
Direct Nanopore Sequencing of Human Cytomegalovirus Genomes from High-Viral-Load Clinical Samples

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Characterisation
2.2. Illumina Sequencing
2.3. Nanopore Sequencing
2.4. Genotyping Using Illumina and Nanopore Data
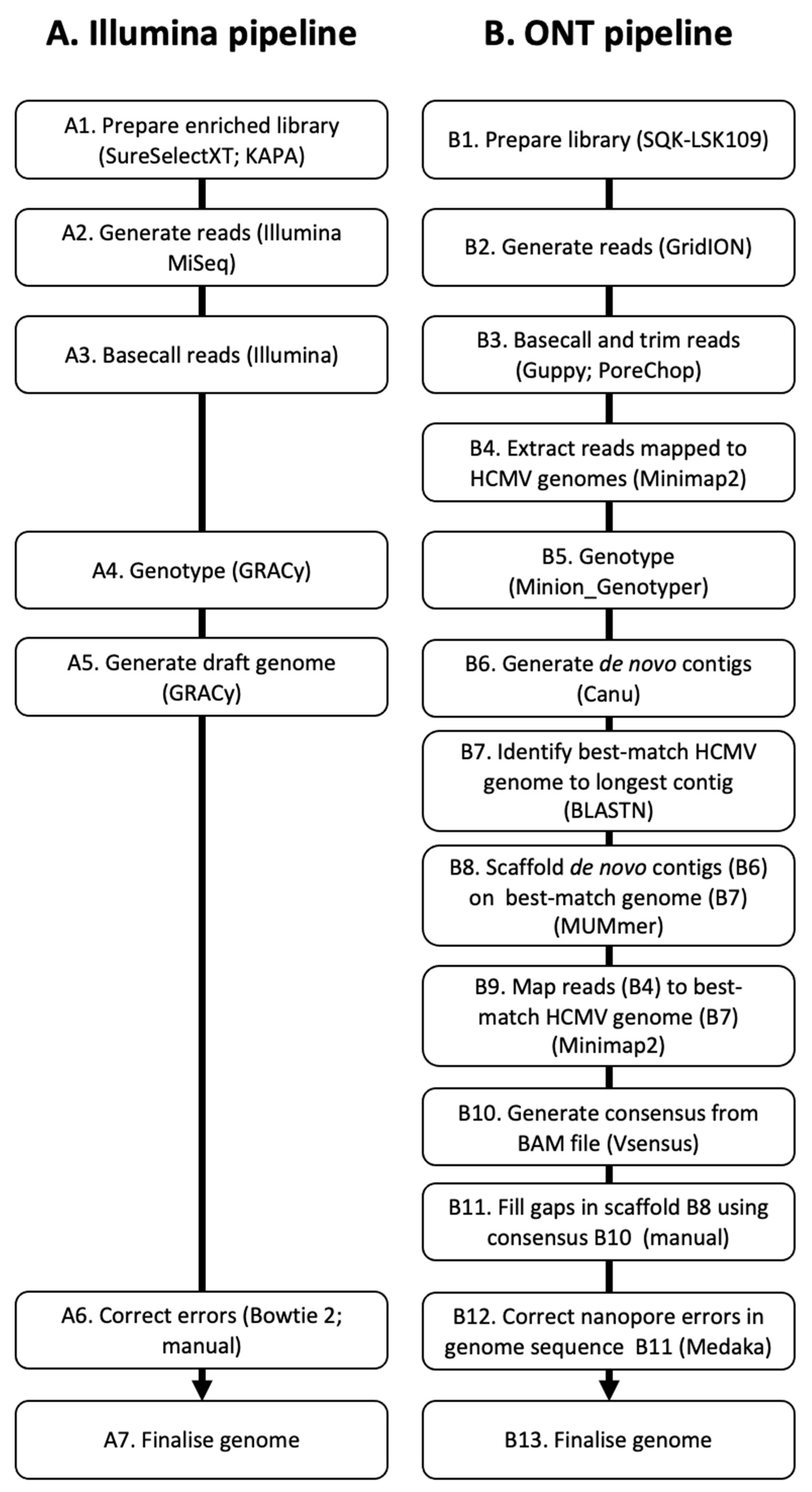
2.5. Genome Determination Using Illumina Data
2.6. Genome Determination Using Nanopore Data
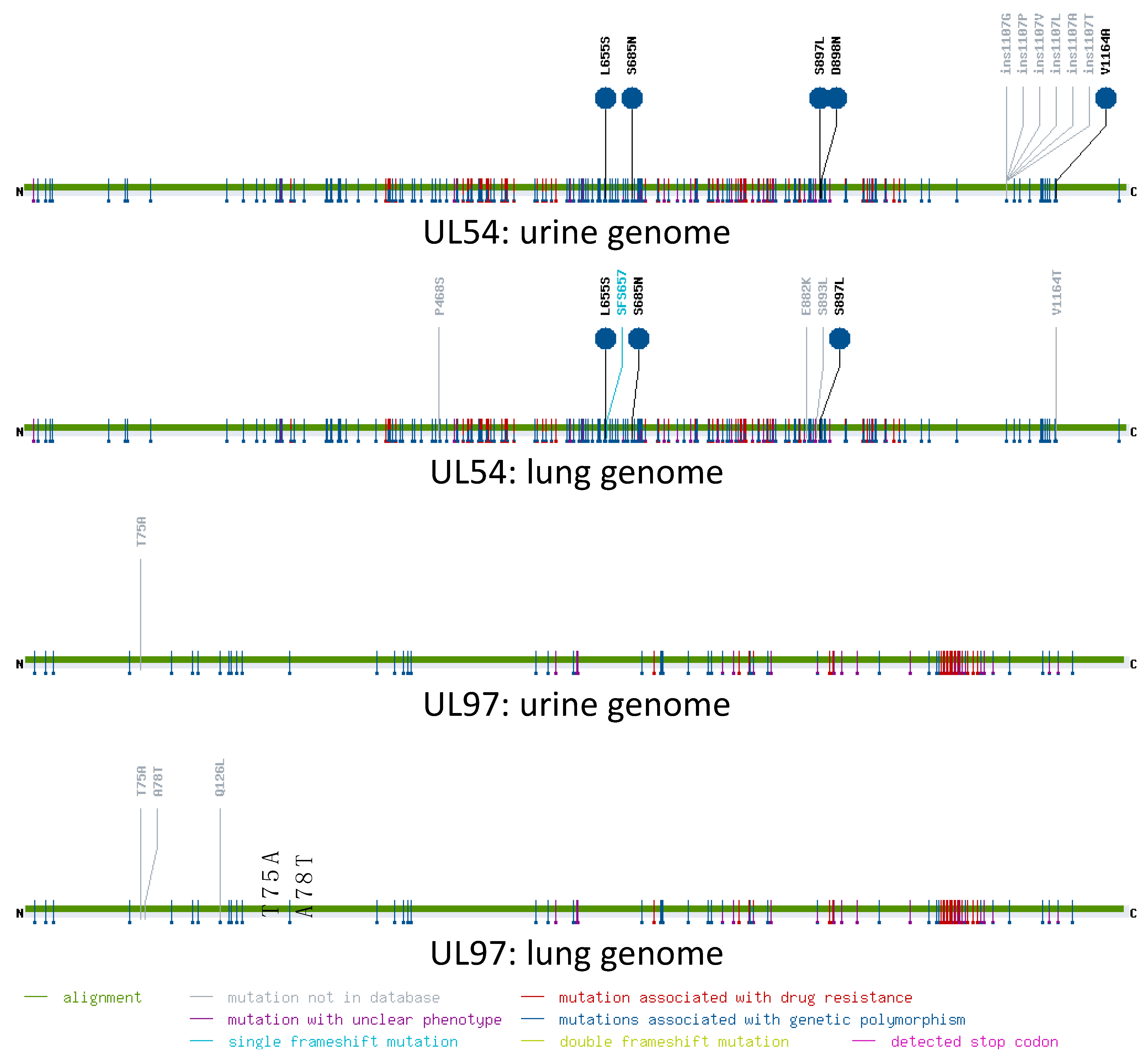
2.7. Analysis of Resistance Mutations
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Asberg, A.; Chou, S.; Danziger-Isakov, L.; Humar, A.; Transplantation Society International CMV Consensus Group. Updated international consensus guidelines on the management of cytomegalovirus in solid-organ transplantation. Transplantation 2013, 96, 333–360. [Google Scholar] [CrossRef]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef]
- Britt, W.J.; Prichard, M.N. New therapies for human cytomegalovirus infections. Antivir. Res. 2018, 159, 153–174. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Poole, UK, 2013; Volume 1, pp. 1–22. [Google Scholar]
- Meyer-König, U.; Vogelberg, C.; Bongarts, A.; Kampa, D.; Delbrück, R.; Wolff-Vorbeck, G.; Kirste, G.; Haberland, M.; Hufert, F.T.; von Laer, D. Glycoprotein B genotype correlates with cell tropism in vivo of human cytomegalovirus infection. J. Med. Virol. 1998, 55, 75–81. [Google Scholar] [CrossRef]
- Barbi, M.; Binda, S.; Caroppo, S.; Primache, V.; Didò, P.; Guidotti, P.; Corbetta, C.; Melotti, D. CMV gB genotypes and outcome of vertical transmission: Study on dried blood spots of congenitally infected babies. J. Clin. Virol. 2001, 21, 75–79. [Google Scholar] [CrossRef]
- Puchhammer-Stöckl, E.; Görzer, I.; Zoufaly, A.; Jaksch, P.; Bauer, C.C.; Klepetko, W.; Popow-Kraupp, T. Emergence of multiple cytomegalovirus strains in blood and lung of lung transplant recipients. Transplantation 2006, 81, 187–194. [Google Scholar] [CrossRef]
- Rasmussen, L.; Geissler, A.; Winters, M. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J. Infect. Dis. 2003, 187, 809–819. [Google Scholar] [CrossRef]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [CrossRef]
- Bradley, A.J.; Lurain, N.S.; Ghazal, P.; Trivedi, U.; Cunningham, C.; Baluchova, K.; Gatherer, D.; Wilkinson, G.W.G.; Dargan, D.J.; Davison, A.J. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J. Gen. Virol. 2009, 90, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.J.; Baluchova, K.; McSharry, B.P.; Tomasec, P.; Emery, V.C.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91, 1535–1546. [Google Scholar] [CrossRef]
- Morey, M.; Fernández-Marmiesse, A.; Castiñeiras, D.; Fraga, J.M.; Couce, M.L.; Cocho, J.A. A glimpse into past, present, and future DNA sequencing. Mol. Genet. Metab. 2013, 110, 3–24. [Google Scholar] [CrossRef]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole-genome sequencing directly from clinical specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef]
- Sijmons, S.; Thys, K.; Mbong Ngwese, M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-throughput analysis of human cytomegalovirus genome diversity highlights the widespread occurrence of gene-disrupting mutations and pervasive recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef]
- Lassalle, F.; Depledge, D.P.; Reeves, M.B.; Brown, A.C.; Christiansen, M.T.; Tutill, H.J.; Williams, R.J.; Einer-Jensen, K.; Holdstock, J.; Atkinson, C.; et al. Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2016, 2, vew017. [Google Scholar] [CrossRef]
- Suárez, N.M.; Wilkie, G.S.; Hage, E.; Camiolo, S.; Holton, M.; Hughes, J.; Maabar, M.; Vattipally, S.B.; Dhingra, A.; Gompels, U.A.; et al. Human cytomegalovirus genomes sequenced directly from clinical material: Variation, multiple-strain infection, recombination, and gene loss. J. Infect. Dis. 2019, 220, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Suárez, N.M.; Musonda, K.G.; Escriva, E.; Njenga, M.; Agbueze, A.; Camiolo, S.; Davison, A.J.; Gompels, U.A. Multiple-strain infections of human cytomegalovirus with high genomic diversity are common in breast milk from human immunodeficiency virus-infected women in Zambia. J. Infect. Dis. 2019, 220, 792–801. [Google Scholar] [CrossRef]
- Camiolo, S.; Suárez, N.M.; Chalka, A.; Venturini, C.; Breuer, J.; Davison, A.J. GRACy: A tool for analysing human cytomegalovirus sequence data. Virus Evol. 2020, 7, veaa099. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Jain, M.; Tyson, J.R.; Loose, M.; Ip, C.L.C.; Eccles, D.A.; O’Grady, J.; Malla, S.; Leggett, R.M.; Wallerman, O.; Jansen, H.J.; et al. MinION Analysis and Reference Consortium: Phase 2 data release and analysis of R9.0 chemistry. F1000Research 2017, 6, 760. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- da Silva Filipe, A.; Shepherd, J.G.; Williams, T.; Hughes, J.; Aranday-Cortes, E.; Asamaphan, P.; Ashraf, S.; Balcazar, C.; Brunker, K.; Campbell, A.; et al. Genomic epidemiology reveals multiple introductions of SARS-CoV-2 from mainland Europe into Scotland. Nat. Microbiol. 2021, 6, 112–122. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L.; et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: A prospective genomic surveillance study. Lancet Infect. Dis. 2020, 20, 1263–1272. [Google Scholar] [CrossRef]
- Loman, N.J.; Quinlan, A.R. Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics 2014, 30, 3399–3401. [Google Scholar] [CrossRef]
- Jain, M.; Fiddes, I.T.; Miga, K.H.; Olsen, H.E.; Paten, B.; Akeson, M. Improved data analysis for the MinION nanopore sequencer. Nat. Methods 2015, 12, 351–356. [Google Scholar] [CrossRef]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef]
- Karamitros, T.; van Wilgenburg, B.; Wills, M.; Klenerman, P.; Magiorkinis, G. Nanopore sequencing and full genome de novo assembly of human cytomegalovirus TB40/E reveals clonal diversity and structural variations. BMC Genom. 2018, 19, 577. [Google Scholar] [CrossRef]
- Eckert, S.E.; Chan, J.Z.; Houniet, D.; The Pathseek Consortium; Breuer, J.; Speight, G. Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microb. Genom. 2016, 2, e000087. [Google Scholar] [CrossRef]
- Chorlton, S.D.; Ritchie, G.; Lawson, T.; McLachlan, E.; Romney, M.G.; Matic, N.; Lowe, C.F. Next-generation sequencing for cytomegalovirus antiviral resistance genotyping in a clinical virology laboratory. Antivir. Res. 2021, 192, 105123. [Google Scholar] [CrossRef]
- Lanfear, R.; Schalamun, M.; Kainer, D.; Wang, W.; Schwessinger, B. MinIONQC: Fast and simple quality control for MinION sequencing data. Bioinformatics 2019, 35, 523–525. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Stephen, G.; Cardle, L.; Marshall, D. Tablet: Visualizing next-generation sequence assemblies and mappings. Methods Mol. Biol. 2016, 1374, 253–268. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Chevillotte, M.; von Einem, J.; Meier, B.M.; Lin, F.M.; Kestler, H.A.; Mertens, T. A new tool linking human cytomegalovirus drug resistance mutations to resistance phenotypes. Antivir. Res. 2010, 85, 318–327. [Google Scholar] [CrossRef]
- Dohm, J.C.; Peters, P.; Stralis-Pavese, N.; Himmelbauer, H. Benchmarking of long-read correction methods. NAR Genom. Bioinform. 2020, 2, lqaa037. [Google Scholar] [CrossRef]
- Boom, R.; Sol, C.J.; Schuurman, T.; Van Breda, A.; Weel, J.F.; Beld, M.; Ten Berge, I.J.; Wertheim-Van Dillen, P.M.; De Jong, M.D. Human cytomegalovirus DNA in plasma and serum specimens of renal transplant recipients is highly fragmented. J. Clin. Microbiol. 2002, 40, 4105–4113. [Google Scholar] [CrossRef]
- Suárez, N.M.; Blyth, E.; Li, K.; Ganzenmueller, T.; Camiolo, S.; Avdic, S.; Withers, B.; Linnenweber-Held, S.; Gwinner, W.; Dhingra, A.; et al. Whole-genome approach to assessing human cytomegalovirus dynamics in transplant patients undergoing antiviral therapy. Front. Cell. Infect. Microbiol. 2020, 10, 267. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Urine | Lung |
|---|---|---|
| RL5A | G1 | G2 |
| RL6 | G6 | G4 |
| RL12 | G7 | G1B |
| RL13 | G7 | G1 |
| UL1 | G7 | G1 |
| UL9 | G1 | G4 |
| UL11 | G1 | G1 |
| UL20 | G6 | G6 |
| UL73 | G4D | G1 |
| UL74 | G5 | G1A |
| UL120 | G1A | G4B |
| UL146 | G2 | G10 |
| UL139 | G1A | G2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.K.; Lau, B.; Suárez, N.M.; Camiolo, S.; Gunson, R.; Davison, A.J.; Orton, R.J. Direct Nanopore Sequencing of Human Cytomegalovirus Genomes from High-Viral-Load Clinical Samples. Viruses 2023, 15, 1248. https://doi.org/10.3390/v15061248
Li KK, Lau B, Suárez NM, Camiolo S, Gunson R, Davison AJ, Orton RJ. Direct Nanopore Sequencing of Human Cytomegalovirus Genomes from High-Viral-Load Clinical Samples. Viruses. 2023; 15(6):1248. https://doi.org/10.3390/v15061248
Chicago/Turabian StyleLi, Kathy K., Betty Lau, Nicolás M. Suárez, Salvatore Camiolo, Rory Gunson, Andrew J. Davison, and Richard J. Orton. 2023. "Direct Nanopore Sequencing of Human Cytomegalovirus Genomes from High-Viral-Load Clinical Samples" Viruses 15, no. 6: 1248. https://doi.org/10.3390/v15061248