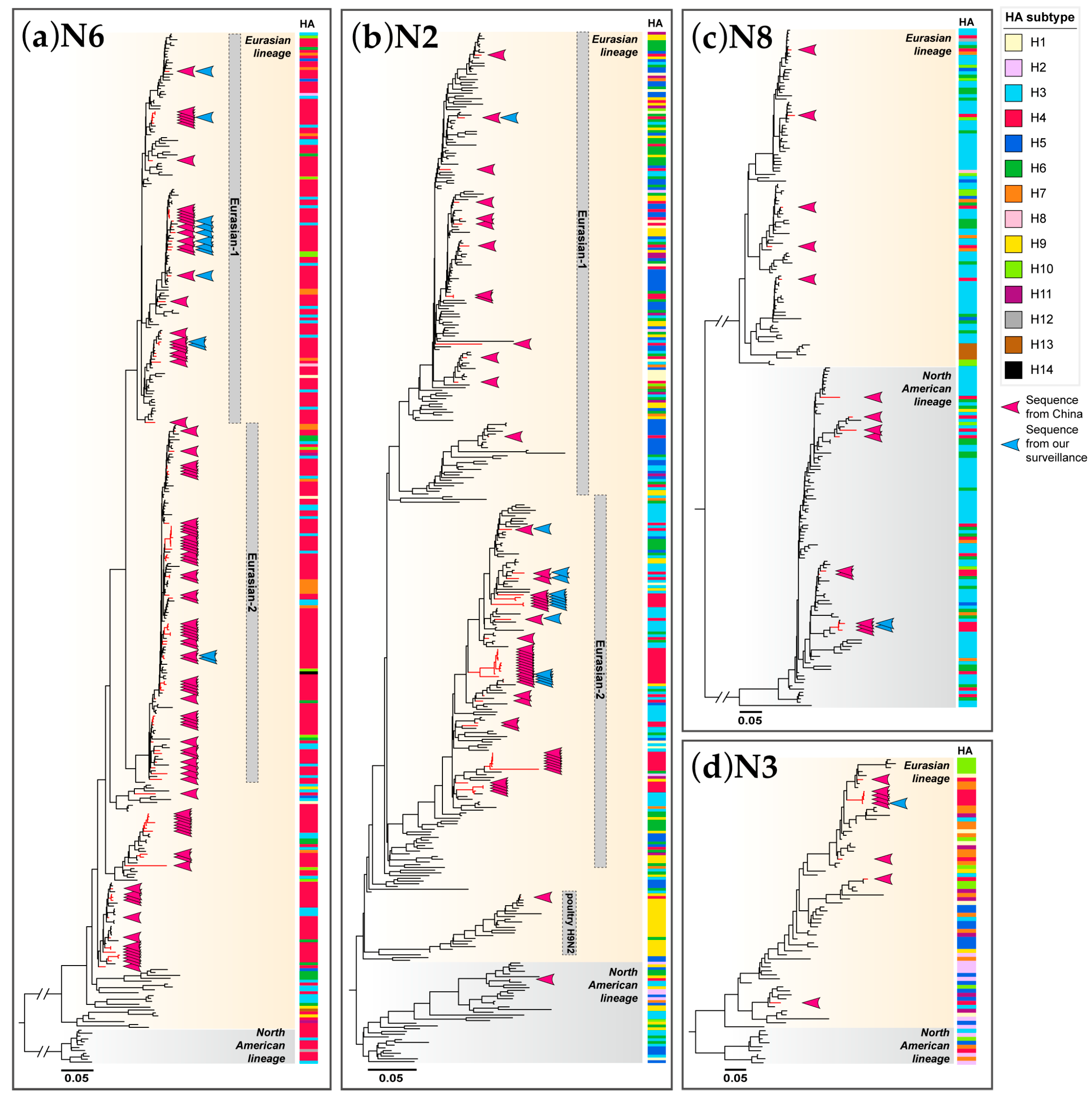
3.2. Phylogenetic Analysis of HA Gene
3.3. Phylogenetic Analysis of NA Genes
3.4. Phylogenetic Analysis of Internal Protein-Coding Genes
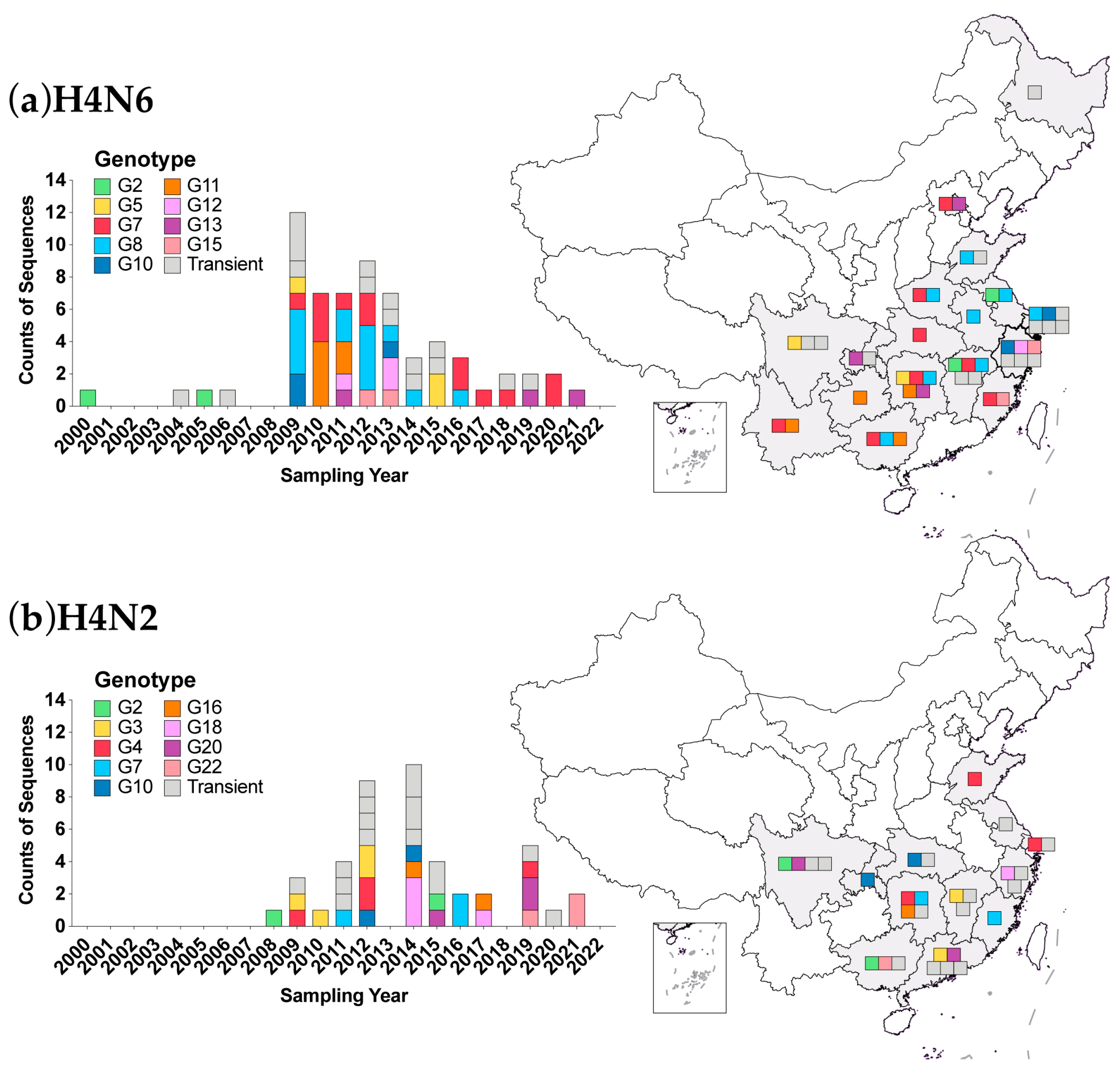
3.5. Genotypic Analysis
3.6. Molecular Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Fouchier, R.A.M.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A.D.M.E. Characterization of a Novel Influenza A Virus Hemagglutinin Subtype (H16) Obtained from Black-Headed Gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.A.; Chen, L.-M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A Distinct Lineage of Influenza A Virus from Bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Saunders-Hastings, P.R.; Krewski, D. Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission. Pathogens 2016, 5, 66. [Google Scholar] [CrossRef]
- Biggerstaff, M.; Cauchemez, S.; Reed, C.; Gambhir, M.; Finelli, L. Estimates of the Reproduction Number for Seasonal, Pandemic, and Zoonotic Influenza: A Systematic Review of the Literature. BMC Infect. Dis. 2014, 14, 480. [Google Scholar] [CrossRef]
- Kawaoka, Y.; Krauss, S.; Webster, R.G. Avian-to-Human Transmission of the PB1 Gene of Influenza A Viruses in the 1957 and 1968 Pandemics. J. Virol. 1989, 63, 4603–4608. [Google Scholar] [CrossRef]
- Lindstrom, S.E.; Cox, N.J.; Klimov, A. Genetic Analysis of Human H2N2 and Early H3N2 Influenza Viruses, 1957–1972: Evidence for Genetic Divergence and Multiple Reassortment Events. Virology 2004, 328, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Krafft, A.E.; Bijwaard, K.E.; Fanning, T.G. Initial Genetic Characterization of the 1918 “Spanish” Influenza Virus. Science 1997, 275, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, W.; Yang, L.; Shu, Y. The Epidemiology, Virology, and Pathogenicity of Human Infections with Avian Influenza Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038620. [Google Scholar] [CrossRef]
- Chen, Y.; Bai, T.; Shu, Y. Poultry to Human Passport: Cross-Species Transmission of Zoonotic H7N9 Avian Influenza Virus to Humans. Zoonoses 2022, 2, 996. [Google Scholar] [CrossRef]
- Wille, M.; Tolf, C.; Latorre-Margalef, N.; Fouchier, R.A.M.; Halpin, R.A.; Wentworth, D.E.; Ragwani, J.; Pybus, O.G.; Olsen, B.; Waldenström, J. Evolutionary Features of a Prolific Subtype of Avian Influenza A Virus in European Waterfowl. Virus Evol. 2022, 8, veac074. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Deng, G.; Shi, J.; Wang, S.; Zhang, Q.; Kong, H.; Gu, C.; Guan, Y.; Suzuki, Y.; Li, Y.; et al. Genetics, Receptor Binding, Replication, and Mammalian Transmission of H4 Avian Influenza Viruses Isolated from Live Poultry Markets in China. J. Virol. 2016, 90, 1455–1469. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tang, L.; Gu, X.; Bo, S.; Ming, L.; Ma, M.; Zhao, C.; Sun, K.; Liu, Y.; He, G. Characterization of Avian Influenza A (H4N2) Viruses Isolated from Wild Birds in Shanghai during 2019 to 2021. Poult. Sci. 2023, 102, 102948. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Huang, T.; Chen, X.; Zhang, J.; Wang, Q.; Zhang, C.; Zhang, T.; Zhou, L.; Shu, L.; Long, C.; et al. Genomic Characterizations of H4 Subtype Avian Influenza Viruses from Live Poultry Markets in Sichuan Province of China, 2014–2015. Sci. China Life Sci. 2018, 61, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Donis, R.O.; Bean, W.J.; Kawaoka, Y.; Webster, R.G. Distinct Lineages of Influenza Virus H4 Hemagglutinin Genes in Different Regions of the World. Virology 1989, 169, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Abolnik, C.; Cornelius, E.; Bisschop, S.P.R.; Romito, M.; Verwoerd, D. Phylogenetic Analyses of Genes from South African LPAI Viruses Isolated in 2004 from Wild Aquatic Birds Suggests Introduction by Eurasian Migrants. Dev. Biol. 2006, 124, 189–199. [Google Scholar]
- Pasick, J.; Pedersen, J.; Hernandez, M.S. Avian Influenza in North America, 2009–2011. Avian Dis. 2012, 56, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Okamatsu, M.; Nishi, T.; Nomura, N.; Yamamoto, N.; Sakoda, Y.; Sakurai, K.; Chu, H.D.; Thanh, L.P.; Van Nguyen, L.; Van Hoang, N.; et al. The Genetic and Antigenic Diversity of Avian Influenza Viruses Isolated from Domestic Ducks, Muscovy Ducks, and Chickens in Northern and Southern Vietnam, 2010–2012. Virus Genes 2013, 47, 317–329. [Google Scholar] [CrossRef]
- Abente, E.J.; Gauger, P.C.; Walia, R.R.; Rajao, D.S.; Zhang, J.; Harmon, K.M.; Killian, M.L.; Vincent, A.L. Detection and Characterization of an H4N6 Avian-Lineage Influenza A Virus in Pigs in the Midwestern United States. Virology 2017, 511, 56–65. [Google Scholar] [CrossRef]
- Gulyaeva, M.; Sobolev, I.; Sharshov, K.; Kurskaya, O.; Alekseev, A.; Shestopalova, L.; Kovner, A.; Bi, Y.; Shi, W.; Shchelkanov, M.; et al. Characterization of Avian-like Influenza A (H4N6) Virus Isolated from Caspian Seal in 2012. Virol. Sin. 2018, 33, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, X.; Li, S.; Guo, X.; Yang, Y.; Jin, M. Complete Genome Sequence of a Novel H4N1 Influenza Virus Isolated from a Pig in Central China. J. Virol. 2012, 86, 13879. [Google Scholar] [CrossRef] [PubMed]
- Karasin, A.I.; Brown, I.H.; Carman, S.; Olsen, C.W. Isolation and Characterization of H4N6 Avian Influenza Viruses from Pigs with Pneumonia in Canada. J. Virol. 2000, 74, 9322–9327. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Qi, W.; Chen, J.; Cao, N.; Zhu, W.; Yuan, L.; Wang, H.; Zhang, G. Complete Genome Sequence of an Avian-Like H4N8 Swine Influenza Virus Discovered in Southern China. J. Virol. 2012, 86, 9542. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Qi, W.; Chen, J.; Zhu, W.; Huang, Z.; Xie, J.; Zhang, G. Seroepidemiological Evidence of Avian Influenza A Virus Transmission to Pigs in Southern China. J. Clin. Microbiol. 2020, 51, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, A.; Takada, A.; Okazaki, K.; Shortridge, K.F.; Kida, H. Seroepidemiological Evidence of Avian H4, H5, and H9 Influenza A Virus Transmission to Pigs in Southeastern China. Vet. Microbiol. 2002, 88, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kayali, G.; Ortiz, E.J.; Chorazy, M.L.; Gray, G.C. Evidence of Previous Avian Influenza Infection among US Turkey Workers. Zoonoses Public Health 2010, 57, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Kayali, G.; Barbour, E.; Dbaibo, G.; Tabet, C.; Saade, M.; Shaib, H.A.; Debeauchamp, J.; Webby, R.J. Evidence of Infection with H4 and H11 Avian Influenza Viruses among Lebanese Chicken Growers. PLoS ONE 2011, 6, e26818. [Google Scholar] [CrossRef]
- Shortridge, K.F. Avian Influenza A Viruses of Southern China and Hong Kong: Ecological Aspects and Implications for Man. Bull. World Health Organ. 1982, 60, 129–135. [Google Scholar]
- Luo, S.; Xie, Z.; Li, M.; Li, D.; Xie, L.; Huang, J.; Zhang, M.; Zeng, T.; Wang, S.; Fan, Q.; et al. Survey of Low Pathogenic Avian Influenza Viruses in Live Poultry Markets in Guangxi Province, Southern China, 2016–2019. Sci. Rep. 2021, 11, 23223. [Google Scholar] [CrossRef]
- Luo, S.; Xie, Z.; Xie, Z.; Xie, L.; Huang, L.; Huang, J.; Deng, X.; Zeng, T.; Wang, S.; Zhang, Y.; et al. Surveillance of Live Poultry Markets for Low Pathogenic Avian Influenza Viruses in Guangxi Province, Southern China, from 2012–2015. Sci. Rep. 2017, 7, 17577. [Google Scholar] [CrossRef]
- Peng, Y.; Xie, Z.; Liu, J.; Pang, Y.; Deng, X.; Xie, Z.; Xie, L.; Fan, Q.; Luo, S. Epidemiological Surveillance of Low Pathogenic Avian Influenza Virus (LPAIV) from Poultry in Guangxi Province, Southern China. PLoS ONE 2013, 8, e77132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St. George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de Novo Short Read Assembly Using de Bruijn Graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Yang, L.; Zhu, W.; Li, X.; Bo, H.; Zhang, Y.; Zou, S.; Gao, R.; Dong, J.; Zhao, X.; Chen, W.; et al. Genesis and Dissemination of Highly Pathogenic H5N6 Avian Influenza Viruses. J. Virol. 2017, 91, e02199-16. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Perez, D.R. Amino Acid 226 in the Hemagglutinin of H9N2 Influenza Viruses Determines Cell Tropism and Replication in Human Airway Epithelial Cells. J. Virol. 2007, 81, 5181–5191. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Qi, J.; Bi, Y.; Zhang, W.; Wang, M.; Zhang, B.; Wang, M.; Liu, J.; Yan, J.; Shi, Y.; et al. Adaptation of Avian Influenza A (H6N1) Virus from Avian to Human Receptor-Binding Preference. EMBO J. 2015, 34, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ibrahim, M.S.; Ellakany, H.F.; Kawashita, N.; Mizuike, R.; Hiramatsu, H.; Sriwilaijaroen, N.; Takagi, T.; Suzuki, Y.; Ikuta, K. Acquisition of Human-Type Receptor Binding Specificity by New H5N1 Influenza Virus Sublineages during Their Emergence in Birds in Egypt. PLoS Pathog. 2011, 7, e1002068. [Google Scholar] [CrossRef] [PubMed]
- McKimm-Breschkin, J.; Trivedi, T.; Hampson, A.; Hay, A.; Klimov, A.; Tashiro, M.; Hayden, F.; Zambon, M. Neuraminidase Sequence Analysis and Susceptibilities of Influenza Virus Clinical Isolates to Zanamivir and Oseltamivir. Antimicrob. Agents Chemother. 2003, 47, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, Y.J.; Park, J.H.; Lee, J.-H.; Baek, Y.H.; Song, M.-S.; Oh, T.-K.; Han, H.-S.; Pascua, P.N.Q.; Choi, Y.-K. Emergence of Amantadine-Resistant H3N2 Avian Influenza A Virus in South Korea. J. Clin. Microbiol. 2008, 46, 3788–3790. [Google Scholar] [CrossRef]
- Abed, Y.; Goyette, N.; Boivin, G. Generation and Characterization of Recombinant Influenza A (H1N1) Viruses Harboring Amantadine Resistance Mutations. Antimicrob. Agents Chemother. 2005, 49, 556–559. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H.; Jiao, P.; Deng, G.; Tian, G.; Li, Y.; Hoffmann, E.; Webster, R.G.; Matsuoka, Y.; Yu, K. Molecular Basis of Replication of Duck H5N1 Influenza Viruses in a Mammalian Mouse Model. J. Virol. 2005, 79, 12058–12064. [Google Scholar] [CrossRef]
- Yamada, S.; Hatta, M.; Staker, B.L.; Watanabe, S.; Imai, M.; Shinya, K.; Sakai-Tagawa, Y.; Ito, M.; Ozawa, M.; Watanabe, T.; et al. Biological and Structural Characterization of a Host-Adapting Amino Acid in Influenza Virus. PLoS Pathog. 2010, 6, e1001034. [Google Scholar] [CrossRef]
- Mok, C.K.P.; Lee, H.H.Y.; Lestra, M.; Nicholls, J.M.; Chan, M.C.W.; Sia, S.F.; Zhu, H.; Poon, L.L.M.; Guan, Y.; Peiris, J.S.M. Amino Acid Substitutions in Polymerase Basic Protein 2 Gene Contribute to the Pathogenicity of the Novel A/H7N9 Influenza Virus in Mammalian Hosts. J. Virol. 2014, 88, 3568–3576. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Kiso, M.; Yasuhara, A.; Ito, M.; Shu, Y.; Kawaoka, Y. Enhanced Replication of Highly Pathogenic Influenza A(H7N9) Virus in Humans. Emerg. Infect. Dis. 2018, 24, 746–750. [Google Scholar] [CrossRef]
- Chen, G.-W.; Shih, S.-R. Genomic Signatures of Influenza A Pandemic (H1N1) 2009 Virus. Emerg. Infect. Dis. 2009, 15, 1897–1903. [Google Scholar] [CrossRef]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent Host Markers in Pandemic and H5N1 Influenza Viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef]
- Miotto, O.; Heiny, A.; Tan, T.W.; August, J.T.; Brusic, V. Identification of Human-to-Human Transmissibility Factors in PB2 Proteins of Influenza A by Large-Scale Mutual Information Analysis. BMC Bioinform. 2008, 9 (Suppl. 1), S18. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Zeng, X.; Guan, D.; Zou, L.; Yi, L.; Liang, L.; Ni, H.; Kang, M.; Zhang, X.; et al. Continuing Reassortment Leads to the Genetic Diversity of Influenza Virus H7N9 in Guangdong, China. J. Virol. 2014, 88, 8297–8306. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jin, T.; Cui, Y.; Pu, X.; Li, J.; Xu, J.; Liu, G.; Jia, H.; Liu, D.; Song, S.; et al. Influenza H7N9 and H9N2 Viruses: Coexistence in Poultry Linked to Human H7N9 Infection and Genome Characteristics. J. Virol. 2014, 88, 3423–3431. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fan, H.; Raghwani, J.; Lam, T.T.-Y.; Li, J.; Pybus, O.G.; Yao, H.-W.; Wo, Y.; Liu, K.; An, X.-P.; et al. Occurrence and Reasortment of Avian Influenza A (H7N9) Viruses Derived from Coinfected Birds in China. J. Virol. 2014, 88, 13344–13351. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Bishop, M.A.; Trovão, N.S.; Ineson, K.M.; Schaefer, A.L.; Puryear, W.B.; Zhou, K.; Foss, A.D.; Clark, D.E.; MacKenzie, K.G.; et al. Ecological Divergence of Wild Birds Drives Avian Influenza Spillover and Global Spread. PLoS Pathog. 2022, 18, e1010062. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.-S.; Kang, S.-I.; Lee, Y.-N.; Lee, E.-K.; Kim, W.-Y.; Lee, Y.-J. Bridging the Local Persistence and Long-Range Dispersal of Highly Pathogenic Avian Influenza Virus (HPAIv): A Case Study of HPAIv-Infected Sedentary and Migratory Wildfowls Inhabiting Infected Premises. Viruses 2022, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X.; et al. The Genesis and Source of the H7N9 Influenza Viruses Causing Human Infections in China. Nature 2013, 502, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; He, S.; Walker, D.; Zhou, N.; Perez, D.R.; Mo, B.; Li, F.; Huang, X.; Webster, R.G.; Webby, R.J. The Influenza Virus Gene Pool in a Poultry Market in South Central China. Virology 2003, 305, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Chen, Q.; Wang, Q.; Chen, J.; Jin, T.; Wong, G.; Quan, C.; Liu, J.; Wu, J.; Yin, R.; et al. Genesis, Evolution and Prevalence of H5N6 Avian Influenza Viruses in China. Cell Host Microbe 2016, 20, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Peng, X.; Peng, X.; Cheng, L.; Lu, X.; Jin, C.; Xie, T.; Yao, H.; Wu, N. Genetic Characterization of Natural Reassortant H4 Subtype Avian Influenza Viruses Isolated from Domestic Ducks in Zhejiang Province in China from 2013 to 2014. Virus Genes 2015, 51, 347–355. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.; Zhang, Y.; Yang, J.; Yang, L.; Li, X.; Bo, H.; Liu, J.; Tan, M.; Zhu, W.; Wang, D.; et al. Cross-Species Transmission Potential of H4 Avian Influenza Viruses in China: Epidemiological and Evolutionary Study. Viruses 2024, 16, 353. https://doi.org/10.3390/v16030353
Lin S, Zhang Y, Yang J, Yang L, Li X, Bo H, Liu J, Tan M, Zhu W, Wang D, et al. Cross-Species Transmission Potential of H4 Avian Influenza Viruses in China: Epidemiological and Evolutionary Study. Viruses. 2024; 16(3):353. https://doi.org/10.3390/v16030353
Chicago/Turabian StyleLin, Shuxia, Ye Zhang, Jiaying Yang, Lei Yang, Xiyan Li, Hong Bo, Jia Liu, Min Tan, Wenfei Zhu, Dayan Wang, and et al. 2024. "Cross-Species Transmission Potential of H4 Avian Influenza Viruses in China: Epidemiological and Evolutionary Study" Viruses 16, no. 3: 353. https://doi.org/10.3390/v16030353