
MDM2 Influences ACE2 Stability and SARS-CoV-2 Uptake
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines
2.2. Virus Stock Preparation
2.3. Virus Infection and Live-Cell Imaging Experiment
2.4. Full Proteome MS Sample Preparation
2.5. LC-MS/MS Data Acquisition
2.6. MS Data Processing and Analysis
2.7. Upstream Regulator Analysis
2.8. VSV-S and VSV Infection
2.9. Virus-like Particle Uptake Assay and Immunofluorescence Staining
2.10. qRT-PCR Analysis
2.11. Western Blotting
3. Results
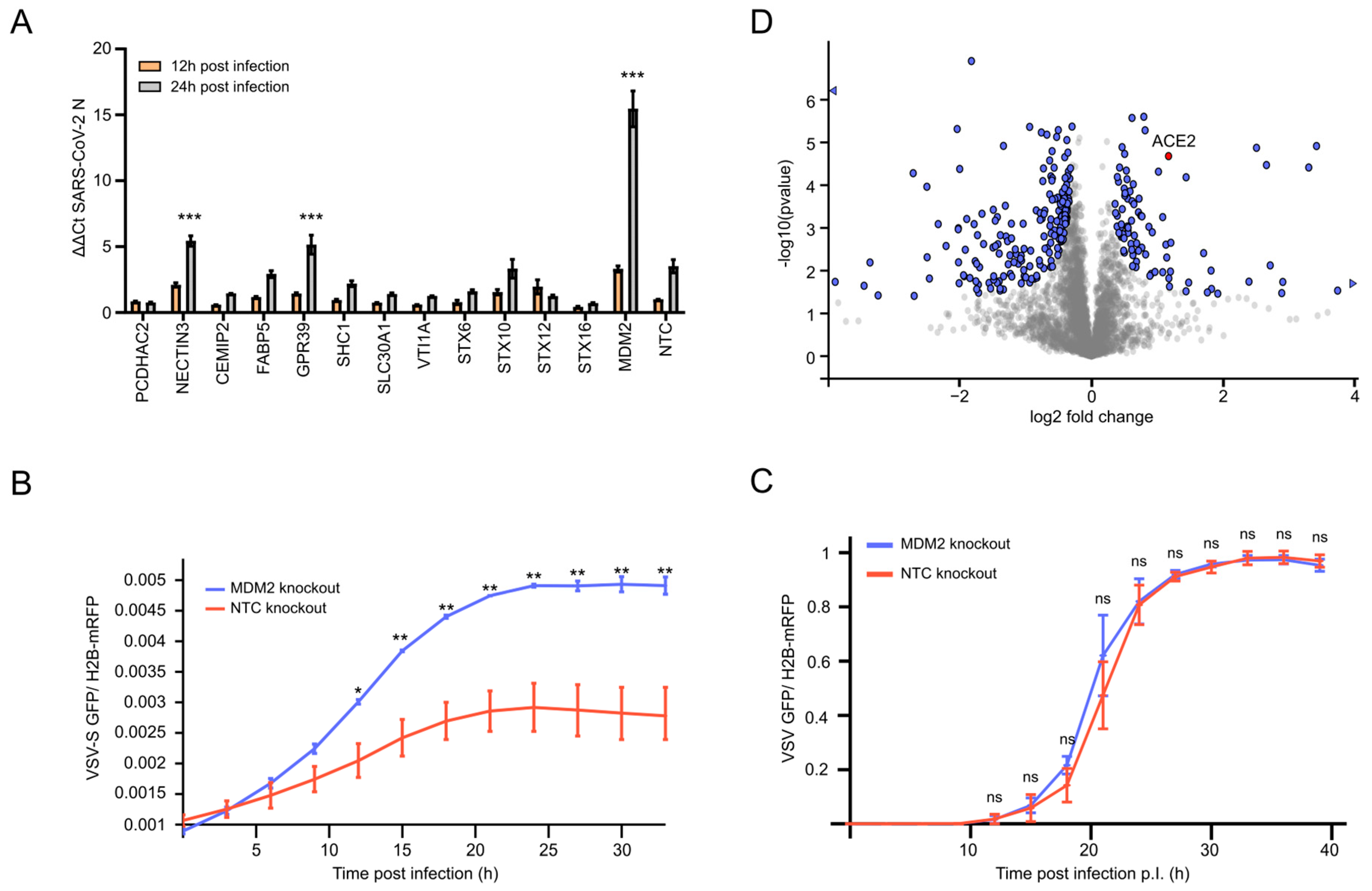
3.1. Selection of Knockout Candidates Based on Multi-Omics Profiling Data
3.2. MDM2 Knockout Specifically Enhances SARS-CoV-2 Infection
3.3. MDM2 Knockout Stabilizes ACE2 and Increases SARS-CoV-2 Replication
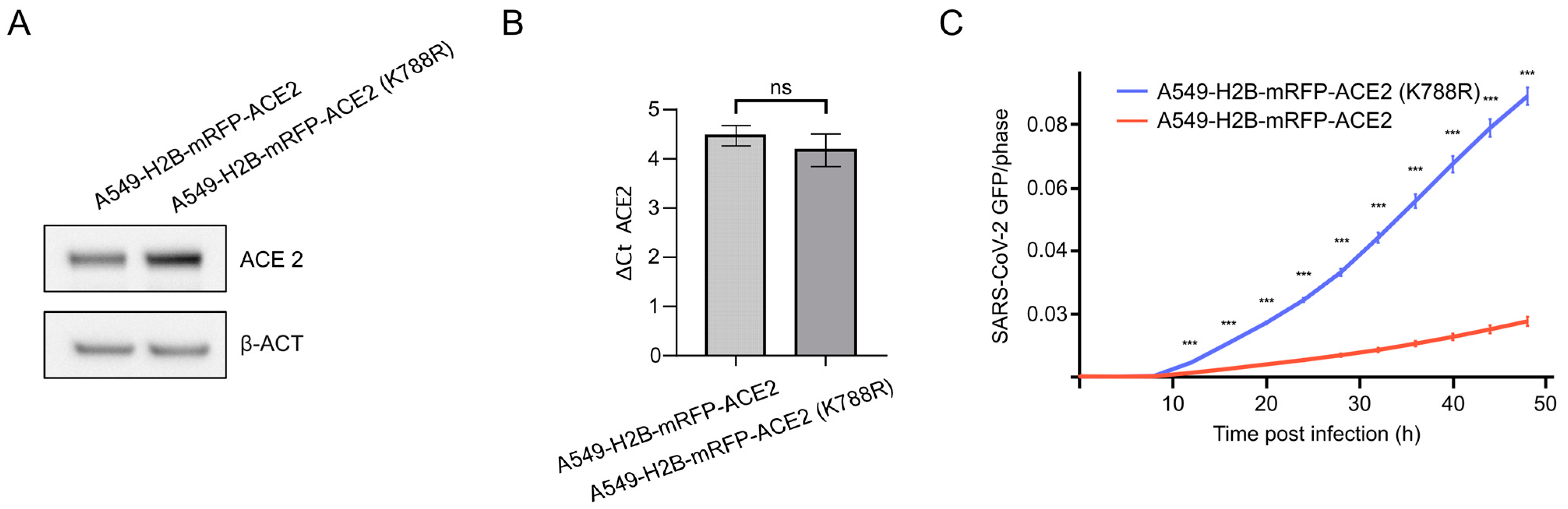
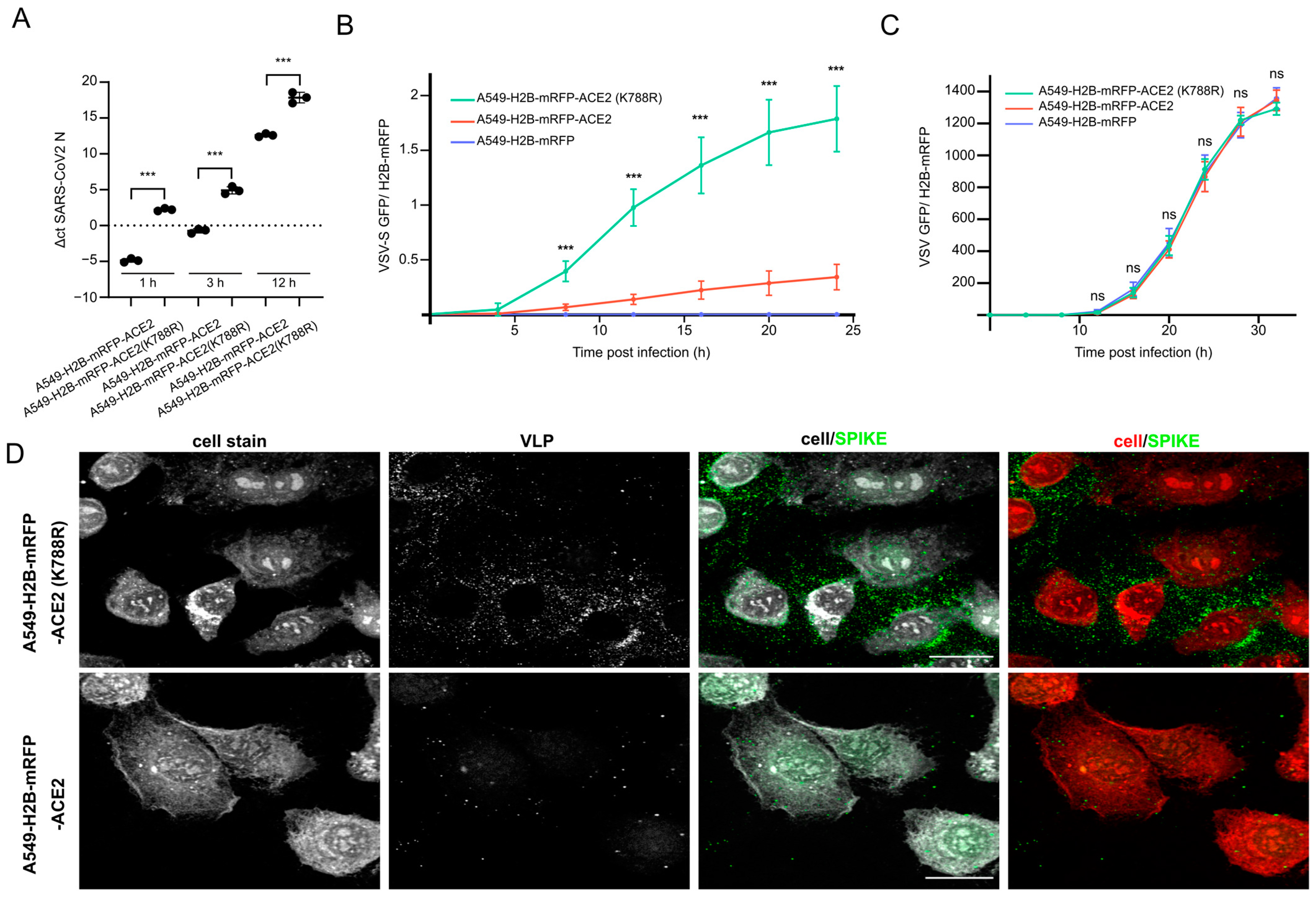
3.4. Lysine 788 within ACE2 Mediates ACE2 Stability and SARS-CoV-2 Replication
3.5. ACE2-K788R Increases SARS-CoV-2 Particle Uptake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Ardes, D.; Boccatonda, A.; Rossi, I.; Guagnano, M.T.; Santilli, F.; Cipollone, F.; Bucci, M. COVID-19 and RAS: Unravelling an Unclear Relationship. Int. J. Mol. Sci. 2020, 21, 3003. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.-N.; Yan, R.; Wang, G.; Zhang, Y.; Zhang, Z.-R.; Li, Y.; Ou, J.; Chu, W.; Liang, Z.; et al. ACE2-targeting monoclonal antibody as potent and broad-spectrum coronavirus blocker. Signal Transduct. Target. Ther. 2021, 6, 315. [Google Scholar] [CrossRef]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.-Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, S.-B.; Zhou, Z.-J.; Li, J.-Y.; Lv, J.-Z.; Hu, B.; Yuan, S.; Qiu, Y.; Ge, X.-Y. Key mutations on spike protein altering ACE2 receptor utilization and potentially expanding host range of emerging SARS-CoV-2 variants. J. Med. Virol. 2023, 95, e28116. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X.-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef] [PubMed]
- Jocher, G.; Grass, V.; Tschirner, S.K.; Riepler, L.; Breimann, S.; Kaya, T.g.; Oelsner, M.; Hamad, M.S.; Hofmann, L.I.; Blobel, C.P.; et al. ADAM10 and ADAM17 promote SARS-CoV-2 cell entry and spike protein-mediated lung cell fusion. EMBO Rep. 2022, 23, e54305. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Bergant, V.; Yamada, S.; Grass, V.; Tsukamoto, Y.; Lavacca, T.; Krey, K.; Mühlhofer, M.-T.; Wittmann, S.; Ensser, A.; Herrmann, A.; et al. Attenuation of SARS-CoV-2 replication and associated inflammation by concomitant targeting of viral and host cap 2’-O-ribose methyltransferases. EMBO J. 2022, 41, e111608. [Google Scholar] [CrossRef]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5’-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence factor NSs of rift valley fever virus recruits the F-box protein FBXO3 to degrade subunit p62 of general transcription factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef]
- Roessler, J.; Pich, D.; Albanese, M.; Wratil, P.R.; Krähling, V.; Hellmuth, J.C.; Scherer, C.; von Bergwelt-Baildon, M.; Becker, S.; Keppler, O.T.; et al. Quantitation of SARS-CoV-2 neutralizing antibodies with a virus-free, authentic test. PNAS Nexus 2022, 1, pgac045. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654–1665. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic expression of angiotensin-converting enzyme (ACE) and ACE2 in human renal tissue and confounding effects of hypertension on the ACE to ACE2 ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, S.; Zhao, D.; Yan, J.; Chen, J.; Yang, C.; Zheng, G. MNX1 reduces sensitivity to anoikis by activating TrkB in human glioma cells. Mol. Med. Rep. 2018, 18, 3271–3279. [Google Scholar] [CrossRef] [PubMed]
- Schaecher, S.R.; Mackenzie, J.M.; Pekosz, A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. 2007, 81, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhao, Q.; Rao, J.; Zeng, F.; Yuan, S.; Ji, M.; Sun, X.; Li, J.; Yang, J.; Cui, J.; et al. SARS-CoV-2 Accessory Protein ORF7b Mediates Tumor Necrosis Factor-α-Induced Apoptosis in Cells. Front. Microbiol. 2021, 12, 654709. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.Y.H.; Wang, Y.; Tang, B.L. The syntaxins. Genome Biol. 2021, 2, reviews3012.1. [Google Scholar]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef]
- Miyazono, K.; Olofsson, A.; Colosetti, P.; Heldin, C.H. A role of the latent TGF-beta 1-binding protein in the assembly and secretion of TGF-beta 1. EMBO J. 1991, 10, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; AlHilali, S.; Al-Swailem, S.; Secchiero, P.; Voltan, R. Therapeutic potential of the MDM2 inhibitor Nutlin-3 in counteracting SARS-CoV-2 infection of the eye through p53 activation. Front. Med. 2022, 9, 902713. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Tisato, V.; Secchiero, P. Rationale for Considering Oral Idasanutlin as a Therapeutic Option for COVID-19 Patients. Front. Pharmacol. 2020, 11, 1156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.-T. LY6E Restricts Entry of Human Coronaviruses, Including Currently Pandemic SARS-CoV-2. J. Virol. 2020, 94, e00562-20. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Nishitsuji, H.; Sugiyama, M.; Nishida, N.; Mizokami, M.; Shimotohno, K. Orchestration of Intracellular Circuits by G Protein-Coupled Receptor 39 for Hepatitis B Virus Proliferation. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef]
- Malavolta, M.; Costarelli, L.; Giacconi, R.; Basso, A.; Piacenza, F.; Pierpaoli, E.; Provinciali, M.; Ogo, O.A.; Ford, D. Changes in Zn homeostasis during long term culture of primary endothelial cells and effects of Zn on endothelial cell senescence. Exp. Gerontol. 2017, 99, 35–45. [Google Scholar] [CrossRef]
- Moskovskich, A.; Goldmann, U.; Kartnig, F.; Lindinger, S.; Konecka, J.; Fiume, G.; Girardi, E.; Superti-Furga, G. The transporters SLC35A1 and SLC30A1 play opposite roles in cell survival upon VSV virus infection. Sci. Rep. 2019, 9, 10471. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512-20. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, J.; Wang, C.; Jain, P.P.; Xiong, M.; Shi, X.; Lei, Y.; Chen, S.; Yin, Q.; Thistlethwaite, P.A.; et al. MDM2-Mediated Ubiquitination of Angiotensin-Converting Enzyme 2 Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2020, 142, 1190–1204. [Google Scholar] [CrossRef]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef]
- Gu, Y.; Princely Abudu, Y.; Kumar, S.; Bissa, B.; Choi, S.W.; Jia, J.; Lazarou, M.; Eskelinen, E.-L.; Johansen, T.; Deretic, V. Mammalian Atg8 proteins regulate lysosome and autolysosome biogenesis through SNAREs. EMBO J. 2019, 38, e101994. [Google Scholar] [CrossRef]
- Chen, Y.; Gan, B.Q.; Tang, B.L. Syntaxin 16: Unraveling cellular physiology through a ubiquitous SNARE molecule. J. Cell. Physiol. 2010, 225, 326–332. [Google Scholar] [CrossRef]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Pazos, M.; Delgado, I.; Murk, W.; Mungamuri, S.K.; Lee, S.W.; García-Sastre, A.; Moran, T.M.; Aaronson, S.A. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J. Immunol. 2011, 187, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Q.; Zhang, H.; Liang, F.; Zhang, C.; Wang, J.; Chen, Z.; Wu, R.; Yu, H.; Sun, B.; et al. Degradation of SARS-CoV-2 receptor ACE2 by the E3 ubiquitin ligase Skp2 in lung epithelial cells. Front. Med. 2021, 15, 252–263. [Google Scholar] [CrossRef]
- Amirfakhryan, H.; Safari, F. Outbreak of SARS-CoV2: Pathogenesis of infection and cardiovascular involvement. Hellenic J. Cardiol. 2021, 62, 13–23. [Google Scholar] [CrossRef]
- Saito, R.; Rocanin-Arjo, A.; You, Y.-H.; Darshi, M.; van Espen, B.; Miyamoto, S.; Pham, J.; Pu, M.; Romoli, S.; Natarajan, L.; et al. Systems biology analysis reveals role of MDM2 in diabetic nephropathy. JCI Insight 2016, 1, e87877. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liu, X.; Hu, B.; Li, D.; Chen, L.; Li, Y.; Tu, Y.; Xiong, S.; Wang, G.; Deng, J.; et al. Mechanisms of SARS-CoV-2 Infection-Induced Kidney Injury: A Literature Review. Front. Cell. Infect. Microbiol. 2022, 12, 838213. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Wang, Q.; Xing, Y.; Feng, L.; Kong, J.; Peng, C.; Zhang, L.; Yang, H.; Lu, M. Identification of proteasome and caspase inhibitors targeting SARS-CoV-2 M. Signal Transduct. Target. Ther. 2021, 6, 214. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wu, Y.; Xiao, T.; Qi, F.; Fan, L.; Zhang, S.; Zhou, J.; He, Y.; Gao, X.; Zeng, H.; et al. Multiomics approach reveals the ubiquitination-specific processes hijacked by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 312. [Google Scholar] [CrossRef] [PubMed]
- Große, M.; Setz, C.; Rauch, P.; Auth, J.; Morokutti-Kurz, M.; Temchura, V.; Schubert, U. Inhibitors of Deubiquitinating Enzymes Interfere with the SARS-CoV-2 Papain-like Protease and Block Virus Replication In Vitro. Viruses 2022, 14, 1404. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emslander, Q.; Krey, K.; Hamad, S.; Maidl, S.; Oubraham, L.; Hesse, J.; Henrici, A.; Austen, K.; Mergner, J.; Grass, V.; et al. MDM2 Influences ACE2 Stability and SARS-CoV-2 Uptake. Viruses 2023, 15, 1763. https://doi.org/10.3390/v15081763
Emslander Q, Krey K, Hamad S, Maidl S, Oubraham L, Hesse J, Henrici A, Austen K, Mergner J, Grass V, et al. MDM2 Influences ACE2 Stability and SARS-CoV-2 Uptake. Viruses. 2023; 15(8):1763. https://doi.org/10.3390/v15081763
Chicago/Turabian StyleEmslander, Quirin, Karsten Krey, Sabri Hamad, Susanne Maidl, Lila Oubraham, Joshua Hesse, Alexander Henrici, Katharina Austen, Julia Mergner, Vincent Grass, and et al. 2023. "MDM2 Influences ACE2 Stability and SARS-CoV-2 Uptake" Viruses 15, no. 8: 1763. https://doi.org/10.3390/v15081763