
Towards Next-Generation Sequencing for HIV-1 Drug Resistance Testing in a Clinical Setting


Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples and Ethics Approval
2.2. Sample Processing
2.3. Population-Sequencing-Based Genotypic Resistance Test (PS-GRT)
2.4. Manual Review of the Electropherograms
2.5. High-Throughput-Sequencing-Based Genotypic Resistance Test (HTS-GRT)
2.5.1. High Fidelity Reverse Transcriptase Polymerase Chain Reaction
2.5.2. Library Preparation and High-Throughput Sequencing
2.6. Manual Review of the Binary Alignment Map Files
2.7. Clinically Actionable Drug Resistance Mutations
2.8. Statistical Analysis
3. Results
3.1. HIV-1 Viral Load and Subtype Coverage
3.2. Agreement between PS-GRT and HTS-GRT
3.2.1. McNemar’s χ2 Test
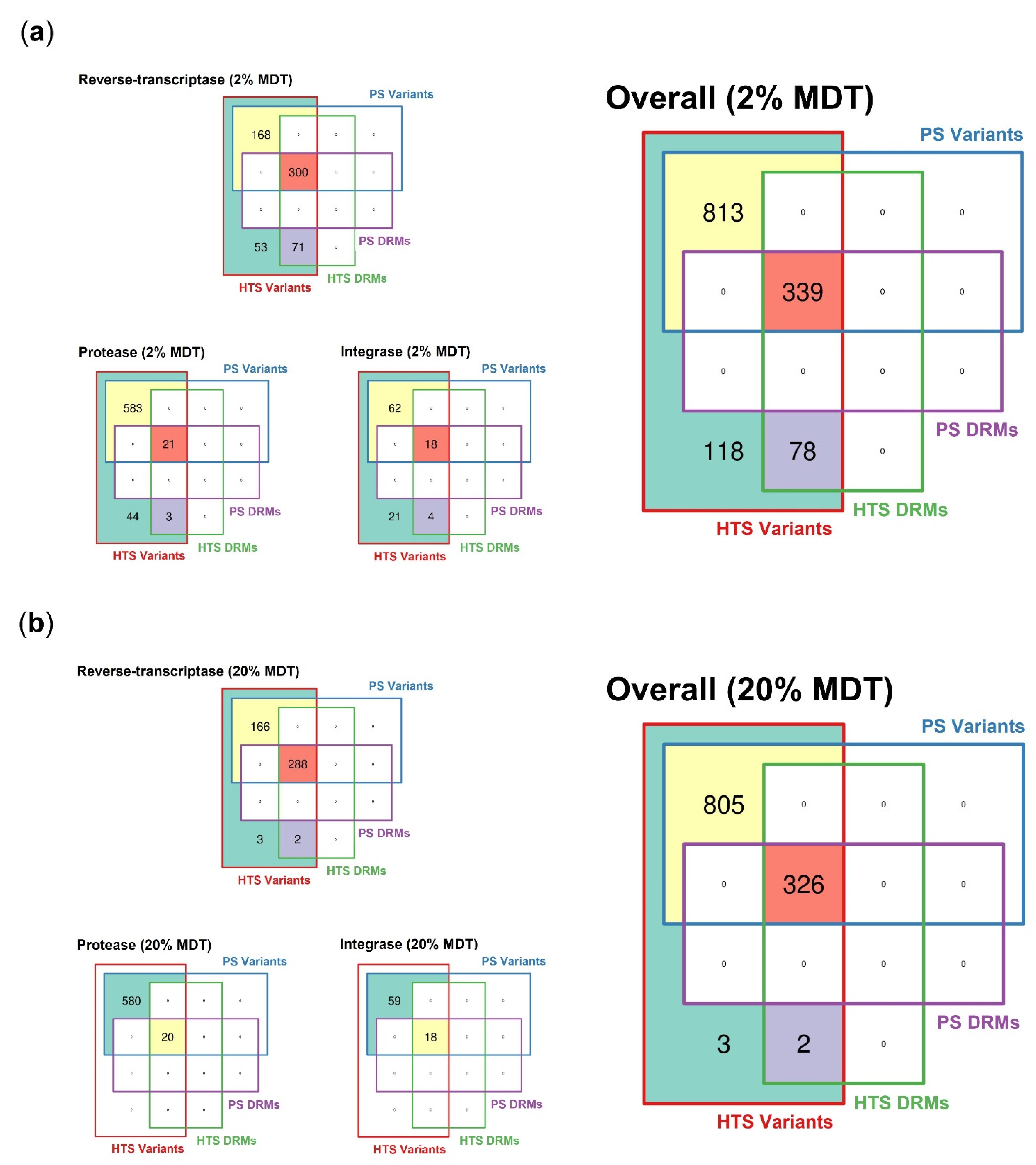
3.2.2. Spectrum of Detected Variants
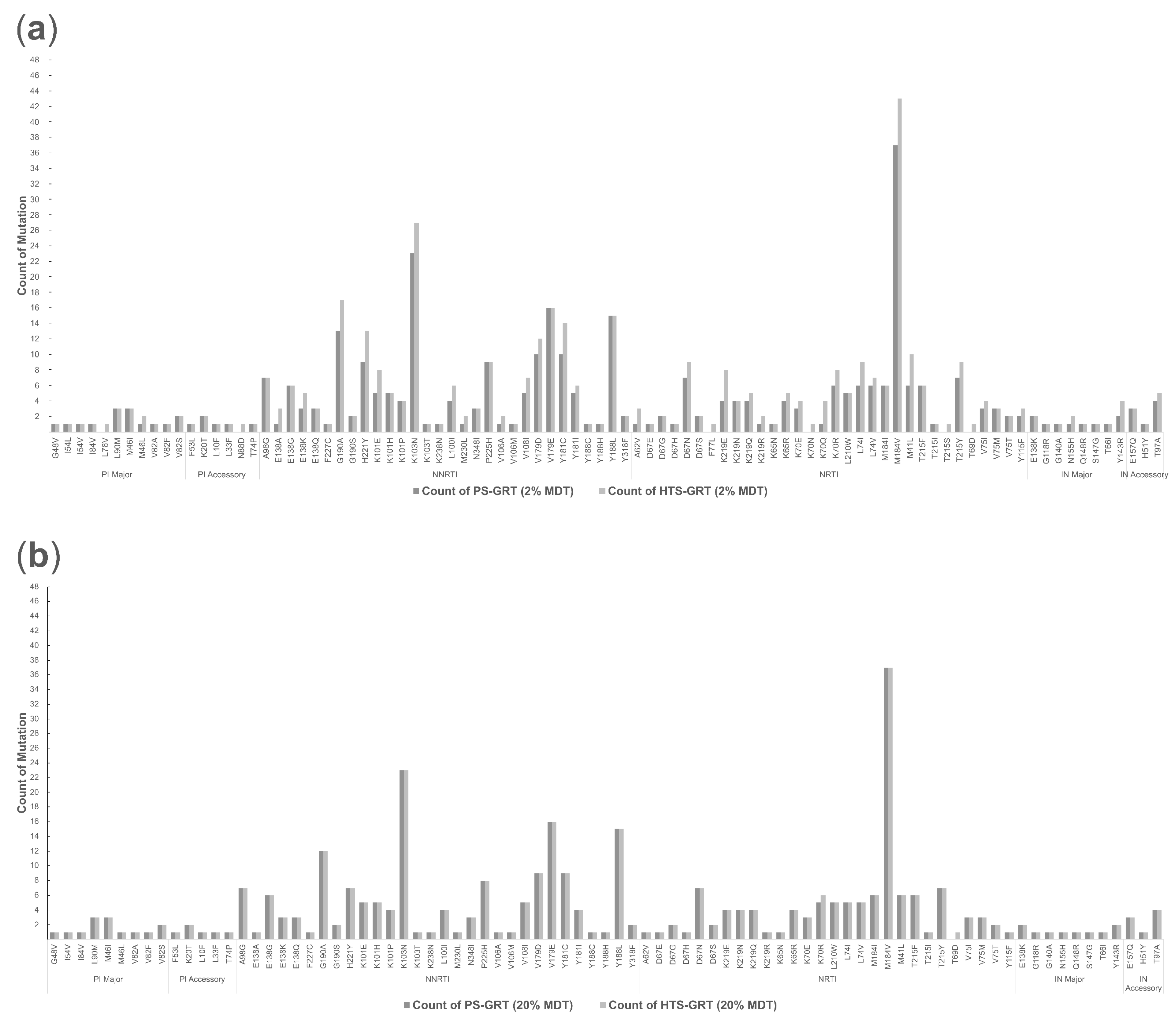
3.2.3. Spectrum of Detected Drug-Resistant Mutations
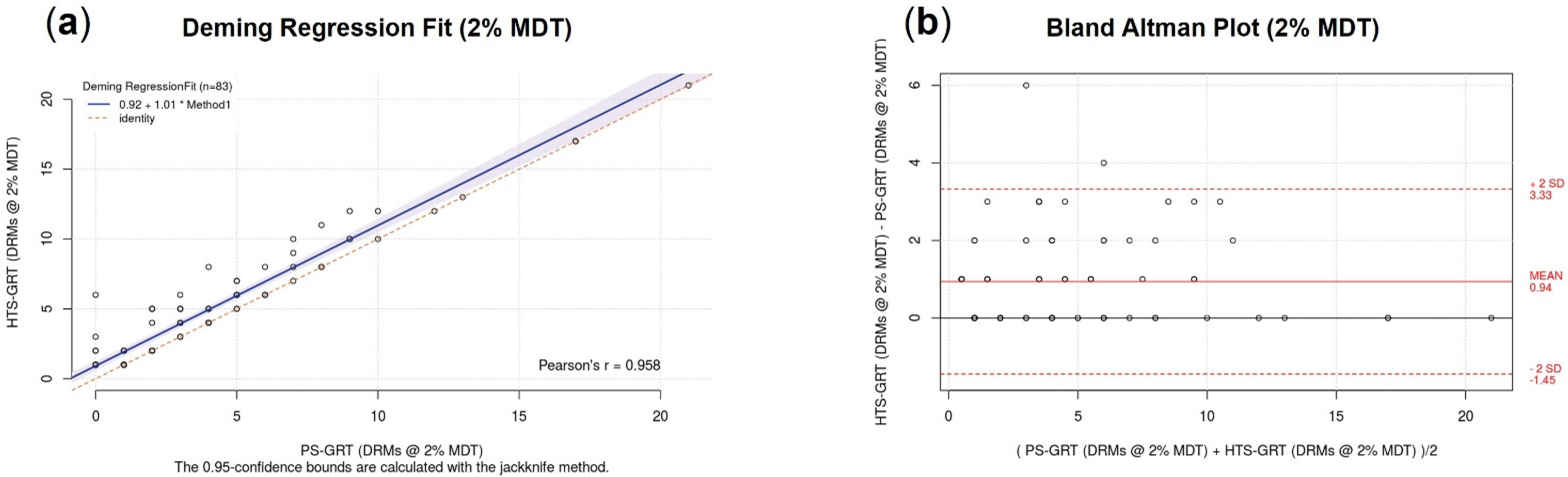
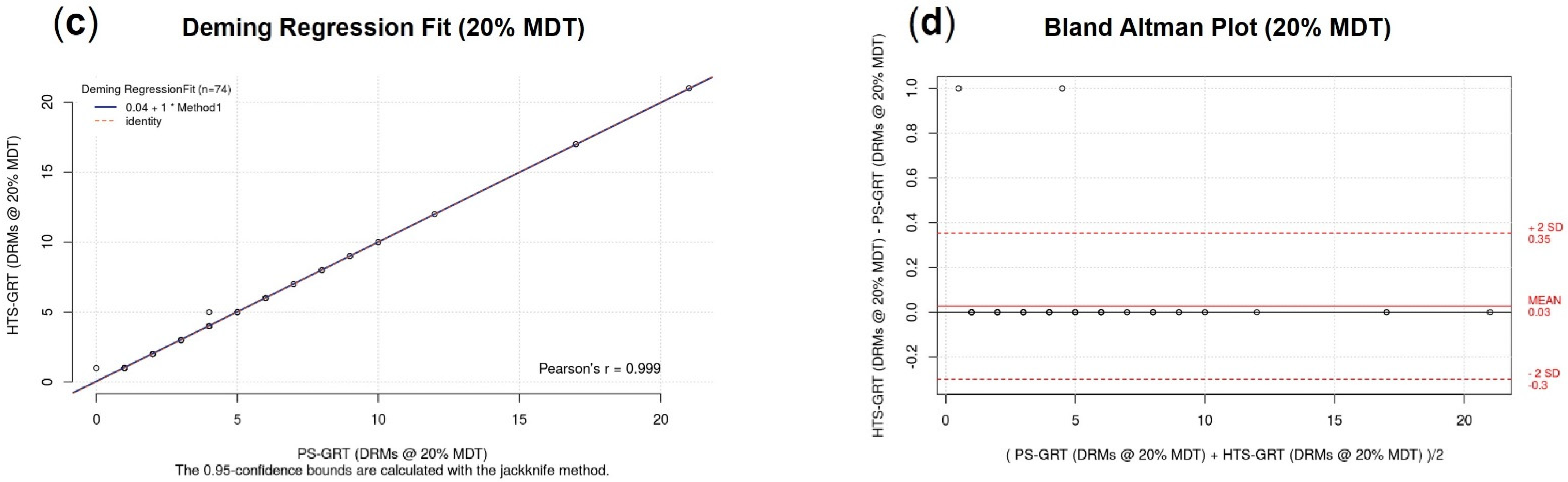
3.2.4. Accuracy
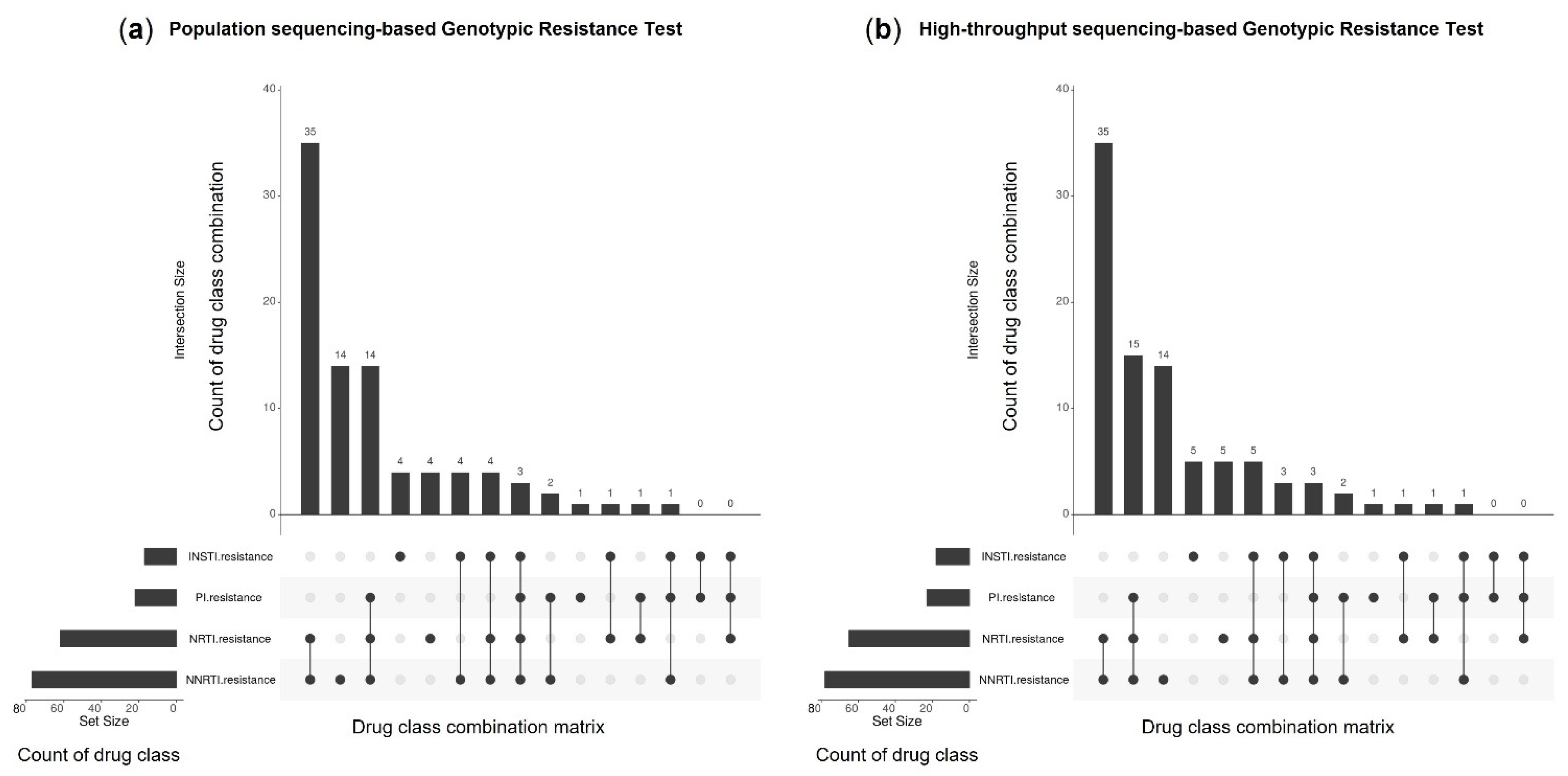
3.2.5. Genotypic Resistance Interpretation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS Data 2021. Available online: https://www.unaids.org/en/resources/documents/2021/2021_unaids_data/ (accessed on 20 August 2022).
- Simon, V.; Ho, D.D.; Karim, Q.A. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 2006, 368, 489–505. [Google Scholar] [CrossRef] [Green Version]
- Baxter, J.D.; Mayers, D.L.; Wentworth, D.N.; Neaton, D.J.; Hoover, M.L.; Winters, M.A.; Mannheimer, S.B.; Thompson, M.A.; Abrams, D.I.; Brizz, B.J.; et al. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS 2000, 14, F83–F93. [Google Scholar] [CrossRef]
- Cingolani, A.; Antinori, A.; Rizzo, M.G.; Murri, R.; Ammassari, A.; Baldini, F.; Giambenedetto, S.D.; Cauda, R.; Luca, A.D. Usefulness of monitoring HIV drug resistance and adherence in individuals failing highly active antiretroviral therapy: A randomized study (ARGENTA). AIDS 2002, 16, 369–379. [Google Scholar] [CrossRef]
- Sax, P.E.; Islam, R.; Walensky, R.P.; Losina, E.; Weinstein, M.C.; Goldie, S.J.; Sadownik, S.N.; Freedberg, K.A. Should Resistance Testing Be Performed for Treatment-Naive HIV-Infected Patients? A Cost-Effectiveness Analysis. Clin. Infect. Dis. 2005, 41, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Mbuagbaw, L.; Ongolo-Zogo, C.; Mendoza, O.C.; Zani, B.; Morfaw, F.; Nyambi, A.; Wang, A.; Kiflen, M.; El-Kechen, H.; Leenus, A.; et al. Guidelines are needed for studies of pre-treatment HIV drug resistance: A methodological study. BMC. Med. Res. Methodol. 2021, 21, 76. [Google Scholar] [CrossRef]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Perelson, A.S.; Neumann, A.U.; Markowitz, M.; Leonard, J.M.; Ho, D.D. HIV-1 dynamics in vivo: Virion clearance rate, infected cell life-span, and viral generation time. Science 1996, 271, 1582–1586. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Rhee, S.; Kantor, R.; Katzenstein, D.A.; Camacho, R.; Morris, L.; Sirivichayakul, S.; Jorgensen, L.; Brigido, L.F.; Schapiro, J.M.; Shafer, R.W.; et al. HIV-1 pol mutation frequency by subtype and treatment experience: Extension of the HIVseq program to seven non-B subtypes. AIDS 2006, 20, 643–651. [Google Scholar] [CrossRef]
- Johnson, J.A.; Li, J.; Wei, X.; Lipscomb, J.; Irlbeck, D.; Craig, C.; Smith, A.; Bennett, D.E.; Monsour, M.; Sandstrom, P.; et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naïve populations and associate with reduced treatment efficacy. PLoS Med. 2008, 5, e158. [Google Scholar] [CrossRef] [PubMed]
- Paar, C.; Palmetshofer, C.; Flieger, K.; Geit, M.; Kaiser, R.; Stekel, H.; Berg, J. Genotypic antiretroviral resistance testing for human immunodeficiency virus type 1 integrase inhibitors by use of the TruGene sequencing system. J. Clin. Microbiol. 2008, 46, 4087–4090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, R.T.; Wurcel, A.; Rosenberg, E.S.; Johnston, M.N.; Hellmann, N.; Bates, M.; Hirsch, M.S.; Walker, B.D. Progressive reversion of human immunodeficiency virus type 1 resistance mutations in vivo after transmission of a multiply drug-resistant virus. Clin. Infect. Dis. 2003, 37, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Iyidogan, P.; Anderson, K.S. Current perspectives on HIV-1 antiretroviral drug resistance. Viruses 2014, 6, 4095–4139. [Google Scholar] [CrossRef] [Green Version]
- Paredes, R.; Clotet, B. Clinical management of HIV-1 resistance. Antivir. Res. 2010, 85, 245–265. [Google Scholar] [CrossRef]
- Clercq, E.D. Human viral diseases: What is next for antiviral drug discovery? Curr. Opin. Virol. 2012, 2, 572–579. [Google Scholar] [CrossRef]
- Gupta-Wright, A.; Fielding, K.; Oosterhout, J.J.; Alufandika, M.; Grint, D.J.; Chimbayo, E.; Heaney, J.; Byott, M.; Nastouli, E.; Mwandumba, H.C.; et al. Virological failure, HIV-1 drug resistance, and early mortality in adults admitted to hospital in Malawi: An observational cohort study. Lancet HIV 2020, 7, e620–e628. [Google Scholar] [CrossRef]
- McCluskey, S.M.; Siedner, M.J.; Marconi, V.C. Management of Virologic Failure and HIV Drug Resistance. Infect. Dis. Clin. N. Am. 2019, 33, 707–742. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Mitsuya, Y.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W. Characterization of mutation spectra with ultra-deep pyrosequencing: Application to HIV-1 drug resistance. Genome Res. 2007, 17, 1195–1201. [Google Scholar] [CrossRef]
- Hirsch, M.S.; Brun-Vézinet, F.; D’Aquila, R.T.; Hammer, S.M.; Johnson, V.A.; Kuritzkes, D.R.; Loveday, C.; Mellors, J.W.; Clotet, B.; Conway, B.; et al. Antiretroviral drug resistance testing in adult HIV-1 infection: Recommendations of an International AIDS Society-USA Panel. JAMA 2000, 283, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.M.; Meyer, A.M.; Winner, D.; Archer, J.; Feyertag, F.; Ruiz-Mateos, E.; Leal, M.; Robertson, D.L.; Schmotzer, C.L.; Quiñones-Mateu, M.E. Sensitive deep-sequencing-based HIV-1 genotyping assay to simultaneously determine susceptibility to protease, reverse transcriptase, integrase, and maturation inhibitors, as well as HIV-1 coreceptor tropism. Antimicrob. Agents Chemother. 2014, 58, 2167–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alidjinou, E.K.; Deldalle, J.; Hallaert, C.; Robineau, O.; Ajana, F.; Choisy, P.; Hober, D.; Bocket, L. RNA and DNA Sanger sequencing versus next-generation sequencing for HIV-1 drug resistance testing in treatment-naive patients. J. Antimicrob. Chemother. 2017, 72, 2823–2830. [Google Scholar] [CrossRef] [Green Version]
- Simen, B.B.; Simons, J.F.; Hullsiek, K.H.; Novak, R.M.; Macarthur, R.D.; Baxter, J.D.; Huang, C.; Lubeski, C.; Turenchalk, G.S.; Braverman, M.S.; et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009, 199, 693–701. [Google Scholar] [CrossRef]
- Varghese, V.; Liu, T.F.; Rhee, S.; Libiran, P.; Trevino, C.; Fessel, W.J.; Shafer, R.W. HIV-1 integrase sequence variability in antiretroviral naïve patients and in triple-class experienced patients subsequently treated with raltegravir. AIDS Res. Hum. Retrovir. 2010, 26, 1323–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Cozzi-Lepri, A.; Noguera-Julian, M.; Giallonardo, F.D.; Schuurman, R.; Däumer, M.; Aitken, S.; Ceccherini-Silberstein, F.; Monforte, A.D.; Geretti, A.M.; Booth, C.L.; et al. Low-frequency drug-resistant HIV-1 and risk of virological failure to first-line NNRTI-based ART: A multicohort European case-control study using centralized ultrasensitive 454 pyrosequencing. J. Antimicrob. Chemother. 2015, 70, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Nicot, F.; Sauné, K.; Raymond, S.; Jeanne, N.; Carcenac, R.; Lefebvre, C.; Cuzin, L.; Marchou, B.; Delobel, P.; Izopet, J. Minority resistant HIV-1 variants and the response to first-line NNRTI therapy. J. Clin. Virol. 2015, 62, 20–24. [Google Scholar] [CrossRef]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The Role of HIV-1 Drug-Resistant Minority Variants in Treatment Failure. J. Infect. Dis. 2017, 216, S847–S850. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Kuritzkes, D.R. Clinical implications of HIV-1 minority variants. Clin. Infect. Dis. 2013, 56, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamers, R.L.; Paredes, R. Next-generation sequencing and HIV drug resistance surveillance. Lancet HIV 2016, 3, e553–e554. [Google Scholar] [CrossRef]
- Li, M.; Liang, S.; Zhou, C.; Chen, M.; Liang, S.; Liu, C.; Zuo, Z.; Liu, L.; Feng, Y.; Song, C.; et al. HIV Drug Resistance Mutations Detection by Next-Generation Sequencing during Antiretroviral Therapy Interruption in China. Pathogens 2021, 10, 264. [Google Scholar] [CrossRef]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-Generation Human Immunodeficiency Virus Sequencing for Patient Management and Drug Resistance Surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef] [Green Version]
- Gianella, S.; Richman, D.D. Minority variants of drug-resistant HIV. J. Infect. Dis. 2010, 202, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.K.; Lee, H.K.; Loh, T.P.; Sethi, S.K.; Koay, E.S.; Tang, J.W. An in-house HIV genotyping assay for the detection of drug resistance mutations in Southeast Asian patients infected with HIV-1. J. Med. Virol. 2012, 84, 394–401. [Google Scholar] [CrossRef] [PubMed]
- RECall (v2.30)–Web Based Sequence Analysis. Available online: https://recall.bccfe.ca/account/login/ (accessed on 20 August 2022).
- HIVdb Program: Sequence Analysis. Available online: https://hivdb.stanford.edu/hivdb/by-sequences/ (accessed on 20 August 2022).
- HIVdb Program: Mutations Analysis. Available online: https://hivdb.stanford.edu/hivdb/by-patterns/ (accessed on 20 August 2022).
- Stoler, N.; Nekrutenko, A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom. Bioinform. 2021, 3, lqab019. [Google Scholar] [CrossRef]
- Metzner, K.J.; Giulieri, S.G.; Knoepfel, S.A.; Rauch, P.; Burgisser, P.; Yerly, S.; Günthard, H.F.; Cavassini, M. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and -adherent patients. Clin. Infect. Dis. 2009, 48, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Halvas, E.K.; Wiegand, A.; Boltz, V.F.; Kearney, M.; Nissley, D.; Wantman, M.; Hammer, S.M.; Palmer, S.; Vaida, F.; Coffin, J.M.; et al. Low frequency nonnucleoside reverse-transcriptase inhibitor-resistant variants contribute to failure of efavirenz-containing regimens in treatment- experienced patients. J. Infect. Dis. 2010, 201, 672–680. [Google Scholar] [CrossRef]
- Delobel, P.; Saliou, A.; Nicot, F.; Dubois, M.; Trancart, S.; Tangre, P.; Aboulker, J.; Taburet, A.; Molina, J.; Massip, P.; et al. Minor HIV-1 variants with the K103N resistance mutation during intermittent efavirenz-containing antiretroviral therapy and virological failure. PLoS ONE 2011, 6, e21655. [Google Scholar] [CrossRef]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Kozal, M.J.; Svarovskaia, E.S.; Johnson, J.A.; Geretti, A.M.; Metzner, K.J.; Jakobsen, M.R.; Hullsiek, K.H.; et al. Impact of minority nonnucleoside reverse transcriptase inhibitor resistance mutations on resistance genotype after virologic failure. J. Infect. Dis. 2013, 207, 893–897. [Google Scholar] [CrossRef] [Green Version]
- Paredes, R.; Lalama, C.N.; Ribaudo, H.J.; Schackman, B.R.; Shikuma, C.; Giguel, F.; Meyer, W.A., 3rd; Johnson, V.A.; Fiscus, S.A.; D’Aquila, R.T.; et al. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J. Infect. Dis. 2010, 201, 662–671. [Google Scholar] [CrossRef] [Green Version]
- May, S.; Adamska, E.; Tang, J. Evaluation of Vela Diagnostics HIV-1 genotyping assay on an automated next generation sequencing platform. J. Clin. Virol. 2020, 127, 104376. [Google Scholar] [CrossRef]
- Parkin, N.T.; Avila-Rios, S.; Bibby, D.F.; Brumme, C.J.; Eshleman, S.H.; Harrigan, P.R.; Howison, M.; Hunt, G.; Ji, H.; Kantor, R.; et al. Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. Viruses 2020, 12, 694. [Google Scholar] [CrossRef]
- Ayitewala, A.; Ssewanyana, I.; Kiyaga, C. Next generation sequencing based in-house HIV genotyping method: Validation report. AIDS Res. Ther. 2021, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, M.A.; Genchi, C.; Lagioia, A.; Talamo, V.; Volpe, A.; Mariggiò, M.A. Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience. Int. J. Mol. Sci. 2022, 23, 2727. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Target Region | Nucleotide Positions φ | Sequence (5′ to 3′) | Orientation | Expected Size |
|---|---|---|---|---|---|
| PS-GRT | PR | 2071 to 2091 | GAR AGA CAG GCT AAT TTT TTA | Forward | 1722 |
| RT | 3774 to 3792 | CCC ANT CAG GAA TCC AGG T | Reverse | ||
| IN | 4176 to 4204 | GGA GGA AAT GAA MAA RTA GAT AAA TTA GT | Forward | 885 | |
| IN | 5040 to 5060 | ACC TGC CAT CTG TTT TCC ATA | Reverse | ||
| HTS-GRT | PR | 2071 to 2091 | GAR AGA CAG GCT AAT TTT TTA | Forward | 2990 |
| IN | 5040 to 5060 | ACC TGC CAT CTG TTT TCC ATA | Reverse |
| Characteristics | Count | Median (IQR) |
|---|---|---|
| HIV-1 Viral Load | ||
| With | 44 (43%) | 4.80 log 10 copies/mL (4.35–5.20) |
| Without | 59 (57%) | |
| HIV-1 Subtype | ||
| B | 20 (19%) | |
| C | 3 (3%) | |
| G | 2 (2%) | |
| F2 | 1 (1%) | |
| CRF01_AE | 70 (68%) | |
| CRF33_01B | 2 (2%) | |
| CRF07_BC | 1 (1%) | |
| CRF48_01B | 1 (1%) | |
| CRF51_01B | 1 (1%) | |
| CRF52_01B | 1 (1%) | |
| CRF54_01B | 1 (1%) |
| Characteristics | Count | Unique Codon Positions | Frequency Level (Median and IQR) | McNemar’s χ2 (p-Value) |
|---|---|---|---|---|
| 2% Mutation Detection Threshold: | ||||
| gag-pol region | ||||
| HTS-GRT (Detected) | 1348 | 86 | 100% (80–100%) | |
| PS-GRT (Detected) | 1152 | 72 | 100% (99–100%) | 194.01 (<0.05) |
| PS-GRT (Missed) | 196 | 68 | 4% (2–9%) | |
| PR | ||||
| HTS-GRT (Detected) | 651 | 29 | 100% (99–100%) | |
| PS-GRT (Detected) | 604 | 26 | 100% (99–100%) | 45.02 (<0.05) |
| PS-GRT (Missed) | 47 | 20 | 3% (2–5%) | |
| RT | ||||
| HTS-GRT (Detected) | 592 | 37 | 99% (27–100%) | |
| PS-GRT (Detected) | 468 | 36 | 100% (90–100%) | 122.01 (<0.05) |
| PS-GRT (Missed) | 124 | 35 | 4% (3–10%) | |
| IN | ||||
| HTS-GRT (Detected) | 105 | 20 | 99% (16–100%) | |
| PS-GRT (Detected) | 80 | 17 | 99.50% (96.25–100%) | 23.04 (<0.05) |
| PS-GRT (Missed) | 25 | 13 | 5% (2–10%) | |
| 20% Mutation Detection Threshold: | ||||
| gag-pol region | ||||
| HTS-GRT (Detected) | 1136 | 80 | 100% (99–100%) | |
| PS-GRT (Detected) | 1131 | 80 | 100% (99–100%) | 3.20 (0.07) |
| PS-GRT (Missed) | 5 | 3 | 26% (23–31%) | |
| PR | ||||
| HTS-GRT (Detected) | 600 | 27 | 100% (99–100%) | |
| PS-GRT (Detected) | 600 | 27 | 100% (99–100%) | - |
| PS-GRT (Missed) | 0 | - | - | |
| RT | ||||
| HTS-GRT (Detected) | 459 | 36 | 100% (92–100%) | |
| PS-GRT (Detected) | 454 | 36 | 100% (100%) | 3.20 (0.07) |
| PS-GRT (Missed) | 5 | 3 | 26% (23–31%) | |
| IN | ||||
| HTS-GRT (Detected) | 77 | 17 | 100% (98–100%) | |
| PS-GRT (Detected) | 77 | 17 | 100% (98–100%) | - |
| PS-GRT (Missed) | 0 | - | - | |
| Characteristics | Count | Unique Codon Positions | Frequency Level (Median and IQR) | McNemar’s χ2 (p-Value) |
|---|---|---|---|---|
| 2% Mutation Detection Threshold: | ||||
| gag-pol region | ||||
| HTS-GRT (Detected) | 417 | 57 | 100% (80–100%) | |
| PS-GRT (Detected) | 339 | 50 | 100% (92–100%) | 76.01 (<0.05) |
| PS-GRT (Missed) | 78 | 30 | 5% (3–11%) | |
| PR | ||||
| HTS-GRT (Detected) | 24 | 13 | 100% (99–100%) | |
| PS-GRT (Detected) | 21 | 11 | 100% (99–100%) | 1.33 (0.25) |
| PS-GRT (Missed) | 3 | 3 | 3% (2–3.50%) | |
| RT | ||||
| HTS-GRT (Detected) | 371 | 33 | 99% (29–100%) | |
| PS-GRT (Detected) | 300 | 30 | 100% (91–100%) | 69.01 (<0.05) |
| PS-GRT (Missed) | 71 | 24 | 5% (3–11%) | |
| IN | ||||
| HTS-GRT (Detected) | 22 | 11 | 99% (45.75–100%) | |
| PS-GRT (Detected) | 18 | 11 | 100% (98.25–100%) | 2.25 (0.13) |
| PS-GRT (Missed) | 4 | 3 | 10.50% (8.25–11%) | |
| 20% Mutation Detection Threshold: | ||||
| gag-pol region | ||||
| HTS-GRT (Detected) | 328 | 53 | 100% (95.75–100%) | |
| PS-GRT (Detected) | 326 | 52 | 100% (96–100%) | 0.50 (0.48) |
| PS-GRT (Missed) | 2 | 2 | 28.50% (27.25–29.75%) | |
| PR | ||||
| HTS-GRT (Detected) | 20 | 11 | 100% (99–100%) | |
| PS-GRT (Detected) | 20 | 11 | 100% (99–100%) | - |
| PS-GRT (Missed) | 0 | - | - | |
| RT | ||||
| HTS-GRT (Detected) | 290 | 31 | 100% (94–100%) | |
| PS-GRT (Detected) | 288 | 30 | 100% (94.75–100%) | 0.50 (0.48) |
| PS-GRT (Missed) | 2 | 2 | 28.50% (27.25–29.75%) | |
| IN | ||||
| HTS-GRT (Detected) | 18 | 11 | 100% (98.25–100%) | |
| PS-GRT (Detected) | 18 | 11 | 100% (98.25–100%) | - |
| PS-GRT (Missed) | 0 | - | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teo, C.H.Y.; Norhisham, N.H.B.; Lee, O.F.; Png, S.; Chai, C.N.; Yan, G.; Tang, J.W.-T.; Lee, C.K. Towards Next-Generation Sequencing for HIV-1 Drug Resistance Testing in a Clinical Setting. Viruses 2022, 14, 2208. https://doi.org/10.3390/v14102208
Teo CHY, Norhisham NHB, Lee OF, Png S, Chai CN, Yan G, Tang JW-T, Lee CK. Towards Next-Generation Sequencing for HIV-1 Drug Resistance Testing in a Clinical Setting. Viruses. 2022; 14(10):2208. https://doi.org/10.3390/v14102208
Chicago/Turabian StyleTeo, Calesta Hui Yi, Nurul Hannah Binte Norhisham, Ogestelli Fabia Lee, Siyu Png, Chean Nee Chai, Gabriel Yan, Julian Wei-Tze Tang, and Chun Kiat Lee. 2022. "Towards Next-Generation Sequencing for HIV-1 Drug Resistance Testing in a Clinical Setting" Viruses 14, no. 10: 2208. https://doi.org/10.3390/v14102208