
Development of HIV-Resistant CAR T Cells by CRISPR/Cas-Mediated CAR Integration into the CCR5 Locus
 , , and
, , and 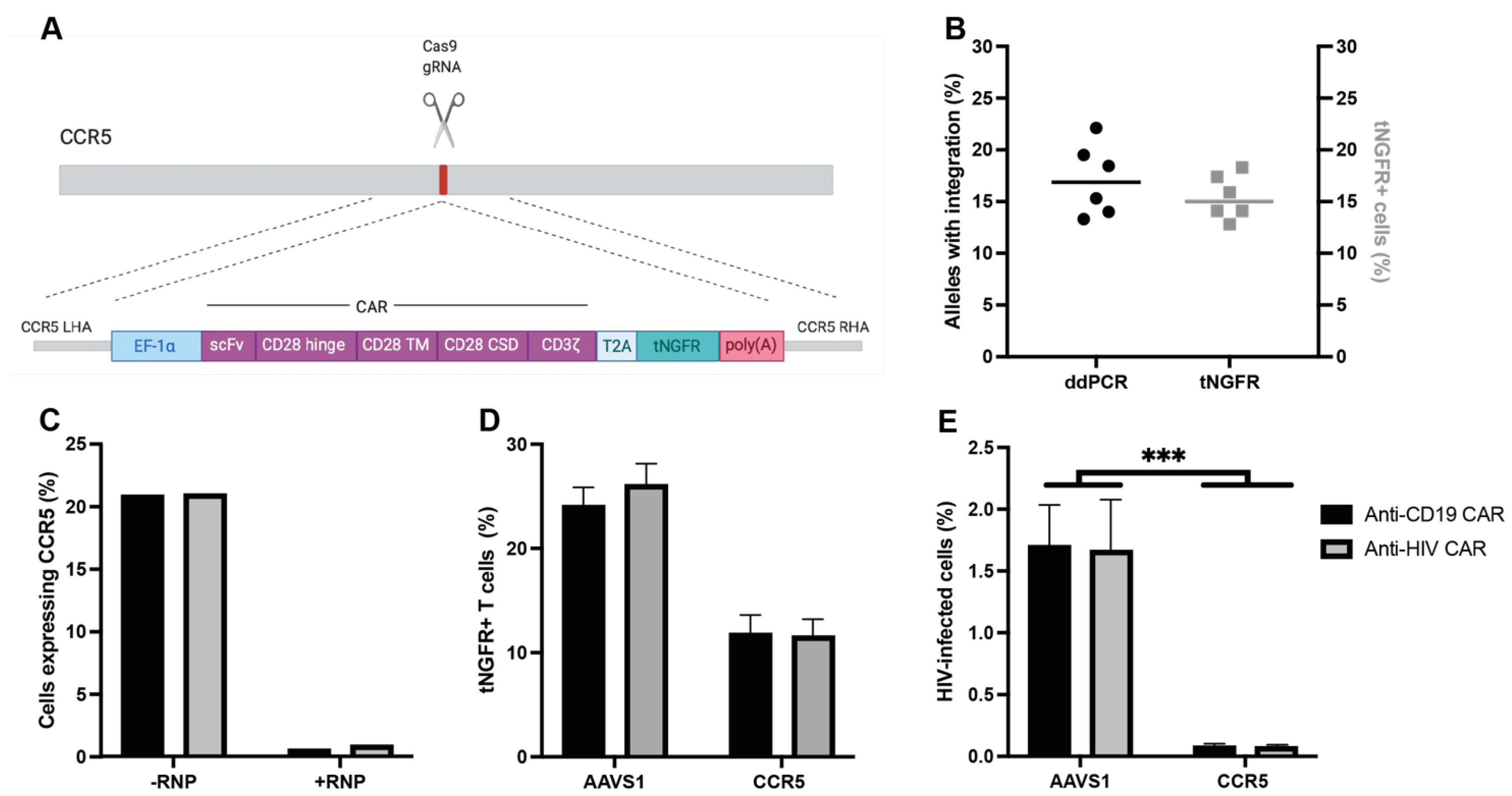{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. CAR Design, Vector Cloning and AAV Production
2.2. Cell Lines and Virus
2.3. CAR T Cell Production
2.4. INDEL Quantification and Allele Editing Frequencies
2.5. Infection of CAR T Cells
2.6. Flow Cytometry and Antibodies
2.7. Cytotoxicity Assays
2.8. Humanized Mouse Model of HIV Infection
2.9. Statistical Analysis
3. Results
3.1. Integration of a CAR into CCR5 Leads to CCR5 Disruption and Resistance to HIV Infection
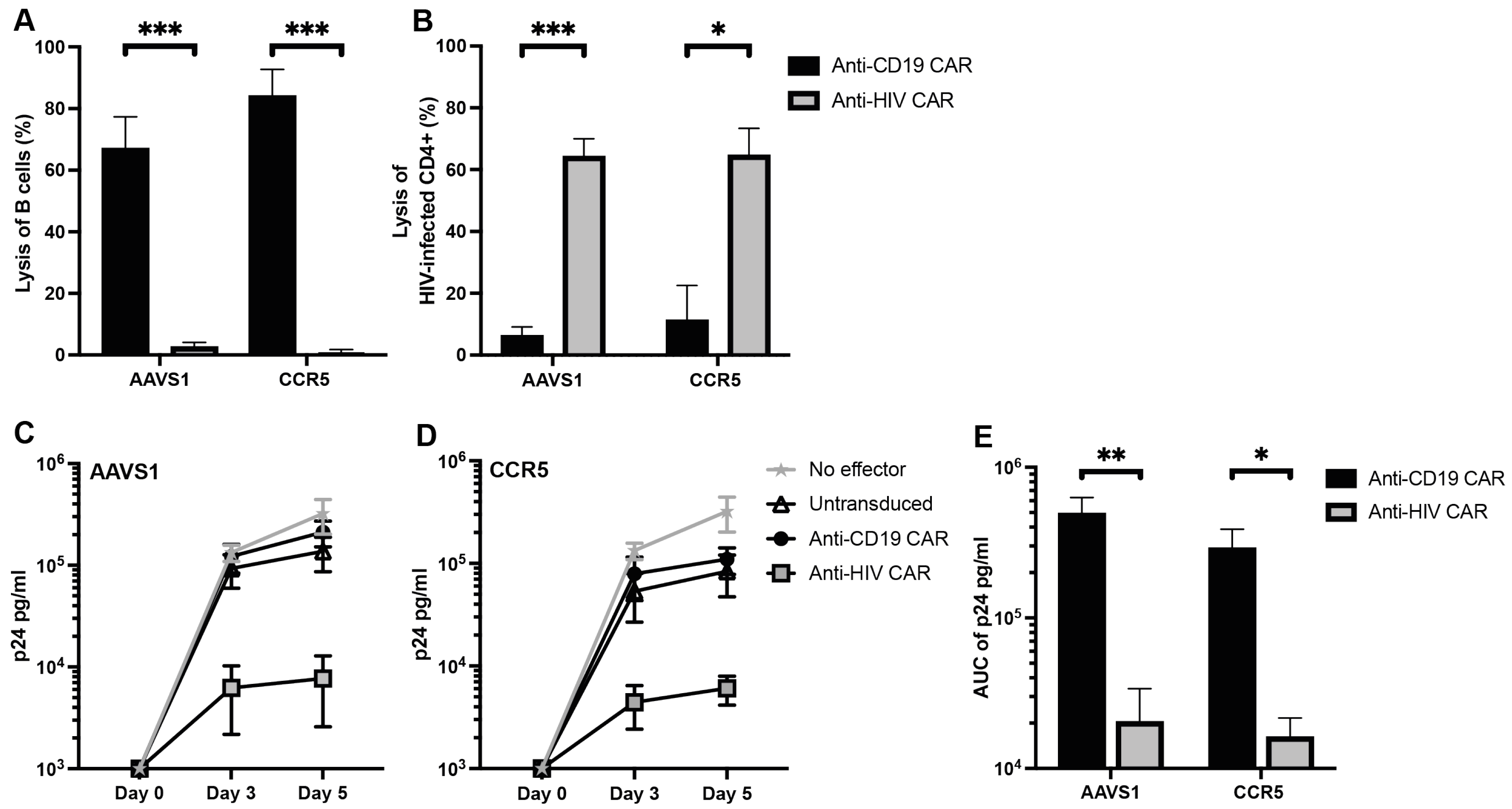
3.2. Anti-CD19 CAR T Cells Selectively Kill B Cells and Anti-HIV CAR T Cells Selectively Kill HIV-Infected Cells In Vitro
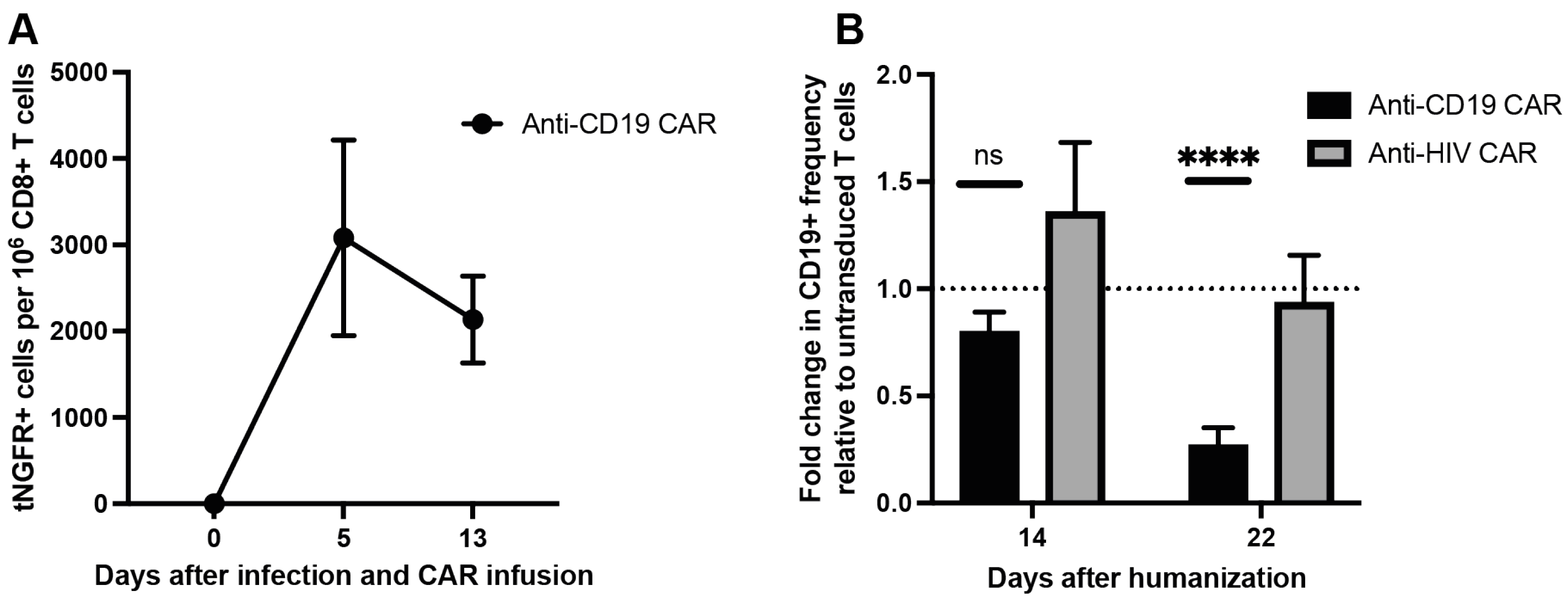
3.3. Anti-CD19 CAR T Cells Kill B Cells in PBMC Humanized Mice
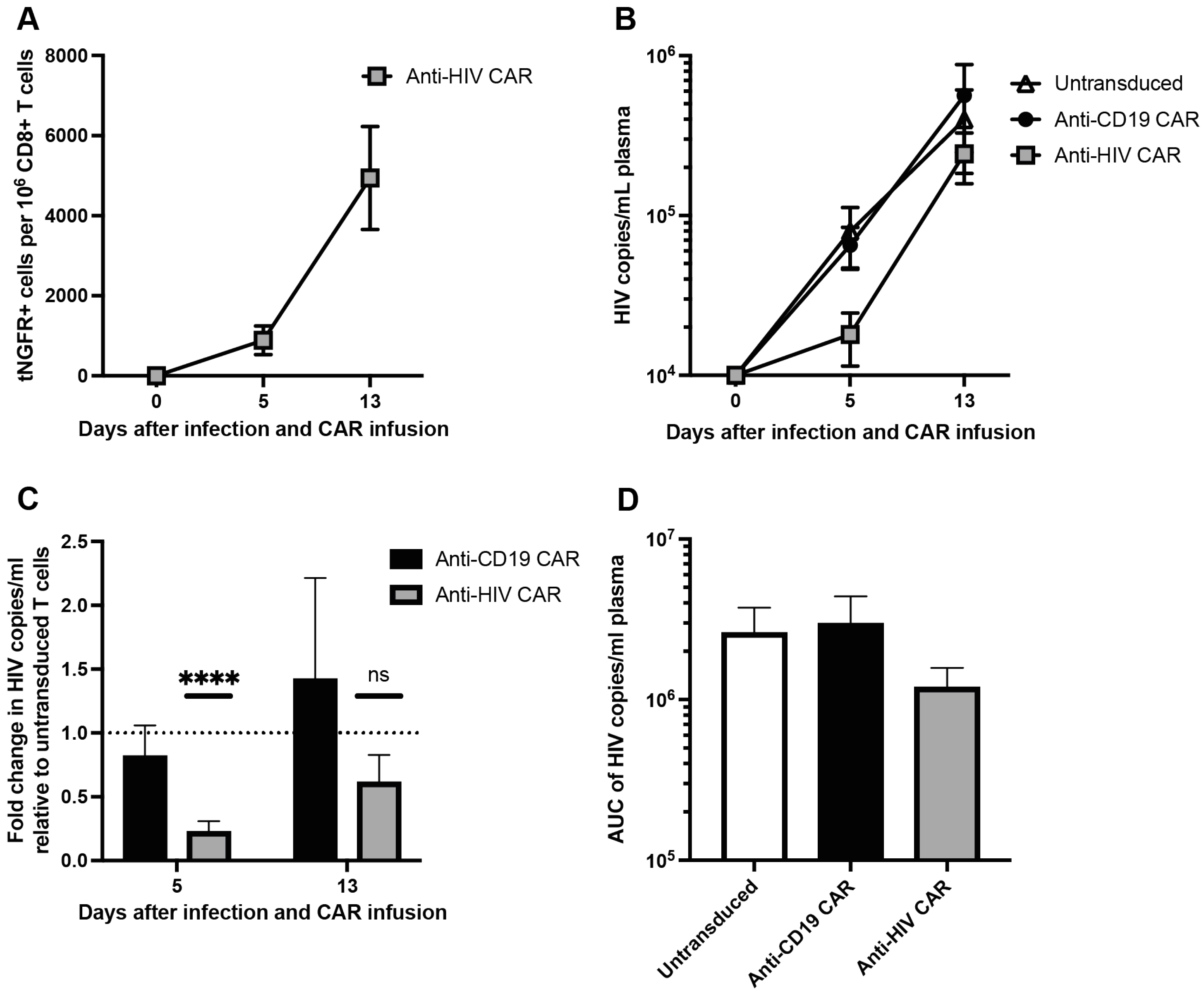
3.4. Anti-HIV CAR T Cells Expand and Transiently Limit HIV Infection in Humanized Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Hege, K.; Riley, J.L. CAR Talk: How Cancer-Specific CAR T Cells Can Instruct How to Build CAR T Cells to Cure HIV. Front. Immunol. 2019, 10, 2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus–infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.-T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-Long Safety and Function of Retroviral-Modified Chimeric Antigen Receptor T Cells. Sci. Transl. Med. 2012, 4, 132ra53. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Patel, B.; Ghanem, M.H.; Bundoc, V.; Zheng, Z.; Morgan, R.A.; Rosenberg, S.A.; Dey, B.; Berger, E.A. Novel CD4-Based Bispecific Chimeric Antigen Receptor Designed for Enhanced Anti-HIV Potency and Absence of HIV Entry Receptor Activity. J. Virol. 2015, 89, 6685–6694. [Google Scholar] [CrossRef] [Green Version]
- Romeo, C.; Seed, B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 1991, 64, 1037–1046. [Google Scholar] [CrossRef]
- Walker, R.E.; Bechtel, C.M.; Natarajan, V.; Baseler, M.; Hege, K.M.; Metcalf, J.A.; Stevens, R.; Hazen, A.; Blaese, R.M.; Chen, C.C.; et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood 2000, 96, 467–474. [Google Scholar] [CrossRef]
- Klein, F.; Mouquet, H.; Dosenovic, P.; Scheid, J.F.; Scharf, L.; Nussenzweig, M.C. Antibodies in HIV-1 Vaccine Development and Therapy. Science 2013, 341, 1199–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunst, J.D.; Pahus, M.H.; Rosas-Umbert, M.; Lu, I.N.; Benfield, T.; Nielsen, H.; Johansen, I.S.; Mohey, R.; Ostergaard, L.; Klastrup, V.; et al. Early intervention with 3BNC117 and romidepsin at antiretroviral treatment initiation in people with HIV-1: A phase 1b/2a, randomized trial. Nat. Med. 2022, 28, 2424–2435. [Google Scholar] [CrossRef]
- Gaebler, C.; Nogueira, L.; Stoffel, E.; Oliveira, T.Y.; Breton, G.; Millard, K.G.; Turroja, M.; Butler, A.; Ramos, V.; Seaman, M.S.; et al. Prolonged viral suppression with anti-HIV-1 antibody therapy. Nature 2022, 606, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Niessl, J.; Baxter, A.E.; Mendoza, P.; Jankovic, M.; Cohen, Y.Z.; Butler, A.L.; Lu, C.L.; Dube, M.; Shimeliovich, I.; Gruell, H.; et al. Combination anti-HIV-1 antibody therapy is associated with increased virus-specific T cell immunity. Nat. Med. 2020, 26, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Caskey, M.; Schoofs, T.; Gruell, H.; Settler, A.; Karagounis, T.; Kreider, E.F.; Murrell, B.; Pfeifer, N.; Nogueira, L.; Oliveira, T.Y.; et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat. Med. 2017, 23, 185–191. [Google Scholar] [CrossRef]
- Ali, A.; Kitchen, S.G.; Chen, I.S.Y.; Ng, H.L.; Zack, J.A.; Yang, O.O. HIV-1-Specific Chimeric Antigen Receptors Based on Broadly Neutralizing Antibodies. J. Virol. 2016, 90, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zou, F.; Lu, L.; Chen, C.; He, D.; Zhang, X.; Tang, X.; Liu, C.; Li, L.; Zhang, H. Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4+ T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive Combined Antiretroviral Therapy. J. Virol. 2016, 90, 9712–9724. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhang, W.; Xia, B.; Jing, S.; Du, Y.; Zou, F.; Li, R.; Lu, L.; Chen, S.; Li, Y.; et al. Broadly neutralizing antibody-derived CAR T cells reduce viral reservoir in individuals infected with HIV-1. J. Clin. Investig. 2021, 131, e150211. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Rocha, B.; Tanchot, C. Towards a cellular definition of CD8+ T-cell memory: The role of CD4+ T-cell help in CD8+ T-cell responses. Curr. Opin. Immunol. 2004, 16, 259–263. [Google Scholar] [CrossRef]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoye, A.A.; Picker, L.J. CD4(+) T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Investig. 2021, 131, e144486. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipping, F.; Newby, G.A.; Eide, C.R.; McElroy, A.N.; Nielsen, S.C.; Smith, K.; Fang, Y.; Cornu, T.I.; Costa, C.; Gutierrez-Guerrero, A.; et al. Disruption of HIV-1 co-receptors CCR5 and CXCR4 in primary human T cells and hematopoietic stem and progenitor cells using base editing. Mol. Ther. 2022, 30, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Ringpis, G.E.; Shimizu, S.; Arokium, H.; Camba-Colon, J.; Carroll, M.V.; Cortado, R.; Xie, Y.; Kim, P.Y.; Sahakyan, A.; Lowe, E.L.; et al. Engineering HIV-1-resistant T-cells from short-hairpin RNA-expressing hematopoietic stem/progenitor cells in humanized BLT mice. PLoS ONE 2012, 7, e53492. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xue, W.; Anderson, D.G. CRISPR-Cas: A tool for cancer research and therapeutics. Nat. Rev. Clin. Oncol. 2019, 16, 281–295. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta-Hanawa, B.; Yamaguchi, T.; Uchida, E. Two-Dimensional Droplet Digital PCR as a Tool for Titration and Integrity Evaluation of Recombinant Adeno-Associated Viral Vectors. Hum. Gene Ther. Methods 2019, 30, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, J.; Rajan, D.; Schindler, M.; Specht, A.; Rucker, E.; Novembre, F.J.; Nerrienet, E.; Muller-Trutwin, M.C.; Peeters, M.; Hahn, B.H.; et al. Nef-mediated enhancement of virion infectivity and stimulation of viral replication are fundamental properties of primate lentiviruses. J. Virol. 2007, 81, 13852–13864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.H.F.; Nielsen, S.S.F.; Olesen, R.; Mack, K.; Dagnaes-Hansen, F.; Uldbjerg, N.; Ostergaard, L.; Sogaard, O.S.; Denton, P.W.; Tolstrup, M. Humanized NOG Mice for Intravaginal HIV Exposure and Treatment of HIV Infection. J. Vis. Exp. 2020, 155, e60723. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Choi, B.S.; Kim, K.C.; Park, K.H.; Lee, H.J.; Cho, Y.K.; Kim, S.I.; Kim, S.S.; Oh, Y.K.; Kim, Y.B. A Simple Mouse Model for the Study of Human Immunodeficiency Virus. AIDS Res. Hum. Retrovir. 2016, 32, 194–202. [Google Scholar] [CrossRef] [Green Version]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “humanized” mice to study human immunity. Curr. Protoc. Immunol. 2008, 81, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, G.; Georgiadis, C.; Gkazi, S.A.; Syed, F.; Zhan, H.; Etuk, A.; Preece, R.; Chu, J.; Kubat, A.; Adams, S.; et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022, 14, eabq3010. [Google Scholar] [CrossRef]
- Foy, S.P.; Jacoby, K.; Bota, D.A.; Hunter, T.; Pan, Z.; Stawiski, E.; Ma, Y.; Lu, W.; Peng, S.; Wang, C.L.; et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 2022, 1–10. [Google Scholar] [CrossRef]
- Wiebking, V.; Lee, C.M.; Mostrel, N.; Lahiri, P.; Bak, R.; Bao, G.; Roncarolo, M.G.; Bertaina, A.; Porteus, M.H. Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing alphabeta T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica 2021, 106, 847–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Paxton, W.A.; Kassam, N.; Ruffing, N.; Rottman, J.B.; Sullivan, N.; Choe, H.; Sodroski, J.; Newman, W.; Koup, R.A.; et al. CCR5 Levels and Expression Pattern Correlate with Infectability by Macrophage-tropic HIV-1, In Vitro. J. Exp. Med. 1997, 185, 1681–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4(+) T cells from HIV-1 infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Morillon, Y.M., 2nd; Sabzevari, A.; Schlom, J.; Greiner, J.W. The Development of Next-generation PBMC Humanized Mice for Preclinical Investigation of Cancer Immunotherapeutic Agents. Anticancer Res. 2020, 40, 5329–5341. [Google Scholar] [CrossRef]
- Tary-Lehmann, M.; Saxon, A. Human mature T cells that are anergic in vivo prevail in SCID mice reconstituted with human peripheral blood. J. Exp. Med. 1992, 175, 503–516. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothemejer, F.H.; Lauritsen, N.P.; Juhl, A.K.; Schleimann, M.H.; König, S.; Søgaard, O.S.; Bak, R.O.; Tolstrup, M. Development of HIV-Resistant CAR T Cells by CRISPR/Cas-Mediated CAR Integration into the CCR5 Locus. Viruses 2023, 15, 202. https://doi.org/10.3390/v15010202
Rothemejer FH, Lauritsen NP, Juhl AK, Schleimann MH, König S, Søgaard OS, Bak RO, Tolstrup M. Development of HIV-Resistant CAR T Cells by CRISPR/Cas-Mediated CAR Integration into the CCR5 Locus. Viruses. 2023; 15(1):202. https://doi.org/10.3390/v15010202
Chicago/Turabian StyleRothemejer, Frederik Holm, Nanna Pi Lauritsen, Anna Karina Juhl, Mariane Høgsbjerg Schleimann, Saskia König, Ole Schmeltz Søgaard, Rasmus O. Bak, and Martin Tolstrup. 2023. "Development of HIV-Resistant CAR T Cells by CRISPR/Cas-Mediated CAR Integration into the CCR5 Locus" Viruses 15, no. 1: 202. https://doi.org/10.3390/v15010202