
Detection of H5N1 High Pathogenicity Avian Influenza Viruses in Four Raptors and Two Geese in Japan in the Fall of 2022
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Diagnosis of Specimens from Wild Bird Cases
2.2. Sequencing
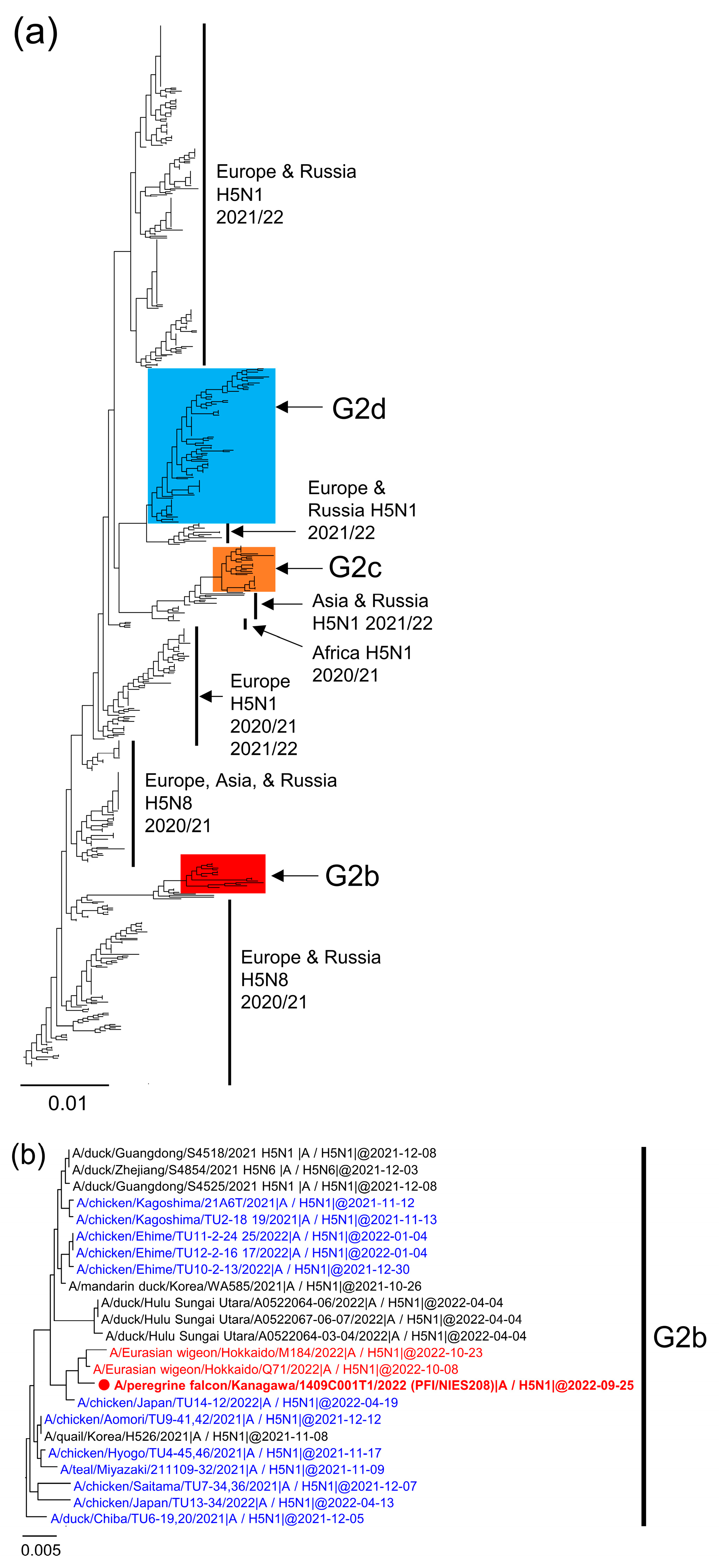
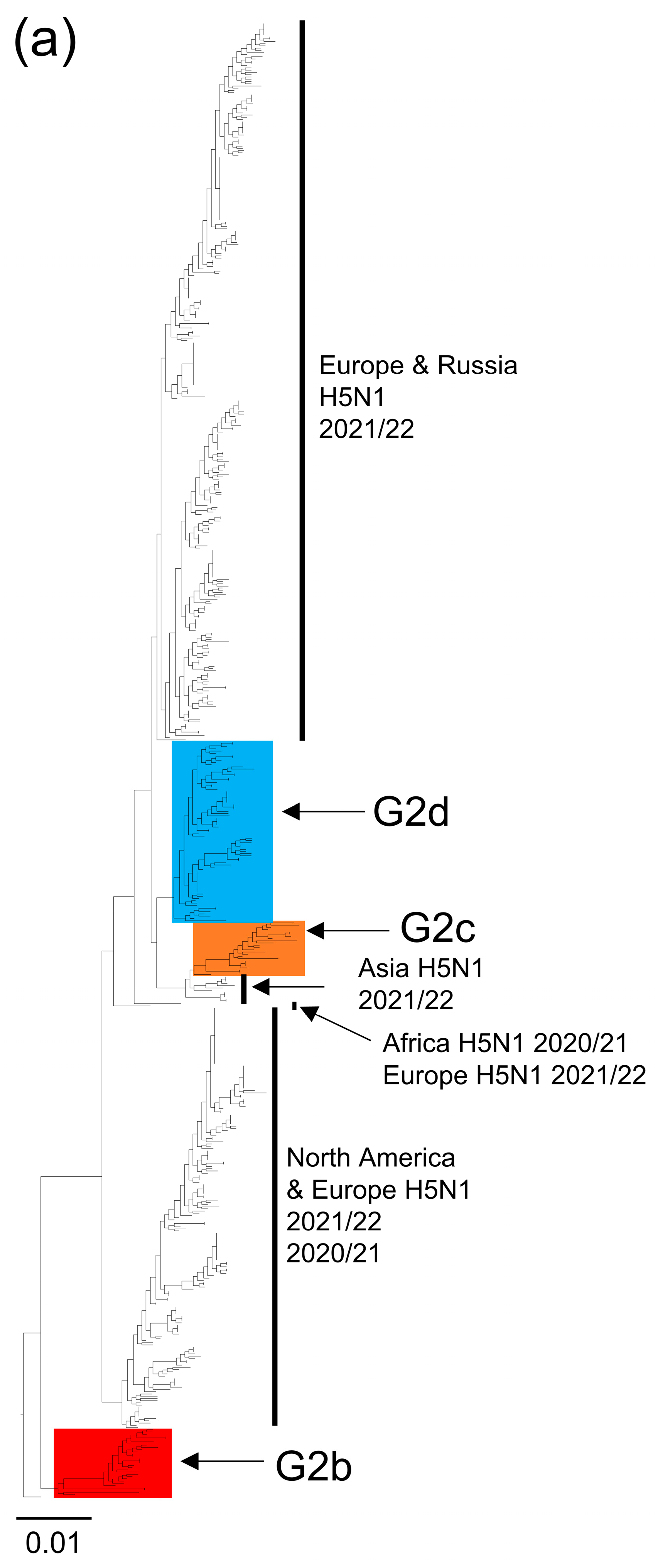
2.3. Phylogenetic Analysis
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef] [PubMed]
- WHO; OIE; FAO; H5N1. Evolution Working Group. Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1). Emerg. Infect. Dis. 2008, 14, e1. [Google Scholar] [CrossRef]
- Okuya, K.; Mine, J.; Tokorozaki, K.; Kojima, I.; Esaki, M.; Miyazawa, K.; Tsunekuni, R.; Sakuma, S.; Kumagai, A.; Takadate, Y.; et al. Genetically Diverse Highly Pathogenic Avian Influenza A(H5N1/H5N8) Viruses among Wild Waterfowl and Domestic Poultry, Japan, 2021. Emerg. Infect. Dis. 2022, 28, 1451–1455. [Google Scholar] [CrossRef]
- Takadate, Y.; Tsunekuni, R.; Kumagai, A.; Mine, J.; Kikutani, Y.; Sakuma, S.; Miyazawa, K.; Uchida, Y. Different Infectivity and Transmissibility of H5N8 and H5N1 High Pathogenicity Avian Influenza Viruses Isolated from Chickens in Japan in the 2021/2022 Season. Viruses 2023, 15, 265. [Google Scholar] [CrossRef] [PubMed]
- Isoda, N.; Onuma, M.; Hiono, T.; Sobolev, I.; Lim, H.Y.; Nabeshima, K.; Honjyo, H.; Yokoyama, M.; Shestopalov, A.; Sa-koda, Y. Detection of New H5N1 High Pathogenicity Avian Influenza Viruses in Winter 2021–2022 in the Far East, Which Are Genetically Close to Those in Europe. Viruses 2022, 14, 2168. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.-G.; Lee, Y.-N.; Lee, D.-H.; Shin, J.-I.; Lee, J.-H.; Chung, D.H.; Lee, E.-K.; Heo, G.-B.; Sagong, M.; Kye, S.-J.; et al. Multiple Reassortants of H5N8 Clade 2.3.4.4b Highly Pathogenic Avian Influenza Viruses Detected in South Korea during the Winter of 2020–2021. Viruses 2021, 13, 490. [Google Scholar] [CrossRef] [PubMed]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Mirinaviciute, G.; Niqueux, É.; Stahl, K.; Staubach, C.; Terregino, C.; et al. Avian influenza overview December 2022–March 2023. EFSA J. 2023, 21, e07917. [Google Scholar] [CrossRef] [PubMed]
- Onuma, M.; Kakogawa, M.; Yanagisawa, M.; Haga, A.; Okano, T.; Neagari, Y.; Okano, T.; Goka, K.; Asakawa, M. Characterizing the temporal patterns of avian influenza virus introduction into Japan by migratory birds. J. Vet. Med. Sci. 2017, 79, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Agriculture Forestry and Fisheries. Information on Avian Influenza in Poultry. Available online: https://www.maff.go.jp/j/syouan/douei/tori/ (accessed on 12 July 2023).
- Ministry of the Environment Government of Japan. Information on Avian Influenza in Wild Birds. Available online: https://www.env.go.jp/nature/dobutsu/bird_flu/ (accessed on 12 July 2023).
- National Agriculture and Food Research Organization. Genetic Characterization of H5N1 HPAIV Detected from a Peregrine Falcon in Kanagawa Prefecture in September 2022. Available online: https://www.naro.go.jp/publicity_report/press/laboratory/niah/155326.html (accessed on 2 December 2022).
- Ministry of Agriculture Forestry and Fisheries. Information on High Pathogenicity Avian Influenza Outbreaks in 2022/2023 Season. Available online: https://www.maff.go.jp/j/syouan/douei/tori/220929.html (accessed on 12 July 2023).
- National Agriculture and Food Research Organization. Genetic Characterization of H5N1 and H5N2 HPAIVs Detected from Poultry during 2022/2023 Season. Available online: https://www.naro.go.jp/publicity_report/press/laboratory/niah/157024.html (accessed on 12 July 2023).
- Ministry of the Environment Government of Japan. Information on Avian Influenza in Wild Birds in the 2022/2023 Season. Available online: https://www.env.go.jp/content/000136864.pdf (accessed on 25 July 2023).
- Heine, H.G.; Foord, A.J.; Wang, J.; Valdeter, S.; Walker, S.; Morrissy, C.; Wong, F.Y.; Meehan, B. Detection of highly pathogenic zoonotic influenza virus H5N6 by reverse-transcriptase quantitative polymerase chain reaction. Virol. J. 2015, 12, 18. [Google Scholar] [CrossRef]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Tsai, H.-J.; Chang, C.-M. A complete molecular diagnostic procedure for applications in surveillance and subtyping of avian influenza virus. BioMed Res. Int. 2014, 2014, 653056. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 1999; Volume 41, pp. 95–98. [Google Scholar]
- World Organization for Animal Health. Avian influenza (Including Infection with High Pathogenicity Avian Influenza Viruses). Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.03.04_AI.pdf (accessed on 2 December 2022).
- Ip, H.S.; Uhm, S.; Killian, M.L.; Torchetti, M.K. An Evaluation of Avian Influenza Virus Whole-Genome Sequencing Approaches Using Nanopore Technology. Microorganisms 2023, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 708. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. Information on FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 July 2023).
- Pohlmann, A.; King, J.; Fusaro, A.; Zecchin, B.; Banyard, A.C.; Brown, I.H.; Byrne, A.M.P.; Beerens, N.; Liang, Y.; Heutink, R.; et al. Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021. mBio 2022, 13, e0060922. [Google Scholar] [CrossRef] [PubMed]
- Ministry of the Environment Government of Japan. Survey of Migratory Bird Arrivals in Japan. 2023. Available online: https://www.env.go.jp/nature/dobutsu/bird_flu/migratory/ap_wr_transit/index.html (accessed on 12 July 2023).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Host Species | Prefecture | Date | Results of Virus Isolation | Strain Name (Abbreviation) | Accession Number |
| NIES208 | Peregrine Falcon (Falco peregrinus) | Kanagawa | 25 September | + | A/peregrine falcon/Kanagawa/1409C001T1/2022 (PFI/NIES208) | EPI2223228-33 (GISAID) |
| NIES209 | Greater white-fronted goose (Anser albifrons) | Miyagi | 4 October | + | A/greater white-fronted goose/Miyagi/0410D001/2022 (GWFD/NIES209) | EPI2197765-72 (GISAID) |
| NIES212 | Peregrine Falcon | Fukui | 11 October | - | A/peregrine falcon/Fukui/NIES212/2022 (PFD/NIES212) | LC762442, LC762448, LC774734, LC774738, LC774739, LC774741 (GenBank) |
| NIES214 | Greater white-fronted goose | Miyagi | 14 October | + | A/greater white-fronted goose/Miyagi/NIES214/2022 (GWFD/NIES214) | LC762443, LC762449, LC774733, LC774735-7, LC774740, LC774742 (GenBank) |
| NIES231 | Peregrine Falcon | Niigata | 16 October | + | A/peregrine falcon/Niigata/1510C001T/2022 (PFD/NIES231) | EPI2632285-92 (GISAID) |
| NIES260 | Eastern Buzzard (Buteo japonicus) | Niigata | 21 October | + | A/eastern buzzard/Niigata/1501B001/2022 (EBI/NIES260) | EPI2318005-12 (GISAID) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabeshima, K.; Takadate, Y.; Soda, K.; Hiono, T.; Isoda, N.; Sakoda, Y.; Mine, J.; Miyazawa, K.; Onuma, M.; Uchida, Y. Detection of H5N1 High Pathogenicity Avian Influenza Viruses in Four Raptors and Two Geese in Japan in the Fall of 2022. Viruses 2023, 15, 1865. https://doi.org/10.3390/v15091865
Nabeshima K, Takadate Y, Soda K, Hiono T, Isoda N, Sakoda Y, Mine J, Miyazawa K, Onuma M, Uchida Y. Detection of H5N1 High Pathogenicity Avian Influenza Viruses in Four Raptors and Two Geese in Japan in the Fall of 2022. Viruses. 2023; 15(9):1865. https://doi.org/10.3390/v15091865
Chicago/Turabian StyleNabeshima, Kei, Yoshihiro Takadate, Kosuke Soda, Takahiro Hiono, Norikazu Isoda, Yoshihiro Sakoda, Junki Mine, Kohtaro Miyazawa, Manabu Onuma, and Yuko Uchida. 2023. "Detection of H5N1 High Pathogenicity Avian Influenza Viruses in Four Raptors and Two Geese in Japan in the Fall of 2022" Viruses 15, no. 9: 1865. https://doi.org/10.3390/v15091865