1. Introduction
The ability of combination antiretroviral therapy (cART) to suppress HIV-1 replication safely and effectively has rendered it a manageable chronic condition. However, cART is not a cure and has no direct effect on the HIV-1 DNA reservoir [
1,
2,
3]. Infectious proviruses present in long-lived cells comprise the stable HIV-1 DNA reservoir, which remains the main barrier to cure [
4]. The development of drug resistance related to several factors, including poor adherence and provirus evolution [
5,
6,
7,
8], maintains the risk of viral rebound. Therefore, drug resistance testing in HIV-1 plasma RNA, before treatment initiation or in case of poor treatment response, remains an essential part of informed tailored therapy [
1,
9], but monitoring of archived/low percentages of drug resistance mutations (DRMs) on the DNA level is gaining significance for potential viral rebound despite ART.
Increased use of next-generation sequencing (NGS) for HIV-1 drug resistance (HIVDR) testing on HIV DNA offers enhanced sensitivity not obtainable by traditional Sanger sequencing [
10,
11,
12]. Several DNA HIVDR NGS-based assays are available, with the most prominent clinically used assays reporting amplification success rates of 58.4% to 94% from purified peripheral blood mononuclear cells (PBMCs) [
13,
14]. Other studies reported HIV-1 amplification sensitivity as low as 10 and 131 DNA copies per reaction [
15,
16]. The clear benefit of NGS HIVDR genotyping supports its use in the clinical environment, although low-level resistance results should be interpreted with care [
17,
18]. The clinical use of total DNA HIVDR genotyping has gained interest for monitoring HIV-1 DNA stability in people with suppressed viral loads [
1,
19]. HIVDR genotyping in DNA could prove useful for people that are virologically suppressed and require regimen switching, preliminary investigation of clonal viremia or for monitoring HIV-1 DNA stability (archived resistance) in clinical trials investigating novel therapeutic approaches [
19,
20,
21].
In this study, we present an ultra-sensitive assay for HIV-1 DNA drug resistance genotyping coupled with a robust bioinformatics pipeline. We report our results of targeted drug resistance genotyping covering the protease, reverse transcriptase and integrase regions.
2. Materials and Methods
2.1. Evaluation Specimens
Buffy coat samples (BCS) isolated from 148 adult and 18 paediatric whole blood samples received for routine HIV-1 viral load testing with unknown subtypes were selected for this evaluation. All samples were pseudo-anonymised (patient identifying information was replaced with unique numbers), and the panel was created as part of development of an assay for diagnostic purposes under Human Tissue Authority (HTA) license, for the future use on the stored samples in the D3/Penta 21 paediatric randomised controlled trial (NCT04337450). Patient samples with a range of viral loads and expected variation in HIV-1 DNA load were included in this assessment. Nucleic acid isolation was performed on the QIAsymphony (Qiagen, Hilden, Germany) automated platform using the DSP virus/pathogen kit and human whole blood 1000 protocol, as per manufacturer’s instructions.
In total, 166 residual blood samples collected for routine HIV-1 viral load analysis were selected for this evaluation. Sample subsets for analyses included 30 BCS from adults selected for Sanger vs. NGS comparison. NGS evaluation was performed on 148 adult samples (this includes the initial 30 adult samples) and 18 samples from children submitted for HIV-1 testing (
Table 1).
2.2. Amplification and Sequencing
A nested PCR approach was selected for enhanced sensitivity and specificity. This assay characterises HIV-1 Pol by targeting the protease to reverse transcriptase (PrRT) and integrase (INT) regions separately. PCR reaction mixes contain the following: 1× Qiagen PCR buffer, 2.5 units Qiagen HotStarTaq polymerase, 400 pmol forward and reverse primers, nuclease-free water and 10 µL template. Primers used for first round (locations based on HXB2) PrRT1: GAA GAA ATG ATG ACA GCA TGT CAG GG (1819), PrRT 2: TAA TTT ATC TAC TTG TTC ATT TCC TCC AAT (4173), INT1: TTC TTC CTG CCA TAG GAR ATG CCT AAG (4143), INT2: AGG AGC AGA AAC TTW CTA TGT AGA TGG (5571); for the second round, the following were used: PrRT 3: AGA CAG GCT AAT TTT TTA GGG A (2074), PrRT 4: ATG GYT CTT GAT AAA TTT GAT ATG TCC (3559), INT3: TTC RGG ATY AGA AGT AAA YAT AGT AAC AG (4150), INT4: TCC TGT ATG CAR ACC CCA ATA T (5518). The thermocycling conditions were as follows: 1 cycle at 93 °C for 12 min; 1 cycle at 95 °C for 30 s, 65 °C for 45 s, 72 °C for 3 min; 1 cycle at 95 °C for 30 s, 60 °C for 45 s, 72 °C for 3 min; 45 cycles of 95 °C for 30 s, (58 °C for PCR1 and 55 °C for PCR2) for 45 s, 72 °C for 3 min with a final extension of 10 min at 72 °C. PCR products were analysed with gel electrophoresis for PCR products of ~1.4 kb. Positive amplicon library preparation was performed with Illumina Nextera XT DNA library prep kit (Illumina, San Diego, CA, USA), as per manufacturer’s instructions. Sequencing was performed on an Illumina MiSeq system using v3 600-cycle reagent kits.
2.3. Sensitivity
PCR assay sensitivity was evaluated using serially diluted DNA of a quantified 8E5 clonal HIV-1 cell line from 515 copies/µL to below 1 copy/µL. Both PrRT and INT PCR reactions were performed in triplicate at each dilution point. Additionally, real-time-PCR-quantified DNA from a patient sample was also serially diluted (12.7–0.2 copies/µL) to confirm results obtained from 8E5 serial dilution experiments.
We selected a total of 24 BCS, adult (n = 9) and paediatric (n = 15), to determine the HIV-1 DNA assay input thresholds. Human and HIV-1 DNA were quantified according to published methods [
22,
23]. Furthermore, we determined the total HIV-1 DNA loads compared to human DNA copies of selected samples positive for targeted PrRT and INT amplification and sequencing.
2.4. Specificity
Targeted HIV-1 PCR amplification specificity was evaluated using BCS from residual whole blood samples negative for HIV-1 but positive for other bloodborne viruses. This included 7 samples positive for HIV-2, 15 positive for hepatitis B virus (HBV) and 2 for hepatitis C virus (HCV). BCS were processed in the same way as the samples for HIV-1 evaluation.
2.5. Reproducibility
Assay reproducibility was investigated in three ways: amplicon replicates from the same DNA sample, library prep replicates from the same amplicon and mixture experiments to confirm minor variant detection, as described below. These assessments were performed for both PrRT and INT targets.
PCR resampling was evaluated with DNA from the 8E5 HIV-1 cell line and a patient sample. The 8E5 HIV-1 cell line was evaluated with duplicate reactions, and 5 replicates of the patient sample were analysed.
Library preparation and sequence reproducibility assessment comprised 12 PrRT and 5 INT samples. Each amplicon was evaluated in duplicate on separate library preparation and sequence runs for reproducibility evaluation.
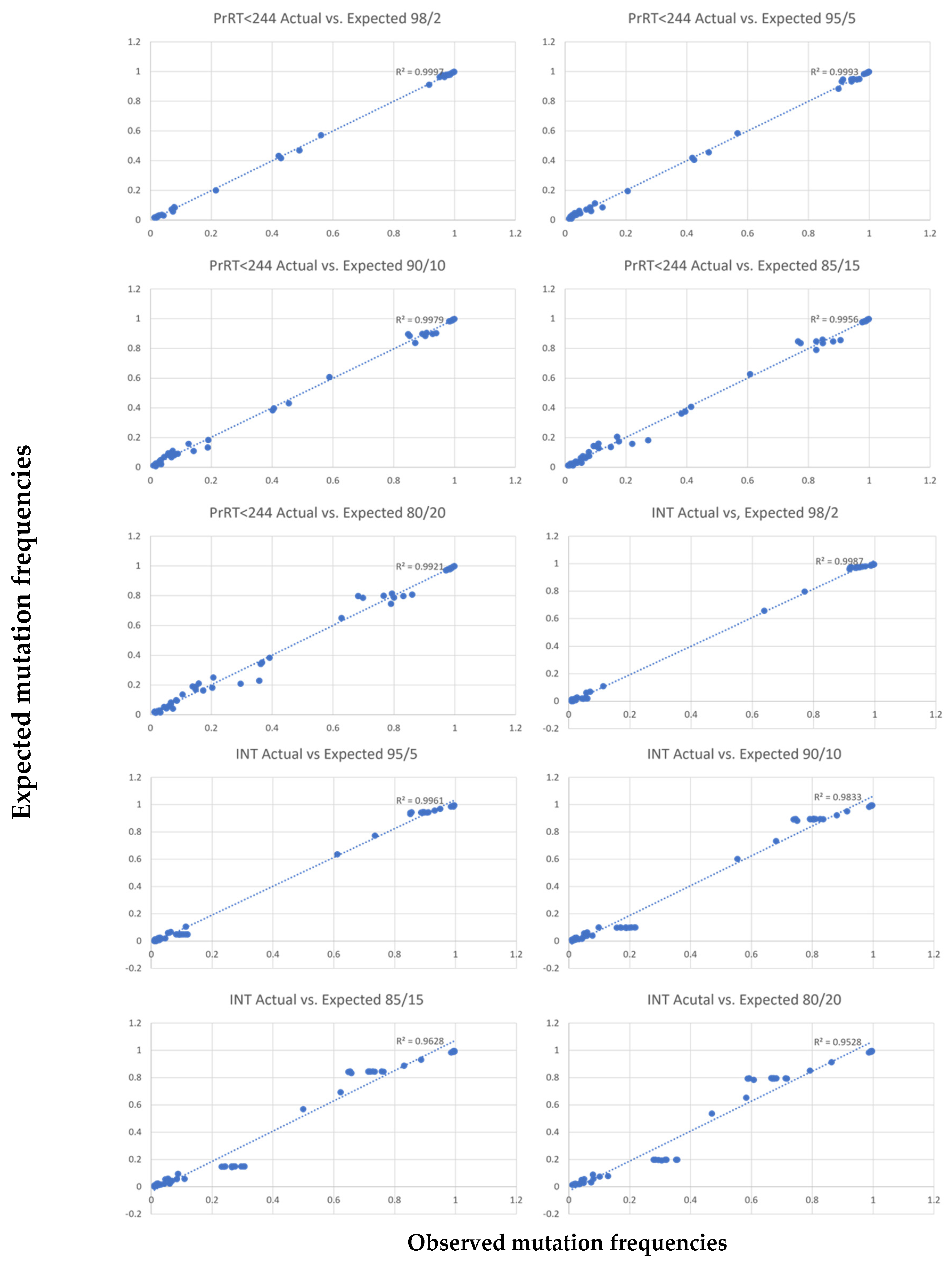
Discrimination of minor variant detection vs. sequencing errors was addressed computationally. Minor variants were evaluated to a threshold of 2%. High region coverage minimised sequencing error impact. Two samples with known mutations were selected for each PCR target and designated as either major or minor samples. Major-to-minor sample combinations were mixed as follows: 80%/20%, 85%/15%, 90%/10%, 95%/5% and 98%/2%, respectively. This allowed for “minor sample” variation analyses at 20%, 15%, 10%, 5% and 2%.
2.6. Bioinformatics Pipeline
Illumina MiSeq data were analysed using an in-house bioinformatics pipeline executed on a high-performance computing cluster. The pipeline uses Trimmomatic v0.40 (Usadel Lab, Dusselforf, Germany) to trim the reads for quality and to remove adapters [
24]. Reads are aligned to the human genome using Bowtie 2 v2.5.1 (
https://bowtie-bio.sourceforge.net/bowtie2/index.shtml, accessed on 21 August 2023) [
25] and filtered to remove host sequences. Reads with hypermutations are detected and removed using a multi-step process: (1) all reads are mapped to HXB2 (GenBank Accession: K03455.1) using Bowtie2 [
26], and a consensus sequence is obtained from the alignment using IVAR v1.4.2 (
https://github.com/andersen-lab/ivar, accessed on 21 August 2023) [
26]; (2) all reads are aligned to the consensus sequence using Bowtie2; (3) pairwise alignments of reads to the consensus sequence are retrieved from the alignment file using sam2fasta (
https://sourceforge.net/projects/sam2fasta/files/, accessed on 21 August 2023) and stored as fasta files; and (4) Hypermut2 [
27] was used to detect hypermutated reads from the fasta file and reads that Hypermut2 assigns a
p-value of <0.05 are filtered. Quasitools Hydra (
https://github.com/phac-nml/quasitools/, accessed on 21 August 2023) is used to assemble and call variants from the remaining reads. QC metrics, variants and sequences are reported [
28].
4. Discussion
There is a growing demand for robust and highly sensitive assays capable of detecting DRMs in HIV-1 DNA. These assays are essential for future studies and clinical trials focused on understanding and targeting viral reservoirs. Furthermore, they could be helpful in clinical practice for treatment optimisation in suppressed patients who require therapy switching. The data presented here demonstrate an evaluation of a sensitive HIV-1 DNA assay for the detection of DRMs in the PrRT and INT regions on the MiSeq NGS platform. Additionally, this study supports the use of BCS, with quicker and easier processing compared to traditional PBMCs.
The evaluation included 166 HIV-1 positive BCS: 148 from adults and 18 from children. The limit of detection evaluation based on the 8E5 control and a patient sample indicates an assay sensitivity of four HIV-1 DNA copies per reaction. Additionally, positive amplification and sequencing were possible for patient samples below this cut-off (down to a single copy per reaction) with background human DNA of up to 318 565 copies per reaction. Therefore, the assay detection threshold is set at four HIV-1 DNA copies per reaction. This level of sensitivity is rarely reported for sequence-based assays [
29,
30,
31].
Sequencing-based assays are inherently less affected by non-specific amplification since off-target sequence results can easily be identified and excluded bioinformatically. Nonetheless, PCR specificity is defined here at 98% with a single HIV-2 positive sample yielding weak amplification, and any non-HIV-1 sequences are filtered out by Hydra as part of the bioinformatics pipeline. Overall amplification and NGS success rate for all evaluated samples were 92% and 99%, respectively, covering six major circulating subtypes and six recombinant forms in HIV-1 group M.
The evaluation of selected positive amplicons in Sanger and NGS platforms showed, as expected [
17,
18], increased sensitivity for DRMs for NGS, with 9/10 discordant results only detected in NGS sequences, supporting the use of NGS in HIV-1 DNA DRM detection.
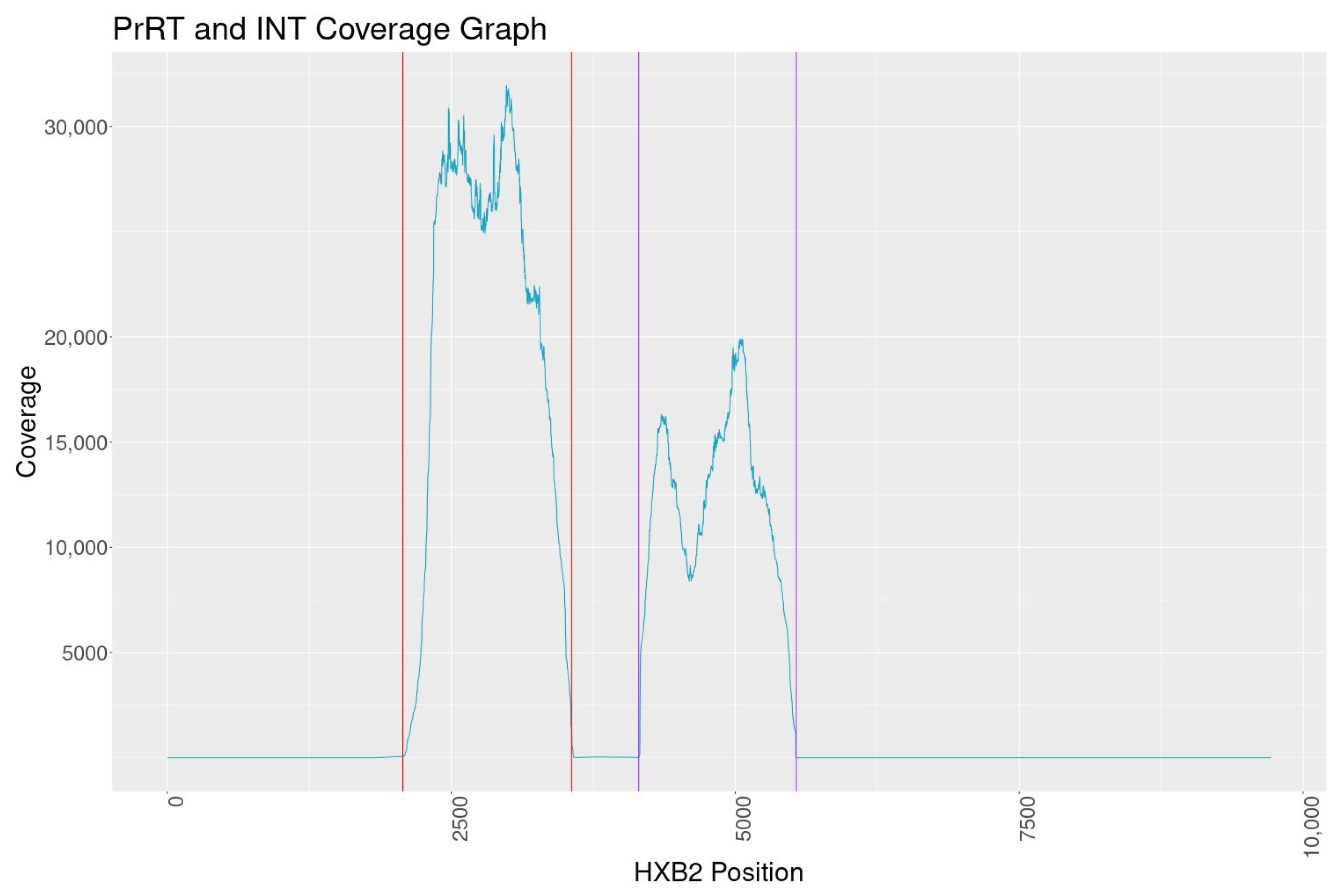
The overall NGS sequence coverage for the targeted PrRT and INT regions exceeds 10,000, with one consistent dip in reads within INT to a minimum depth of 8000 reads. The coverage is well above the minimum of 5000 reads set out by one regional regulatory guidance document for submission of NGS data [
32]. Assay reproducibility demonstrated consistent and nearly identical results when analysing PCR reactions, library prep, MiSeq sequencing and the bioinformatics pipeline independently. Furthermore, dilution experiments indicate minor variant calling can reliably be performed at the 5% mutation frequency cut-off. Based on all findings in this evaluation, a 5% sequence cut-off was selected for DRM characterisation with high confidence of reproducibility.
This assay was evaluated across eight HIV-1 group M subtypes and six CRFs, indicating a broad range of detection capability. In addition, 144 DRMs were characterised across the PrRT and INT regions, including clinically significant DRMs such as M184IV and R263K. HIV-1 DNA sequencing data interpretation poses specific challenges given that in well-suppressed patients, the majority of viruses in latency are defective and/or are hypermutated. Our approach overcomes some of these challenges, as demonstrated by developing a new bioinformatics pipeline, most significantly to identify and remove hypermutated reads prior to assembly and analyses. The amplification and sensitive variant identification by this assay allow for in-depth HIV-1 DNA analyses and drug resistance genotyping. As the clinical relevance of minor HIV-1 DNA variants is still being explored, we echo the message that this data should be interpreted with care in a clinical setting [
17,
18].
Here, we present a highly sensitive and specific Pan-HIV-1 assay for PrRT- and INT-targeted drug resistance genotyping from buffy coats. In addition, the robust bioinformatics pipeline is purpose-built and offers reproducible variant and DRM calling. This assay is optimal for monitoring archived HIV-1 drug resistance in HIV-1-suppressed individuals and in clinical trials of novel therapeutic approaches.


 ,
, 


{kind=link}
{kind=link}