
MiR-96-5p Suppresses Progression of Arsenite-Induced Human Keratinocyte Proliferation and Malignant Transformation by Targeting Denticleless E3 Ubiquitin Protein Ligase Homolog
 and
and 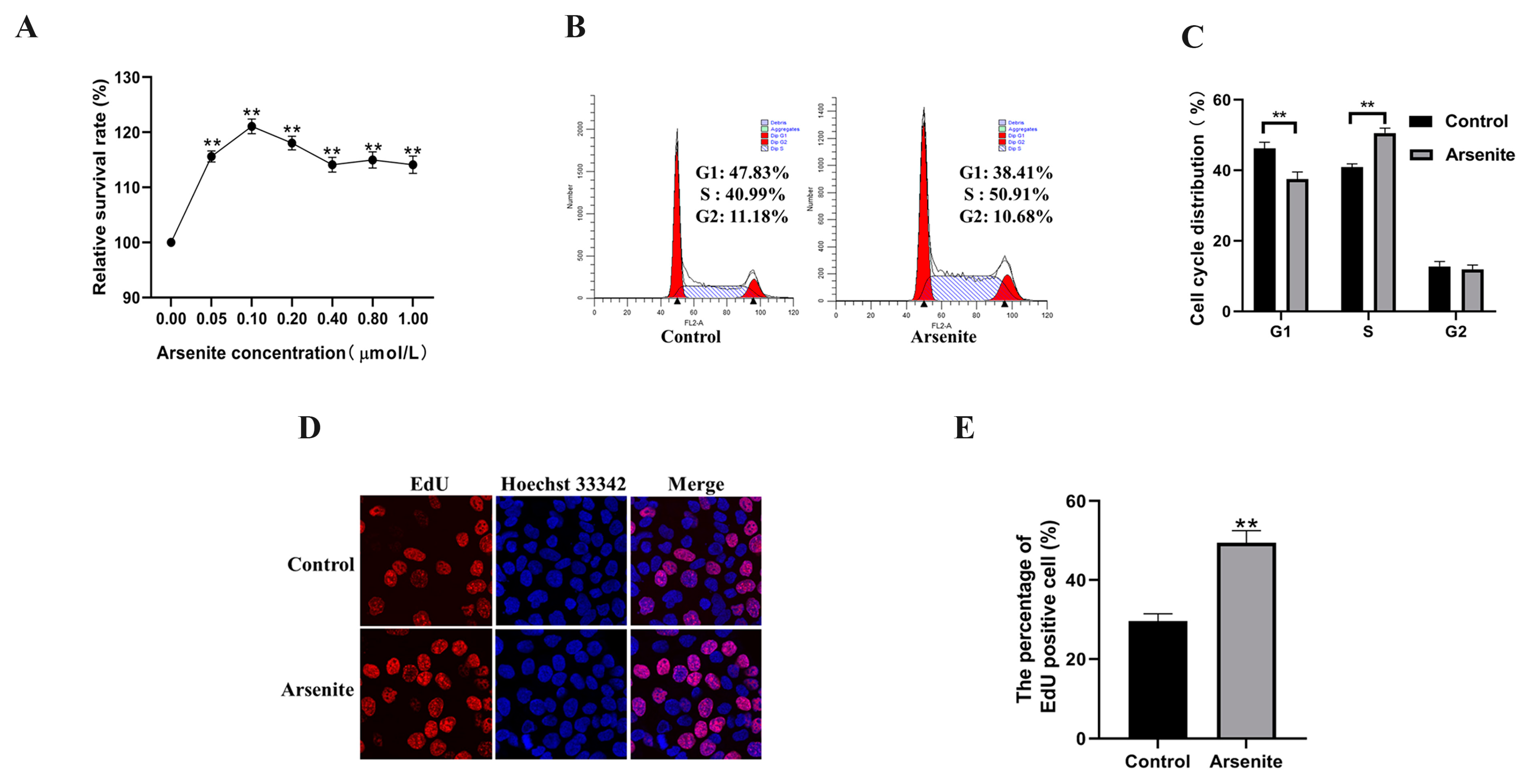{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microarray Data Analysis
2.2. Cell Culture and Treatment
2.3. Cell Transfection
2.4. Cell Viability Assay
2.5. EdU Incorporation Assay
2.6. Cell Cycle Assay
2.7. Immunofluorescence
2.8. Cell Growth Curve
2.9. Flat Plate Colony Formation Assay
2.10. Soft-Agar Colony Formation Assay
2.11. RT-qPCR
2.12. Luciferase Reporter Assay
2.13. In Vivo Tumor Model
2.14. Western Blot
2.15. Statistical Analysis
3. Results
3.1. Effects of a Low Level of Arsenite on HaCaT Cells Viability and Proliferation
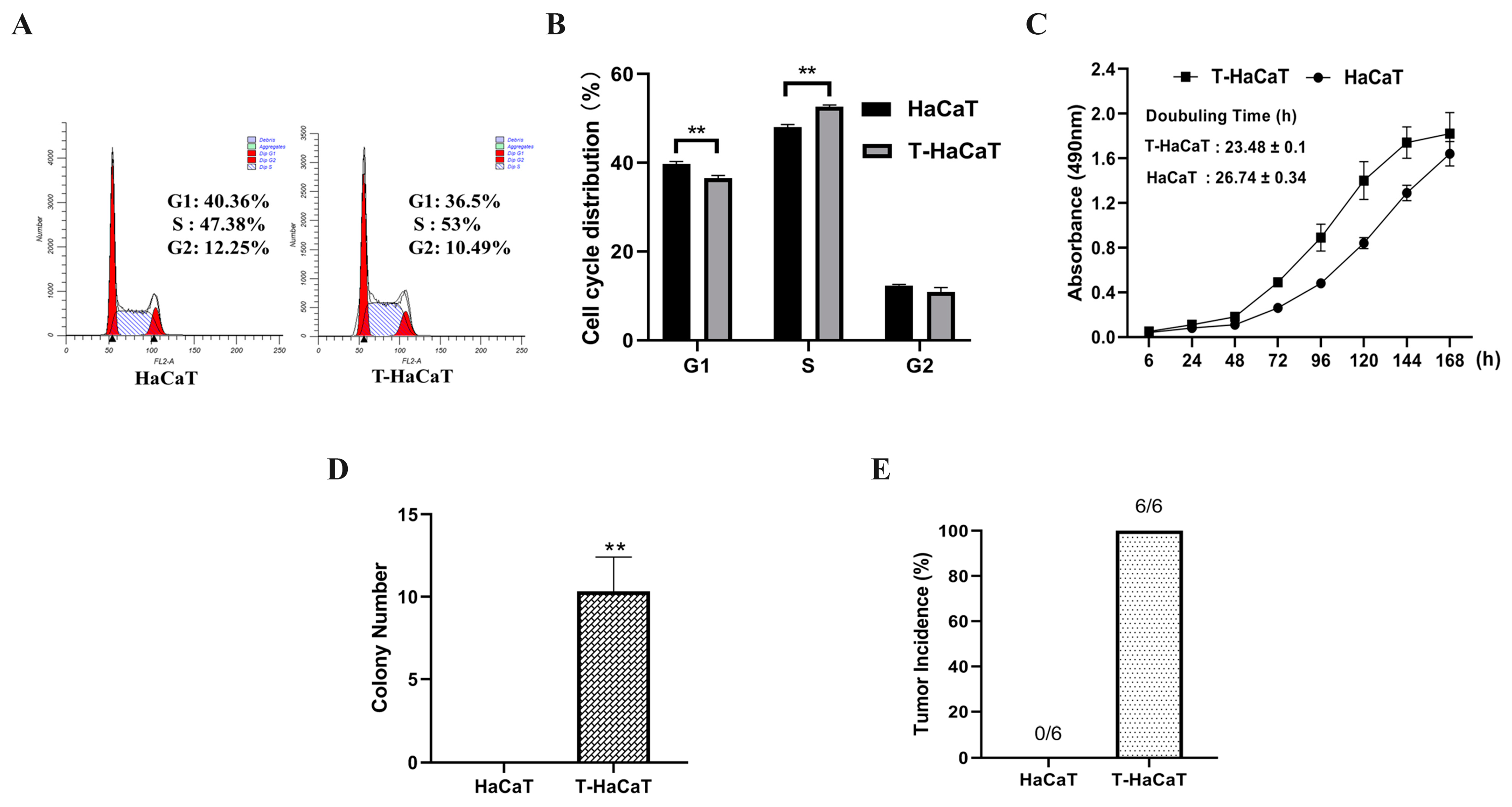
3.2. Arsenite-Induced Malignant Transformation Model of HaCaT Cells Was Successfully Constructed
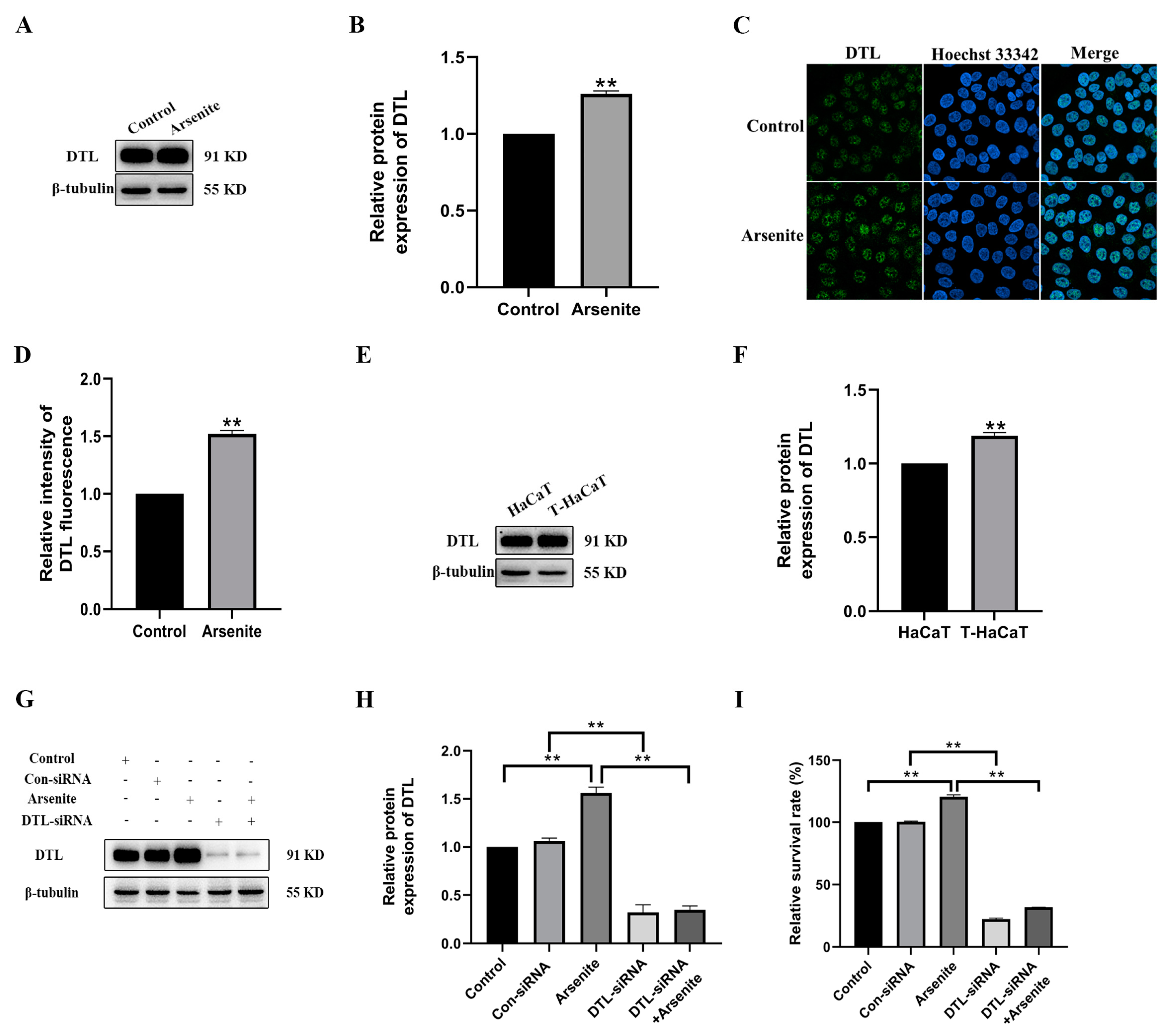
3.3. DTL Activation Involved in the Proliferation of HaCaT Cells Induced by Arsenite
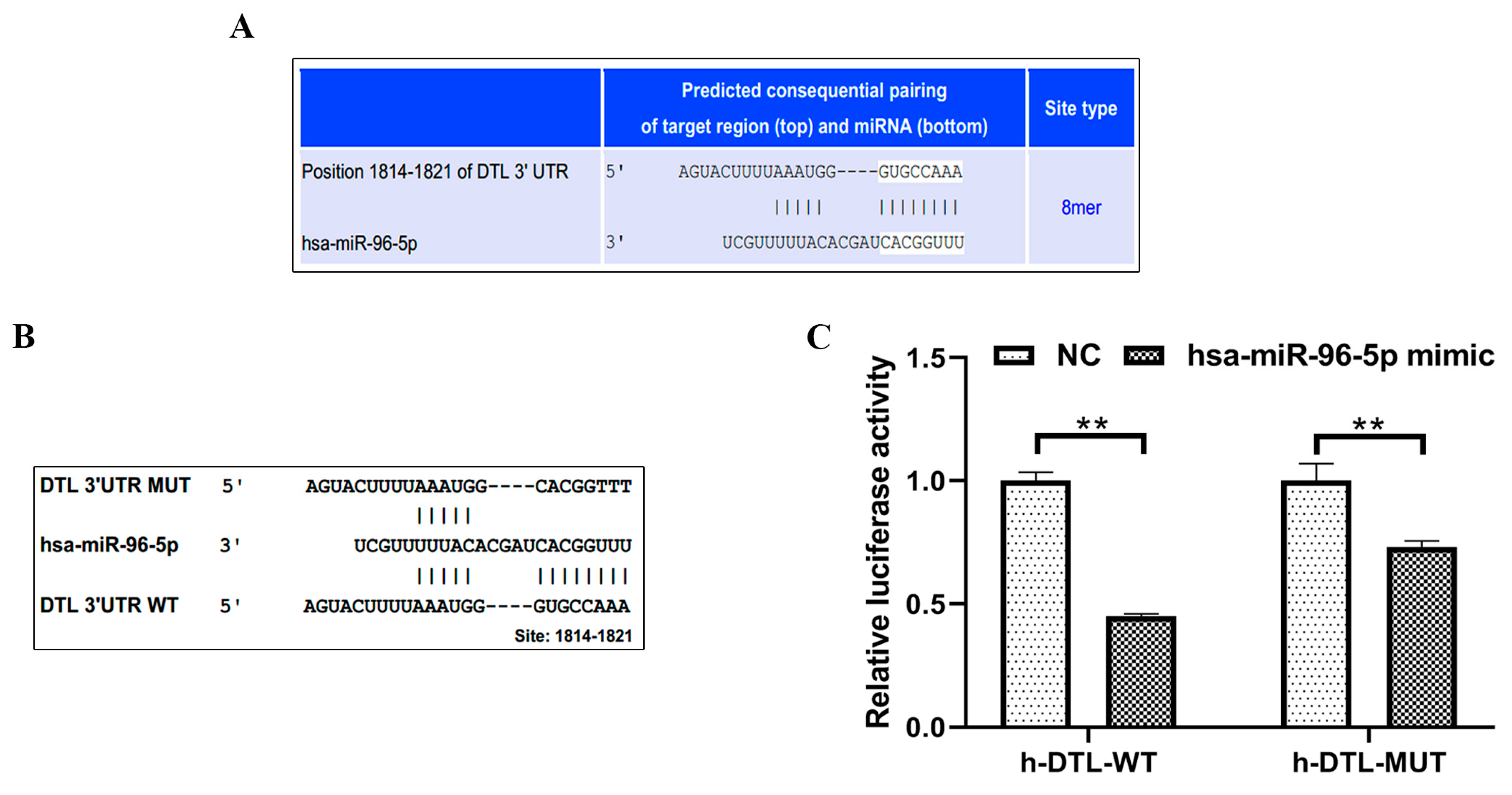
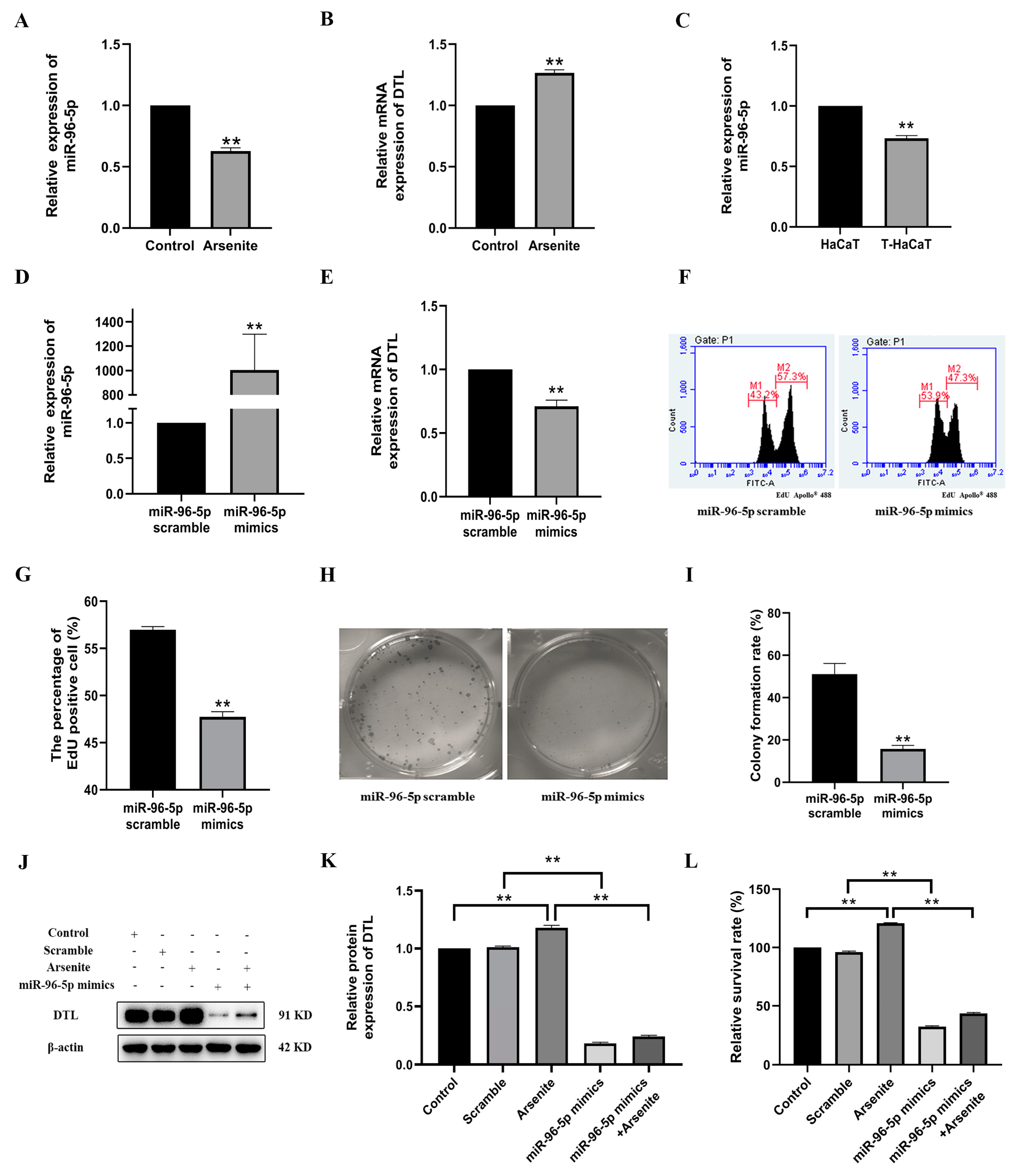
3.4. MiR-96-5p, a Negative Regulator of DTL, Mediated the Proliferation of HaCaT Cells Induced by Arsenite
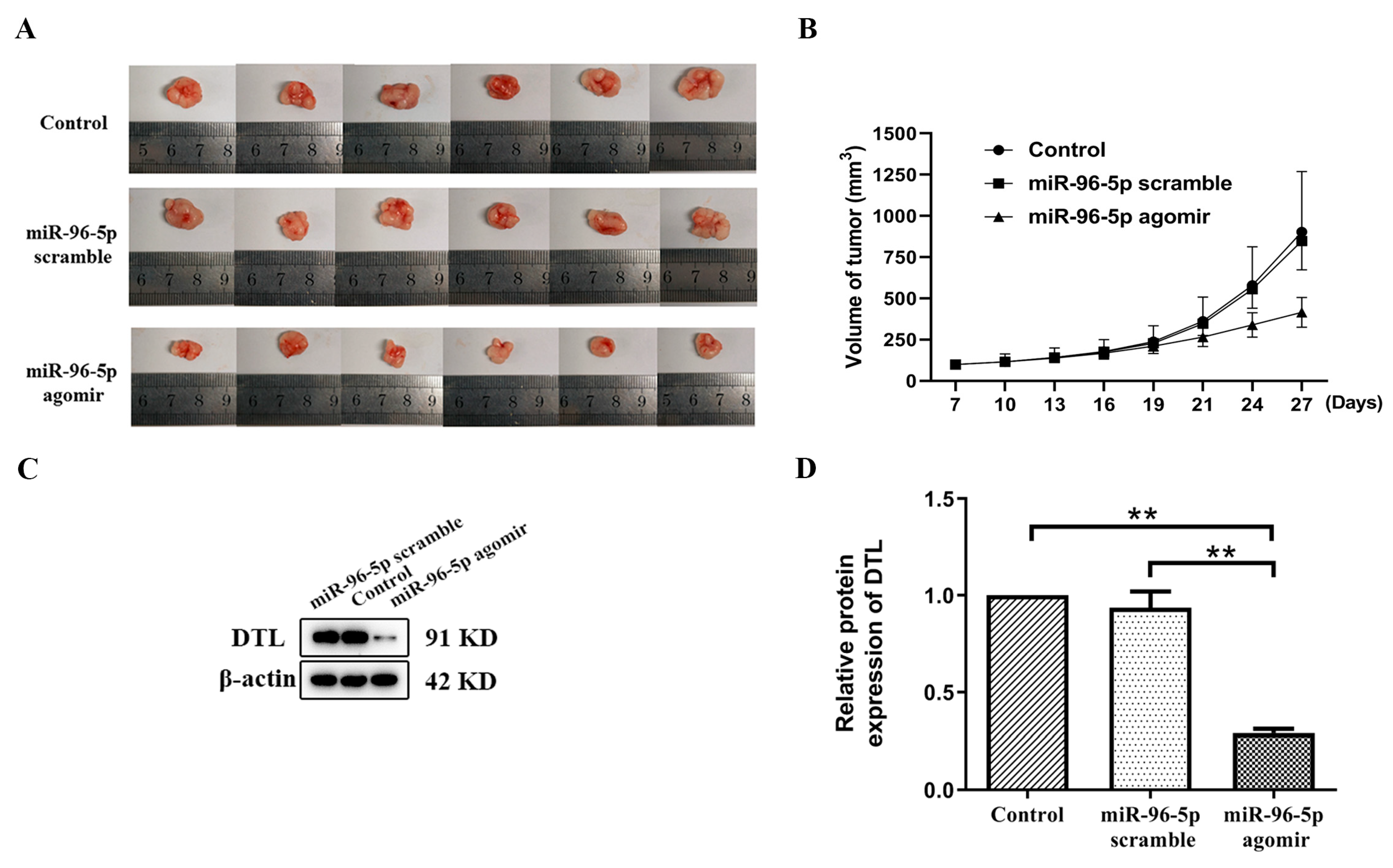
3.5. MiR-96-5p Treatment Suppressed T-HaCaT Cells Growth in Xenograft Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, G.; Sun, D.; Zheng, Y. Health effects of exposure to natural arsenic in groundwater and coal in China: An overview of occurrence. Environ. Health Perspect. 2007, 115, 636–642. [Google Scholar] [CrossRef]
- Naujokas, M.F.; Anderson, B.; Ahsan, H.; Aposhian, H.V.; Graziano, J.H.; Thompson, C.; Suk, W.A. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem. Environ. Health Perspect. 2013, 121, 295–302. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Chatterjee, D.; Singh, K.K.; Giri, A.K. Systems biology approaches to evaluate arsenic toxicity and carcinogenicity: An overview. Int. J. Hyg. Environ. Health 2013, 216, 574–586. [Google Scholar] [CrossRef]
- Huang, L.; Wu, H.; van der Kuijp, T.J. The health effects of exposure to arsenic-contaminated drinking water: A review by global geographical distribution. Int. J. Environ. Health Res. 2015, 25, 432–452. [Google Scholar] [CrossRef]
- Lamm, S.H.; Boroje, I.J.; Ferdosi, H.; Ahn, J. A review of low-dose arsenic risks and human cancers. Toxicology 2021, 456, 152768. [Google Scholar] [CrossRef]
- Smith, A.H.; Hopenhayn-Rich, C.; Bates, M.N.; Goeden, H.M.; Hertz-Picciotto, I.; Duggan, H.M.; Wood, R.; Kosnett, M.J.; Smith, M.T. Cancer risks from arsenic in drinking water. Environ. Health Perspect. 1992, 97, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef] [PubMed]
- Dastgiri, S.; Mosaferi, M.; Fizi, M.A.; Olfati, N.; Zolali, S.; Pouladi, N.; Azarfam, P. Arsenic exposure, dermatological lesions, hypertension, and chromosomal abnormalities among people in a rural community of northwest Iran. J. Health Popul. Nutr. 2010, 28, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Isokpehi, R.D.; Udensi, U.K.; Anyanwu, M.N.; Mbah, A.N.; Johnson, M.O.; Edusei, K.; Bauer, M.A.; Hall, R.A.; Awofolu, O.R. Knowledge building insights on biomarkers of arsenic toxicity to keratinocytes and melanocytes. Biomark. Insights 2012, 7, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Zhao, L.; Wang, Y.; Lu-Yao, G.; Liu, L.Z. Epigenetic Dysregulations in Arsenic-Induced Carcinogenesis. Cancers 2022, 14, 4502. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Lema, C.; Kirken, R.A.; Maldonado, R.A.; Varela-Ramirez, A.; Aguilera, R.J. Arsenic-exposed keratinocytes exhibit differential microRNAs expression profile; potential implication of miR-21, miR-200a and miR-141 in melanoma pathway. Clin. Cancer Drugs 2015, 2, 138–147. [Google Scholar] [CrossRef]
- Ferragut Cardoso, A.P.; Nail, A.N.; Banerjee, M.; Wise, S.S.; States, J.C. miR-186 induces tetraploidy in arsenic exposed human keratinocytes. Ecotoxicol. Environ. Saf 2023, 256, 114823. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203. [Google Scholar] [CrossRef]
- Mahata, J.; Basu, A.; Ghoshal, S.; Sarkar, J.; Roy, A.; Poddar, G.; Nandy, A.; Banerjee, A.; Ray, K.; Natarajan, A. Chromosomal aberrations and sister chromatid exchanges in individuals exposed to arsenic through drinking water in West Bengal, India. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2003, 534, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011, 2011, 431287. [Google Scholar] [CrossRef]
- Niedzwiecki, M.M.; Hall, M.N.; Liu, X.; Oka, J.; Harper, K.N.; Slavkovich, V.; Ilievski, V.; Levy, D.; van Geen, A.; Mey, J.L. A dose–response study of arsenic exposure and global methylation of peripheral blood mononuclear cell DNA in Bangladeshi adults. Environ. Health Perspect. 2013, 121, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Chen, C.; Luo, F.; Pan, X.; Xu, H.; Yang, P.; Sun, Q.; Liu, X.; Lu, L.; Yang, Q. CircLRP6 regulation of ZEB1 via miR-455 is involved in the epithelial-mesenchymal transition during arsenite-induced malignant transformation of human keratinocytes. Toxicol. Sci. 2018, 162, 450–461. [Google Scholar] [CrossRef]
- Banerjee, M.; Ferragut Cardoso, A.; Al-Eryani, L.; Pan, J.; Kalbfleisch, T.S.; Srivastava, S.; Rai, S.N.; States, J.C. Dynamic alteration in miRNA and mRNA expression profiles at different stages of chronic arsenic exposure-induced carcinogenesis in a human cell culture model of skin cancer. Arch. Toxicol. 2021, 95, 2351–2365. [Google Scholar] [CrossRef]
- Al-Eryani, L.; Waigel, S.; Tyagi, A.; Peremarti, J.; Jenkins, S.F.; Damodaran, C.; States, J.C. Differentially Expressed mRNA Targets of Differentially Expressed miRNAs Predict Changes in the TP53 Axis and Carcinogenesis-Related Pathways in Human Keratinocytes Chronically Exposed to Arsenic. Toxicol. Sci. 2018, 162, 645–654. [Google Scholar] [CrossRef]
- Al-Eryani, L.; Waigel, S.; Jala, V.; Jenkins, S.F.; States, J.C. Cell cycle pathway dysregulation in human keratinocytes during chronic exposure to low arsenite. Toxicol. Appl. Pharmacol. 2017, 331, 130–134. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. CRL4Cdt2: Master coordinator of cell cycle progression and genome stability. Cell Cycle 2011, 10, 241–249. [Google Scholar] [CrossRef]
- Ueki, T.; Nishidate, T.; Park, J.; Lin, M.; Shimo, A.; Hirata, K.; Nakamura, Y.; Katagiri, T. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene 2008, 27, 5672–5683. [Google Scholar] [CrossRef]
- Bartel, D.P.; Chen, C.-Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef]
- Fusenig, N.E.; Boukamp, P. Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol. Carcinog. 1998, 23, 144–158. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, Y.; Wang, H.; Darko, G.M.; Sun, D.; Gao, Y. Identification of miR-200c-3p as a major regulator of SaoS2 cells activation induced by fluoride. Chemosphere 2018, 199, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Freedman, V.H.; Shin, S.-I. Cellular tumorigenicity in nude mice: Correlation with cell growth in semi-solid medium. Cell 1974, 3, 355–359. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular senescence: Aging, cancer, and injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Havens, C.G.; Walter, J.C. Mechanism of CRL4Cdt2, a PCNA-dependent E3 ubiquitin ligase. Genes Dev. 2011, 25, 1568–1582. [Google Scholar] [CrossRef]
- Ress, A.L.; Stiegelbauer, V.; Winter, E.; Schwarzenbacher, D.; Kiesslich, T.; Lax, S.; Jahn, S.; Deutsch, A.; Bauernhofer, T.; Ling, H. MiR-96-5p influences cellular growth and is associated with poor survival in colorectal cancer patients. Mol. Carcinog. 2015, 54, 1442–1450. [Google Scholar] [CrossRef]
- Colita, A.; Tanase, A.D.; Tomuleasa, C.; Colita, A. Hematopoietic Stem Cell Transplantation in Acute Promyelocytic Leukemia in the Era of All-Trans Retinoic Acid (ATRA) and Arsenic Trioxide (ATO). Cancers 2023, 15, 4111. [Google Scholar] [CrossRef]
- Wang, Z.Y. Arsenic compounds as anticancer agents. Cancer Chemother. Pharmacol. 2001, 48 (Suppl. 1), S72–S76. [Google Scholar] [CrossRef]
- Bakhshaiesh, T.O.; Armat, M.; Shanehbandi, D.; Sharifi, S.; Baradaran, B.; Hejazi, M.S.; Samadi, N. Arsenic Trioxide Promotes Paclitaxel Cytotoxicity in Resistant Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 16, 5191–5197. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.J.; Lee, C.H.; Wang, S.L.; Chiou, H.Y.; Chen, C.J.; Seak, C.J.; Wu, I.W.; Hsu, K.H. Low-to-Moderate Arsenic Exposure and Urothelial Tract Cancers with a Long Latent Period of Follow-Up in an Arseniasis Area. J. Epidemiol. Glob Health 2023, 13, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Steinmaus, C.; Yuan, Y.; Bates, M.N.; Smith, A.H. Case-control study of bladder cancer and drinking water arsenic in the western United States. Am. J. Epidemiol. 2003, 158, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Rajput, M.; Kujur, P.K.; Mishra, A.; Singh, R.P. Flavonoids inhibit chronically exposed arsenic-induced proliferation and malignant transformation of HaCaT cells. Photodermatol. Photoimmunol. Photomed. 2018, 34, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.H.; Yang, S.; Wei, J.; Shea, C.R.; Zhong, W.; Wang, F.; Shah, P.; Kibriya, M.G.; Cui, X.; Ahsan, H.; et al. Autophagy of the m(6)A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat. Commun. 2021, 12, 2183. [Google Scholar] [CrossRef] [PubMed]
- Nail, A.N.; McCaffrey, L.M.; Banerjee, M.; Ferragut Cardoso, A.P.; States, J.C. Chronic arsenic exposure suppresses ATM pathway activation in human keratinocytes. Toxicol. Appl. Pharmacol. 2022, 446, 116042. [Google Scholar] [CrossRef]
- Hoesl, C.; Zanuttigh, E.; Fröhlich, T.; Philippou-Massier, J.; Krebs, S.; Blum, H.; Dahlhoff, M. The secretome of skin cancer cells activates the mTOR/MYC pathway in healthy keratinocytes and induces tumorigenic properties. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2020, 118717. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, H.; Wang, L.; Zhao, R.; Guan, D. Overexpression of NRF1-742 or NRF1-772 Reduces Arsenic-Induced Cytotoxicity and Apoptosis in Human HaCaT Keratinocytes. Int. J. Mol. Sci. 2020, 21, 2014. [Google Scholar] [CrossRef]
- Li, J.; Xue, J.; Wang, D.; Dai, X.; Sun, Q.; Xiao, T.; Wu, L.; Xia, H.; Mostofa, G.; Chen, X. Regulation of gasdermin D by miR-379-5p is involved in arsenite-induced activation of hepatic stellate cells and in fibrosis via secretion of IL-1β from human hepatic cells. Metallomics 2019, 11, 483–495. [Google Scholar] [CrossRef]
- Sun, J.-L.; Chen, D.-L.; Hu, Z.-Q.; Xu, Y.-Z.; Fang, H.-S.; Wang, X.-Y.; Kan, L.; Wang, S.-Y. Arsenite promotes intestinal tumor cell proliferation and invasion by stimulating epithelial-to-mesenchymal transition. Cancer Biol. Ther. 2014, 15, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Kumagai, Y.; Sun, G.; Yamauchi, H.; Yoshida, T.; Iso, H.; Endo, A.; Yu, L.; Yuki, K.; Miyauchi, T. Decreased serum concentrations of nitric oxide metabolites among Chinese in an endemic area of chronic arsenic poisoning in inner Mongolia. Free Radic. Biol. Med. 2000, 28, 1137–1142. [Google Scholar] [CrossRef]
- Zlotorynski, E. Chromosome biology: Controlling CENPA mislocalization. Nat. Rev. Mol. Cell Biol. 2014, 15, 368. [Google Scholar]
- Yang, C.; Wu, J.; He, H.; Liu, H. Small molecule NSC1892 targets the CUL4A/4B-DDB1 interactions and causes impairment of CRL4DCAF4 E3 ligases to inhibit colorectal cancer cell growth. Int. J. Biol. Sci. 2020, 16, 1059. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Yang, C.; He, H.; Liu, H. The CARM1-p300-c-Myc-Max (CPCM) transcriptional complex regulates the expression of CUL4A/4B and affects the stability of CRL4 E3 ligases in colorectal cancer. Int. J. Biol. Sci. 2020, 16, 1071. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, Y.; Hao, H.; Wang, Y.; Zhou, Z.; Wang, Z.; Chu, X. Screening Hub genes as prognostic biomarkers of hepatocellular carcinoma by bioinformatics analysis. Cell Transplant. 2019, 28, 76S–86S. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Wang, X.-M.; Xu, D.-Y.; Zhao, W.-J. Bioinformatics analysis of aberrantly methylated-differentially expressed genes and pathways in hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2605. [Google Scholar] [CrossRef]
- Barrio Garcia, S.; Da Via, M.; Garitano-Trojaola, A.; Ruiz-Heredia, Y.; Bittrich, M.; Shi, C.; Zhu, Y.; Lehners, N.; Mai, E.K.; Raab, M.S. IKZF1/3 and CRL4CRBN E3 ubiquitin ligase mutations associate with IMiD resistance in relapsed multiple myeloma. Blood 2017, 130, 270. [Google Scholar]
- Li, T.; Wu, S.; Jia, L.; Cao, W.; Yao, Y.; Zhao, G.; Li, H. CUL4 E3 ligase regulates the proliferation and apoptosis of lung squamous cell carcinoma and small cell lung carcinoma. Cancer Biol. Med. 2020, 17, 357. [Google Scholar] [CrossRef]
- Hu, X.; Meng, Y.; Xu, L.; Qiu, L.; Wei, M.; Su, D.; Qi, X.; Wang, Z.; Yang, S.; Liu, C. Cul4 E3 ubiquitin ligase regulates ovarian cancer drug resistance by targeting the antiapoptotic protein BIRC3. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Shen, J.; Yu, S.; Sun, X.; Yin, M.; Fei, J.; Zhou, J. Identification of key biomarkers associated with development and prognosis in patients with ovarian carcinoma: Evidence from bioinformatic analysis. J. Ovarian Res. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Vanderdys, V.; Allak, A.; Guessous, F.; Benamar, M.; Read, P.W.; Jameson, M.J.; Abbas, T. The Neddylation Inhibitor Pevonedistat (MLN4924) Suppresses and Radiosensitizes Head and Neck Squamous Carcinoma Cells and Tumors. Mol. Cancer Ther. 2018, 17, 368–380. [Google Scholar] [CrossRef]
- Kiran, S.; Dar, A.; Singh, S.K.; Lee, K.Y.; Dutta, A. The deubiquitinase USP46 is essential for proliferation and tumor growth of HPV-transformed cancers. Mol. Cell 2018, 72, 823–835.e825. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, C.; Mallardo, M. The roles of miR-25 and its targeted genes in development of human cancer. Microrna 2016, 5, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, X.; Liu, C.; Xi, X.; Wang, Y.; Wu, X.; Li, H. MiR-490-5p Restrains Progression of Gastric cancer through DTL Repression. Gastroenterol. Res. Pract. 2021, 2021, 2894117. [Google Scholar] [CrossRef]
- Georges, S.A.; Biery, M.C.; Kim, S.-y.; Schelter, J.M.; Guo, J.; Chang, A.N.; Jackson, A.L.; Carleton, M.O.; Linsley, P.S.; Cleary, M.A. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res. 2008, 68, 10105–10112. [Google Scholar] [CrossRef] [PubMed]
- Baraniskin, A.; Birkenkamp-Demtroder, K.; Maghnouj, A.; Zöllner, H.; Munding, J.; Klein-Scory, S.; Reinacher-Schick, A.; Schwarte-Waldhoff, I.; Schmiegel, W.; Hahn, S.A. MiR-30a-5p suppresses tumor growth in colon carcinoma by targeting DTL. Carcinogenesis 2012, 33, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Wu, M.; Qiu, Q.; Zhou, Q.; Li, J.; Yang, J.; Zheng, C.; Luo, A.; Li, X.; Zhang, H.; Cheng, X.; et al. circFBXO7/miR-96-5p/MTSS1 axis is an important regulator in the Wnt signaling pathway in ovarian cancer. Mol. Cancer 2022, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Wang, C.; Wang, S.; Zhang, H.; Zhang, Y. LncRNA STXBP5-AS1 suppressed cervical cancer progression via targeting miR-96-5p/PTEN axis. Biomed. Pharmacother. 2019, 117, 109082. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, K.; Huang, C.; Liu, L.; Zhao, H.; Huo, M.; Zhang, J. Integrated bioinformatics analysis reveals role of the LINC01093/miR-96-5p/ZFAND5/NF-κB signaling axis in hepatocellular carcinoma. Exp. Ther. Med. 2019, 18, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, C.-D.; Liang, Y.; Wu, K.-Z.; Pei, J.-P.; Dai, D.-Q. The comprehensive upstream transcription and downstream targeting regulation network of miRNAs reveal potential diagnostic roles in gastric cancer. Life Sci. 2020, 253, 117741. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, X.; Meng, X. miR-96-5p enhances cell proliferation and invasion via targeted regulation of ZDHHC5 in gastric cancer. Biosci. Rep. 2020, 40, BSR20191845. [Google Scholar] [CrossRef] [PubMed]
- Lian, Z.; Chang, T.; Ma, S.; Li, J.; Zhang, H.; Wang, X.; Liu, R. MiR-96-5p induced NDRG1 deficiency promotes prostate cancer migration and invasion through regulating the NF-κB signaling pathway. Cancer Biomark. 2022, 35, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zheng, Y.; Jiang, Y.; Li, X.; Geng, J.; Shen, Y.; Li, Q.; Wang, X.; Zhao, C.; Chen, Y. The circRNA circPTPRA suppresses epithelial-mesenchymal transitioning and metastasis of NSCLC cells by sponging miR-96-5p. EBioMedicine 2019, 44, 182–193. [Google Scholar] [CrossRef]
- Wang, T.; Xu, Y.; Liu, X.; Zeng, Y.; Liu, L. miR-96-5p is the tumor suppressor in osteosarcoma via targeting SYK. Biochem. Biophys. Res. Commun. 2021, 572, 49–56. [Google Scholar] [CrossRef]
- Li, C.; Du, X.; Tai, S.; Zhong, X.; Wang, Z.; Hu, Z.; Zhang, L.; Kang, P.; Ji, D.; Jiang, X. GPC1 regulated by miR-96-5p, rather than miR-182-5p, in inhibition of pancreatic carcinoma cell proliferation. Int. J. Mol. Sci. 2014, 15, 6314–6327. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Guo, X.; Zhou, L.; Jia, Z.; Peng, Z.; Tang, Y.; Liu, W.; Zhu, B.; Wang, L. GPC 1 exosome and its regulatory mi RNA s are specific markers for the detection and target therapy of colorectal cancer. J. Cell. Mol. Med. 2017, 21, 838–847. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhao, Q.; Yao, J.; Lv, C.; Gao, Y.; Sun, D.; Yang, Y. MiR-96-5p Suppresses Progression of Arsenite-Induced Human Keratinocyte Proliferation and Malignant Transformation by Targeting Denticleless E3 Ubiquitin Protein Ligase Homolog. Toxics 2023, 11, 978. https://doi.org/10.3390/toxics11120978
Li Y, Zhao Q, Yao J, Lv C, Gao Y, Sun D, Yang Y. MiR-96-5p Suppresses Progression of Arsenite-Induced Human Keratinocyte Proliferation and Malignant Transformation by Targeting Denticleless E3 Ubiquitin Protein Ligase Homolog. Toxics. 2023; 11(12):978. https://doi.org/10.3390/toxics11120978
Chicago/Turabian StyleLi, Yan, Qiaoshi Zhao, Jinyin Yao, Chunpeng Lv, Yanhui Gao, Dianjun Sun, and Yanmei Yang. 2023. "MiR-96-5p Suppresses Progression of Arsenite-Induced Human Keratinocyte Proliferation and Malignant Transformation by Targeting Denticleless E3 Ubiquitin Protein Ligase Homolog" Toxics 11, no. 12: 978. https://doi.org/10.3390/toxics11120978