

Myrtenal and Myrtanal as Auxiliaries in the Synthesis of Some C,P-Stereogenic Hydroxyphosphine Oxides and Hydroxyphosphine-Boranes Possessing up to Four Contiguous Centers of Chirality

Abstract
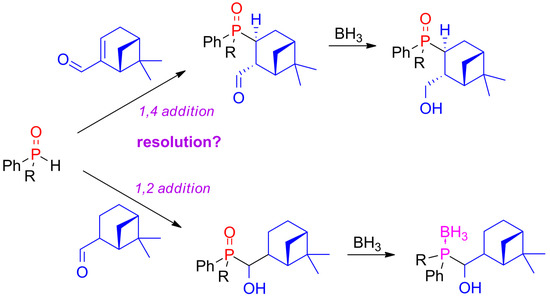
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
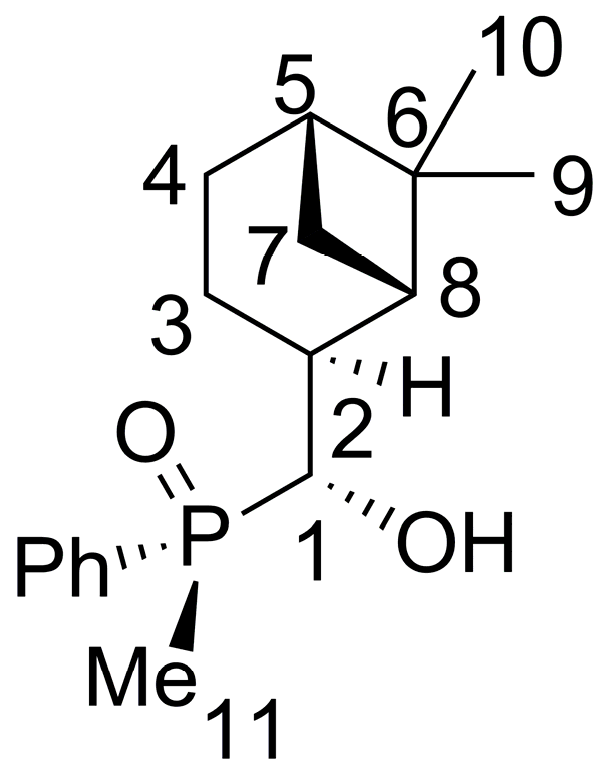{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
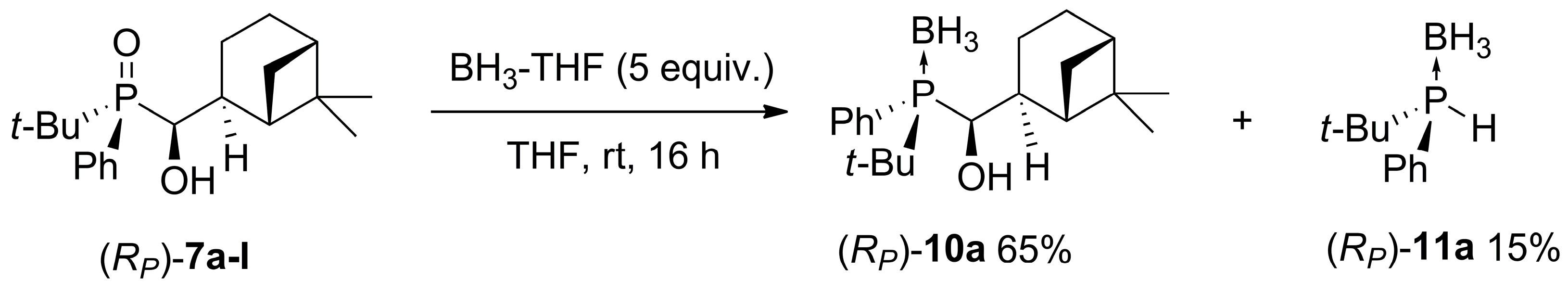{kind=link}
{kind=link}
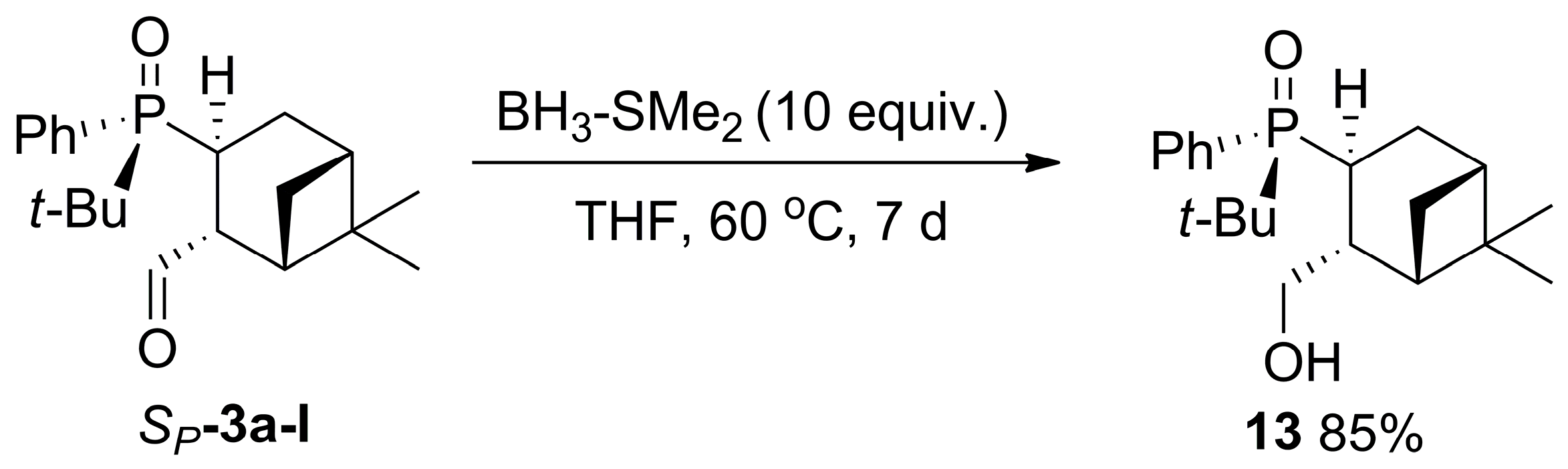{kind=link}
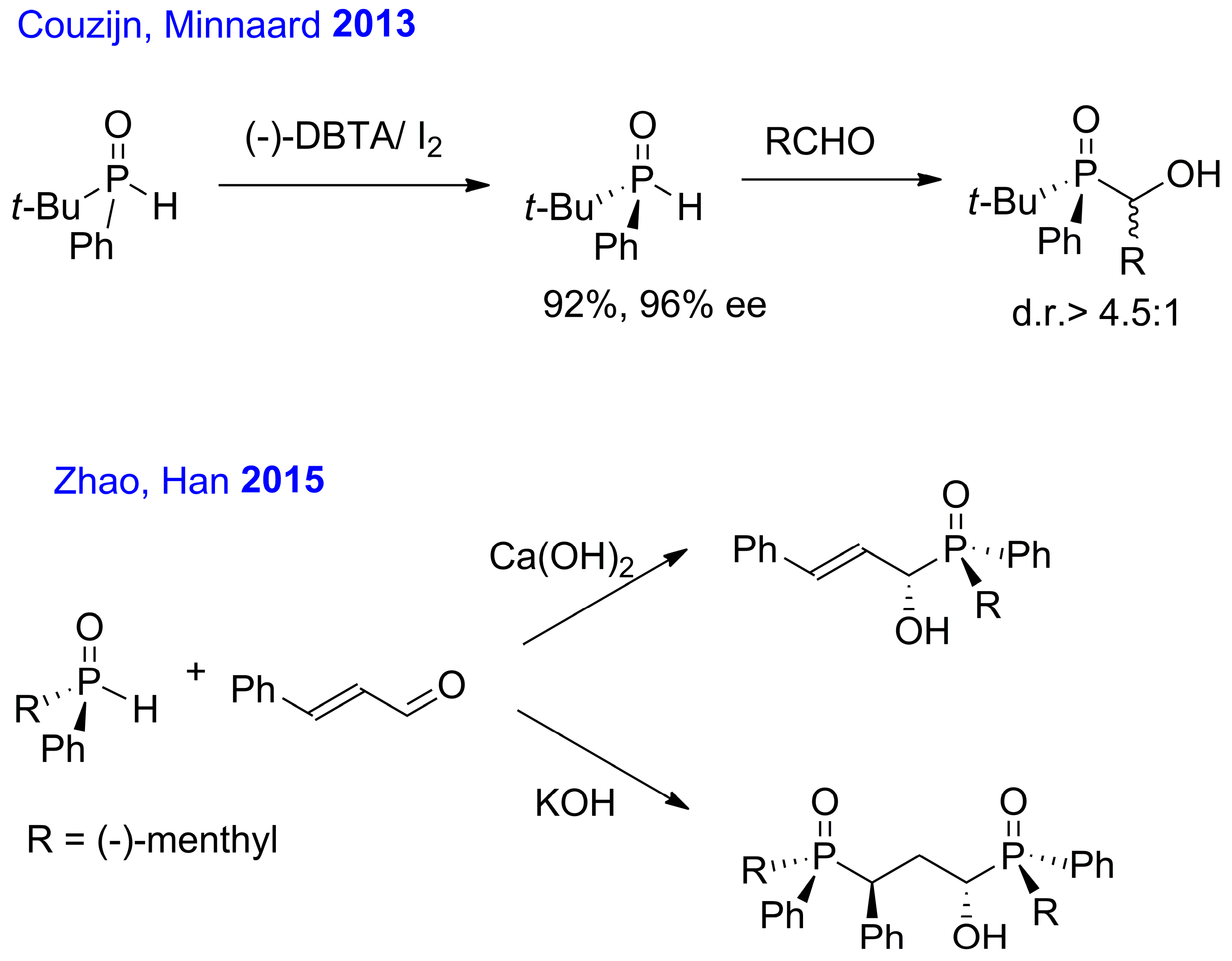
1. Introduction
2. Results and Discussion
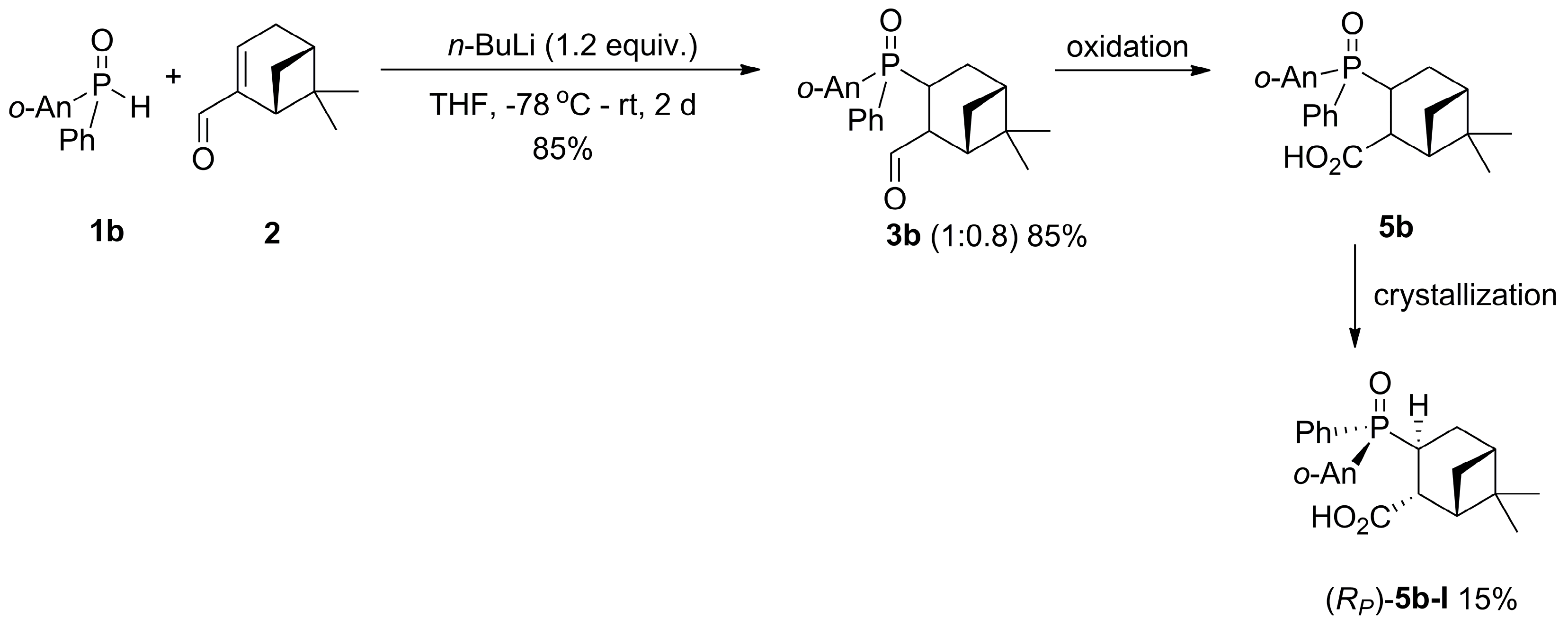
2.1. 1,4-Addition of Secondary Phosphine Oxides to (1R)-Myrtenal
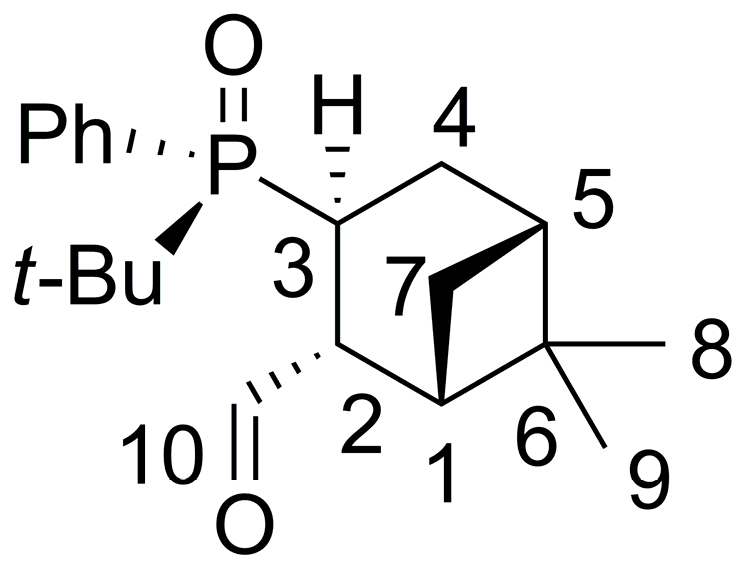
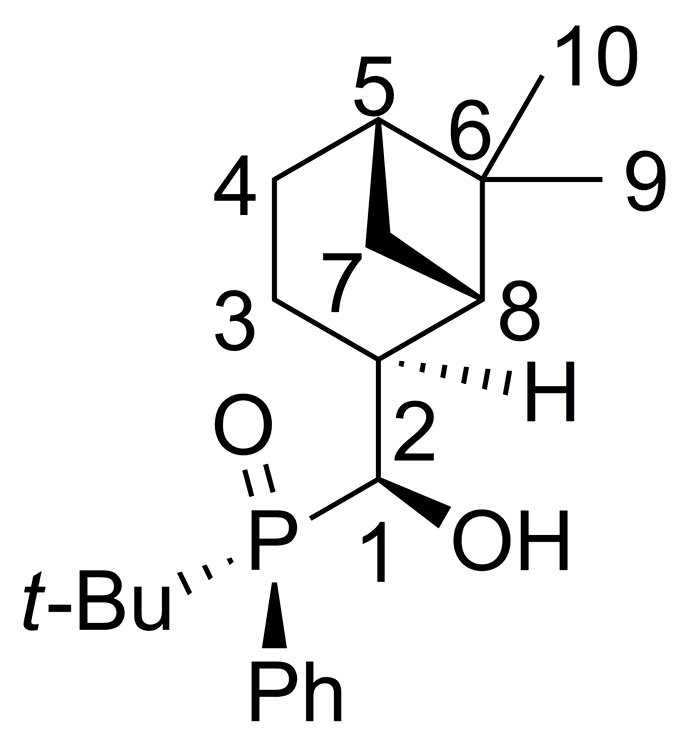
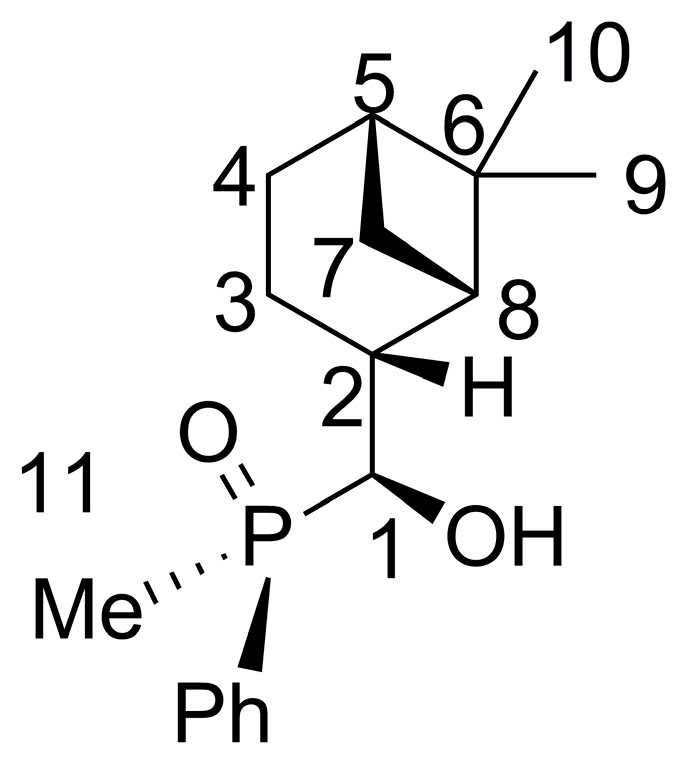
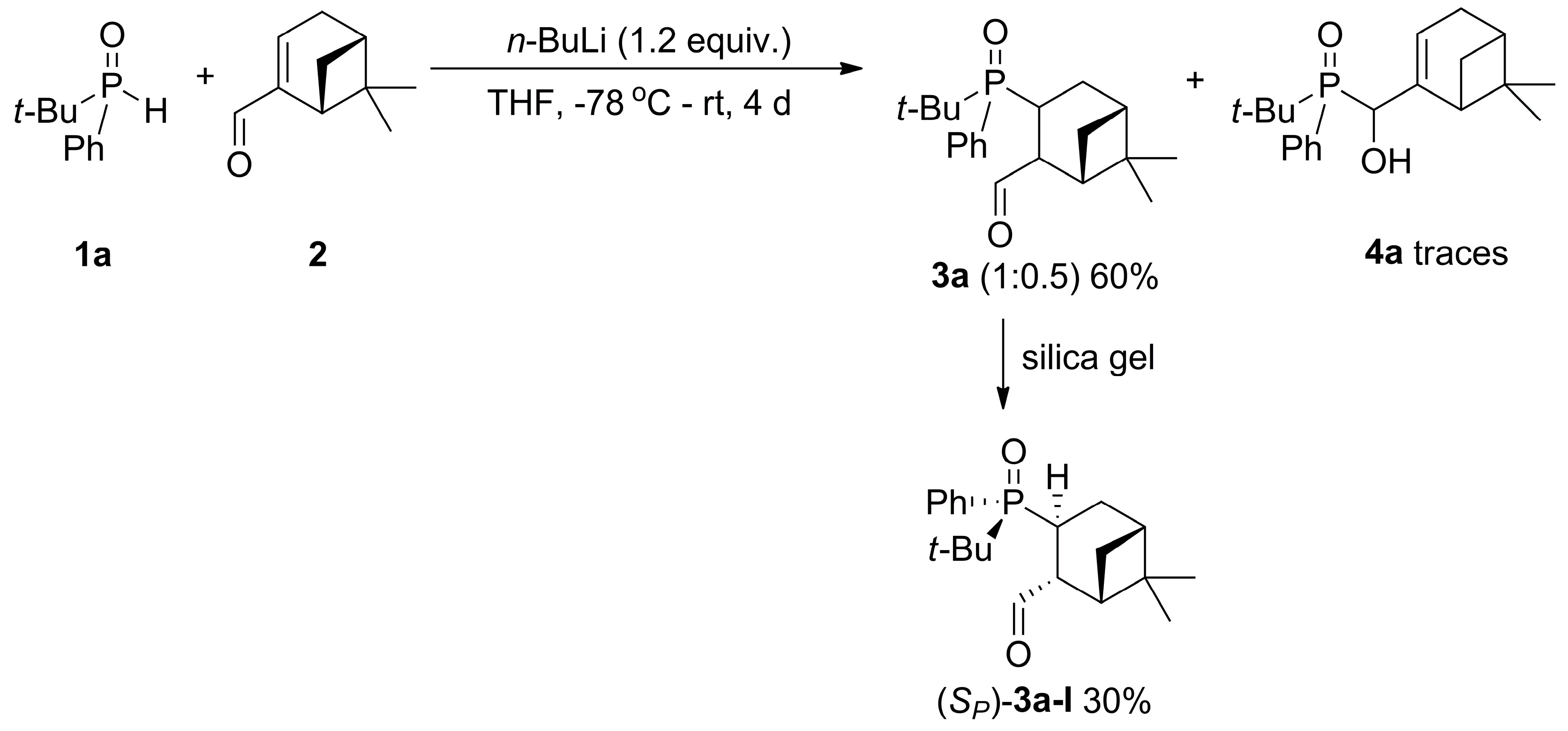
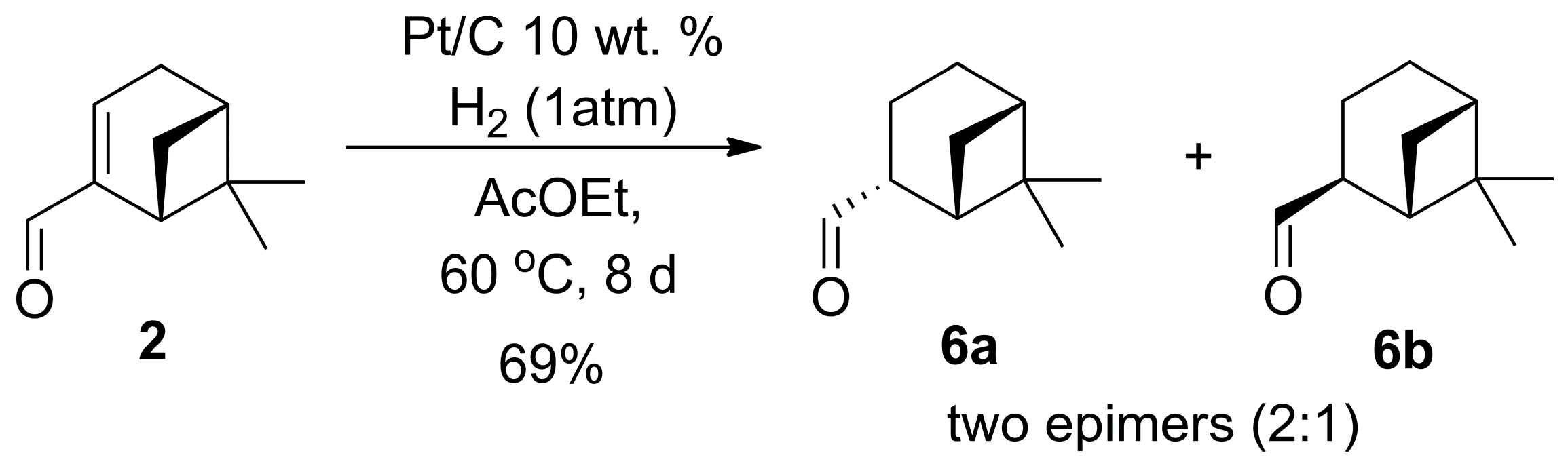
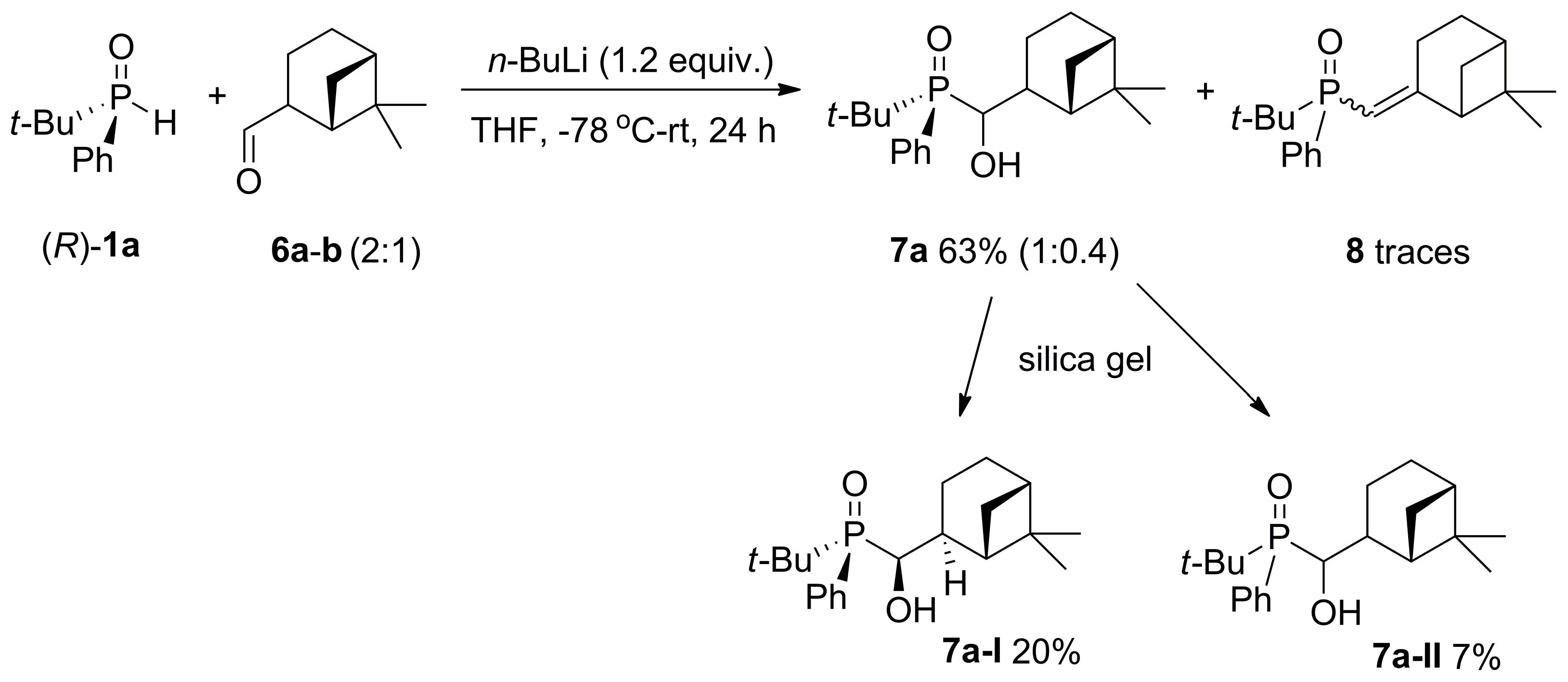
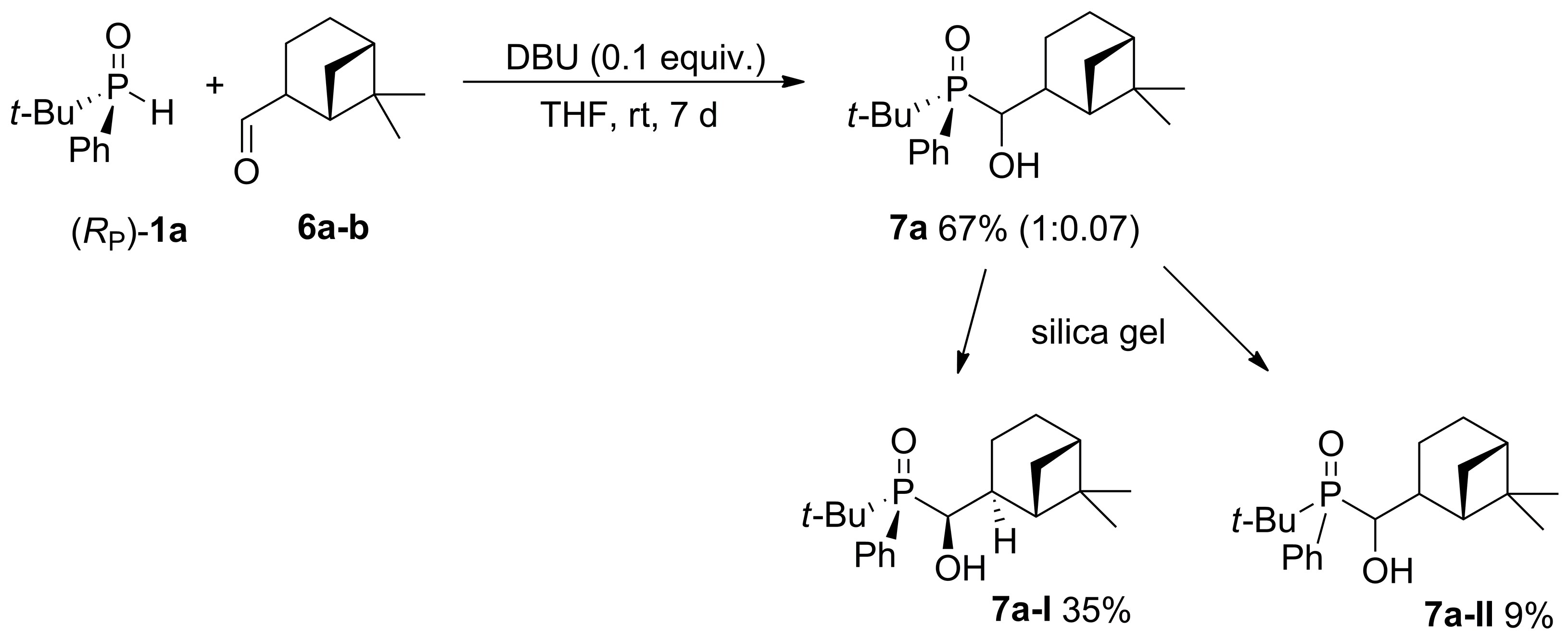
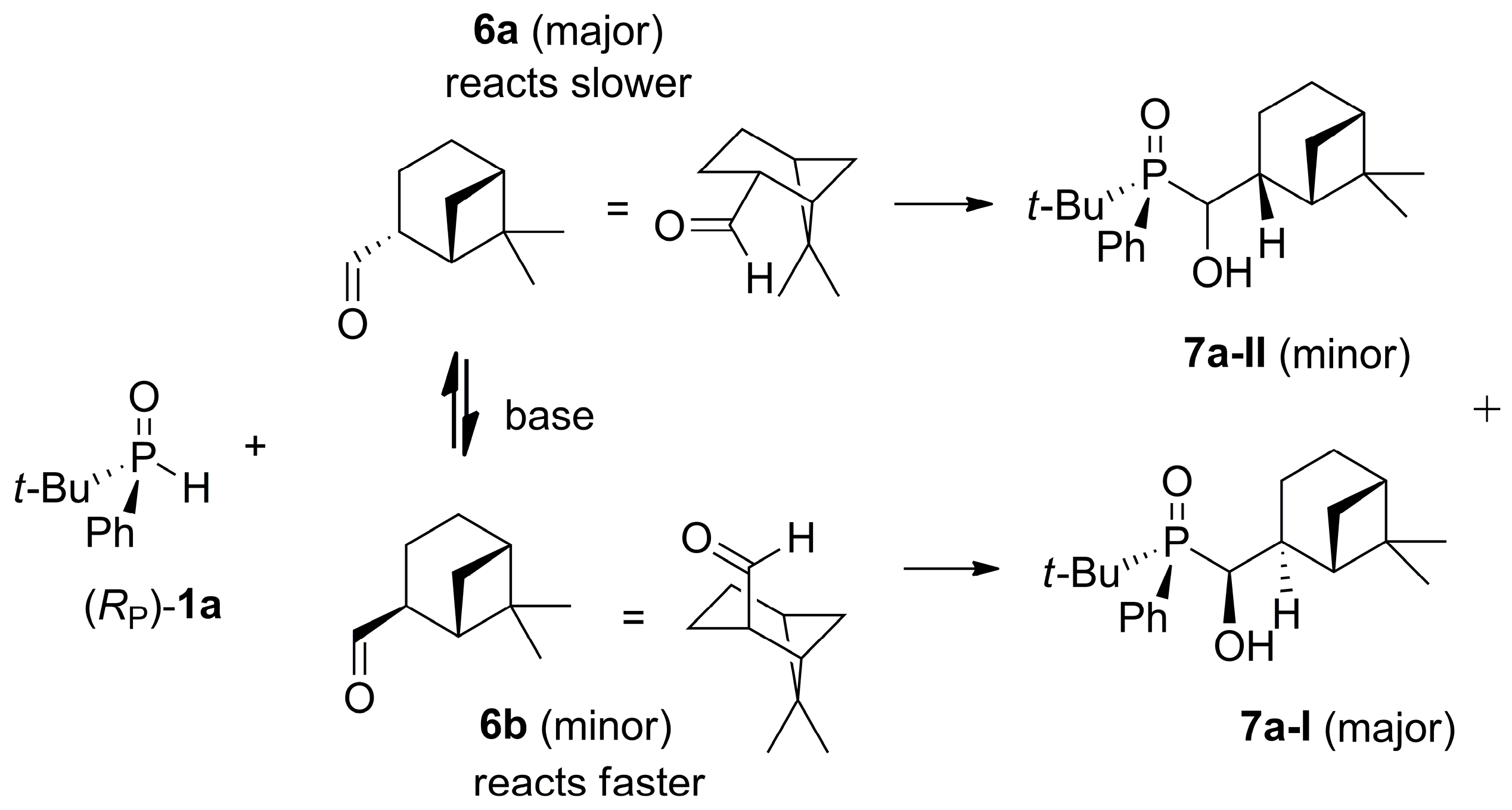
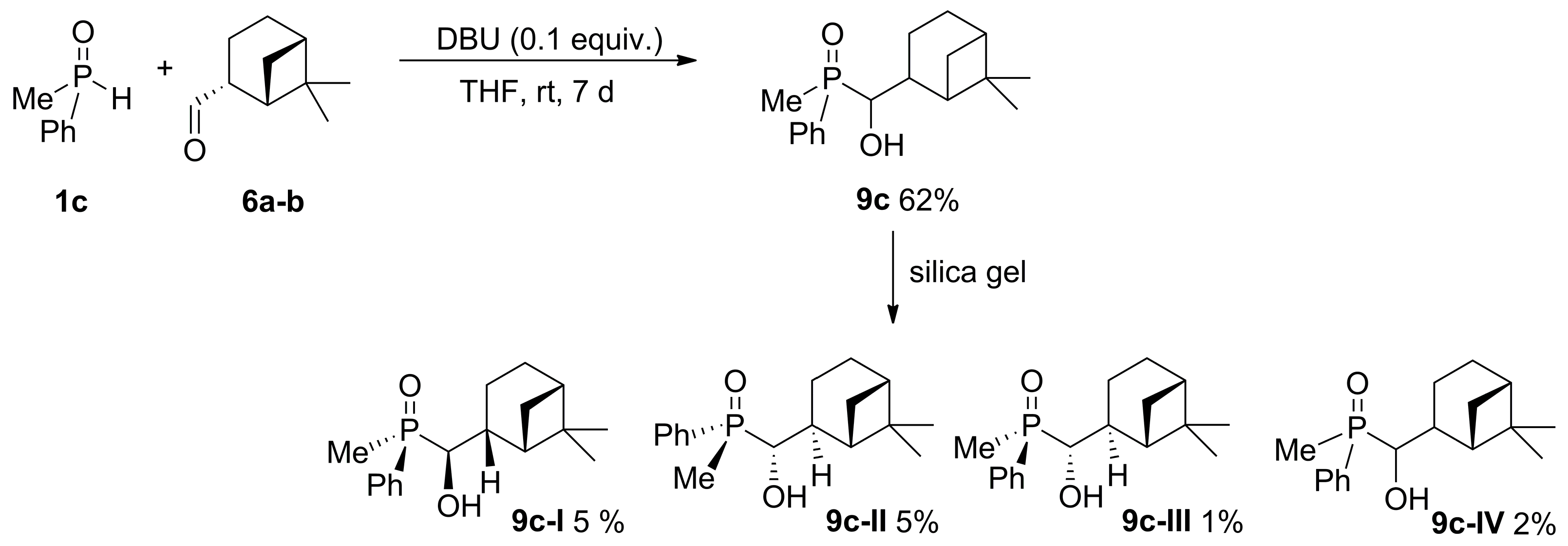
2.2. 1,2-Addition of Secondary Phosphine Oxides to (1R,2R/2S)-Myrtanal
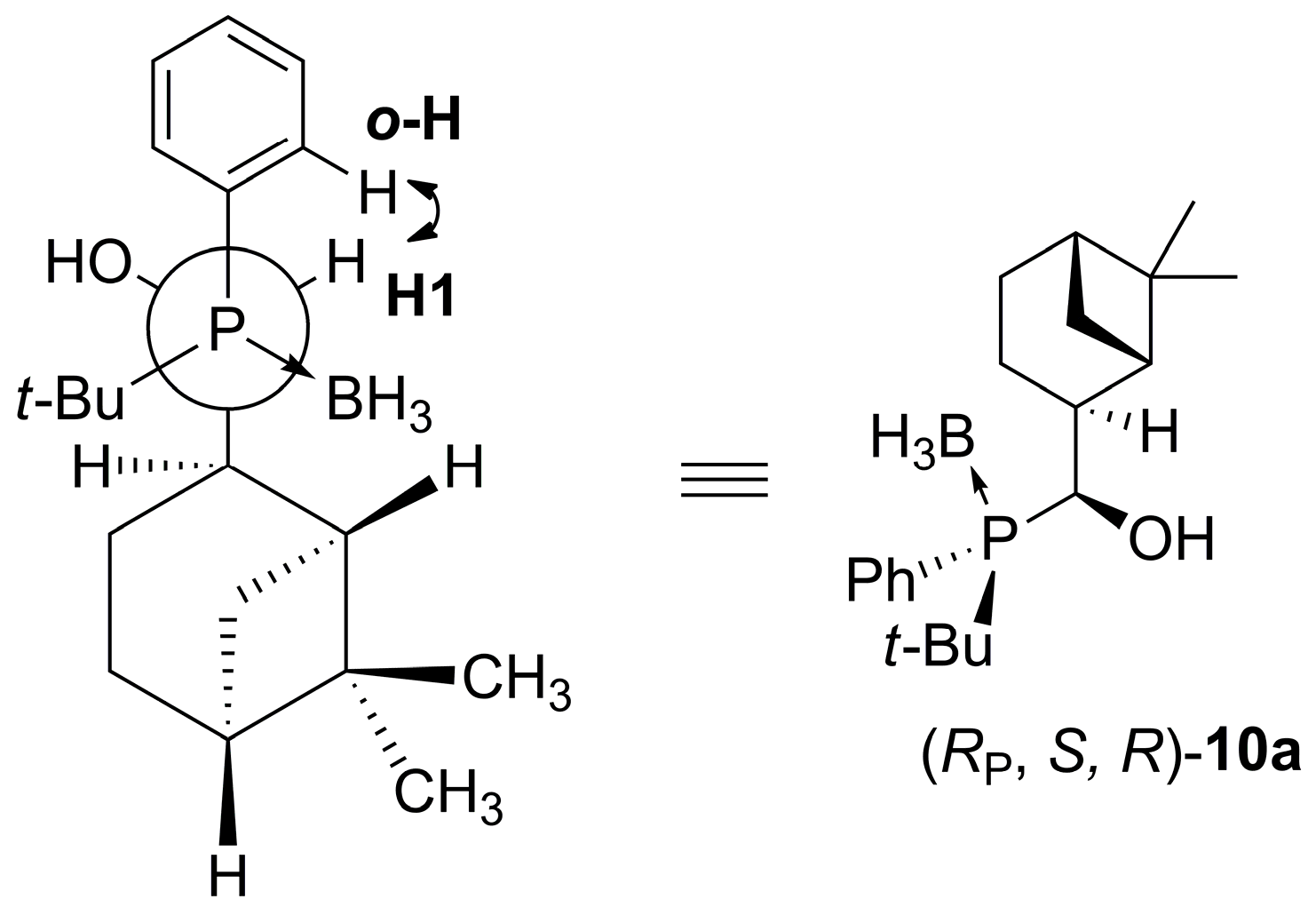
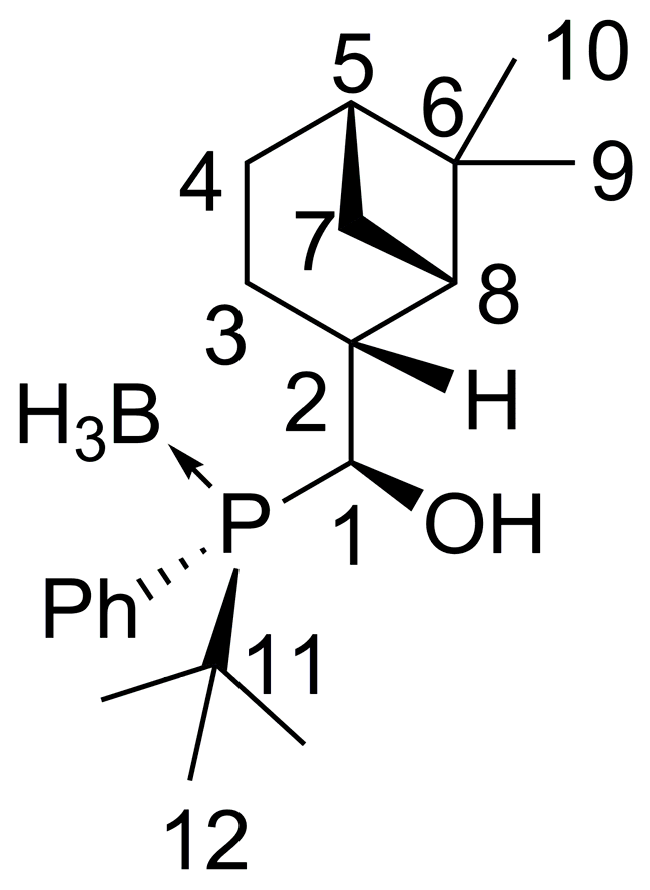
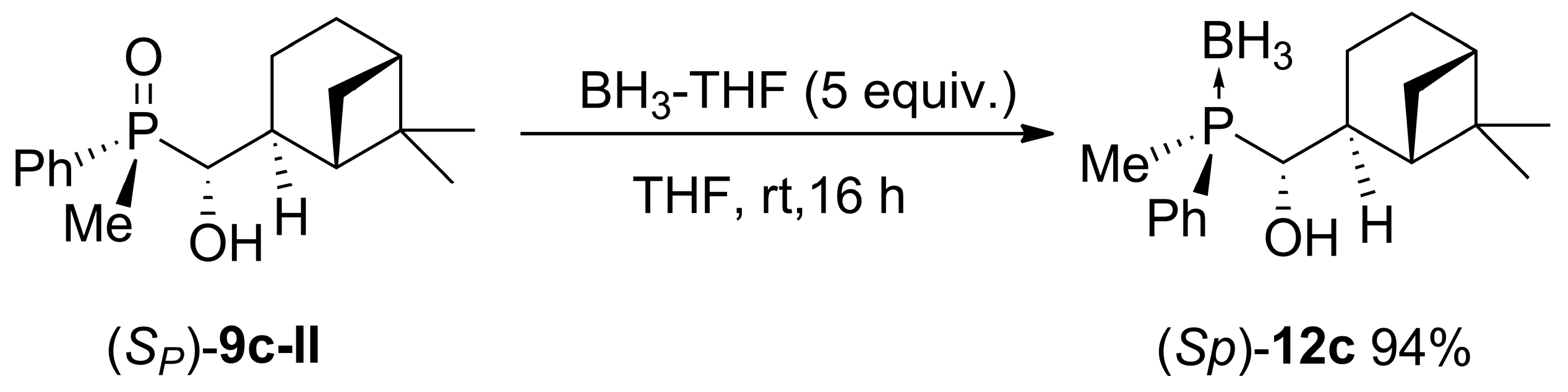
2.3. Synthesis of P-Stereogenic α-Hydroxyphosphine-Boranes
3. Materials and Methods
3.1. General
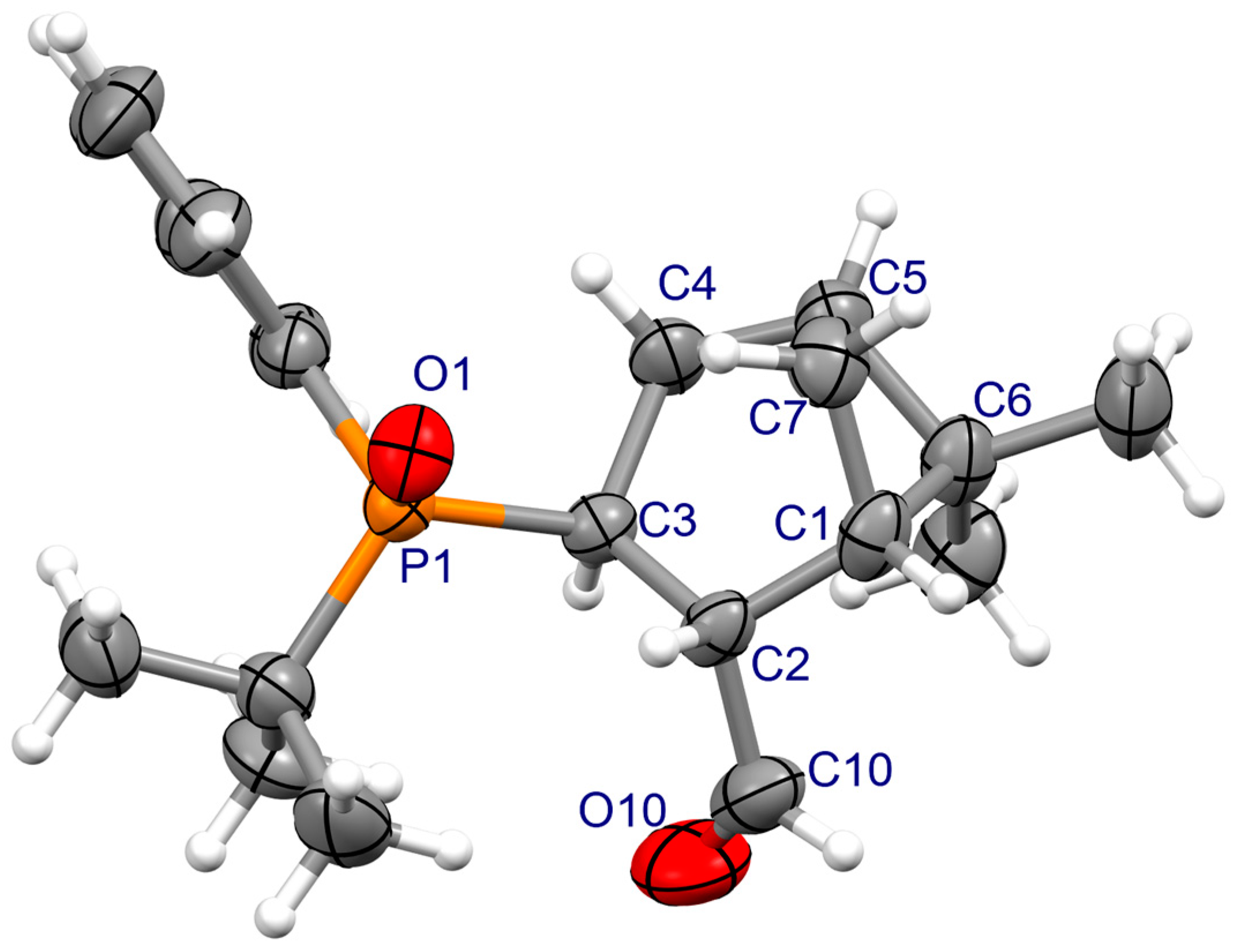
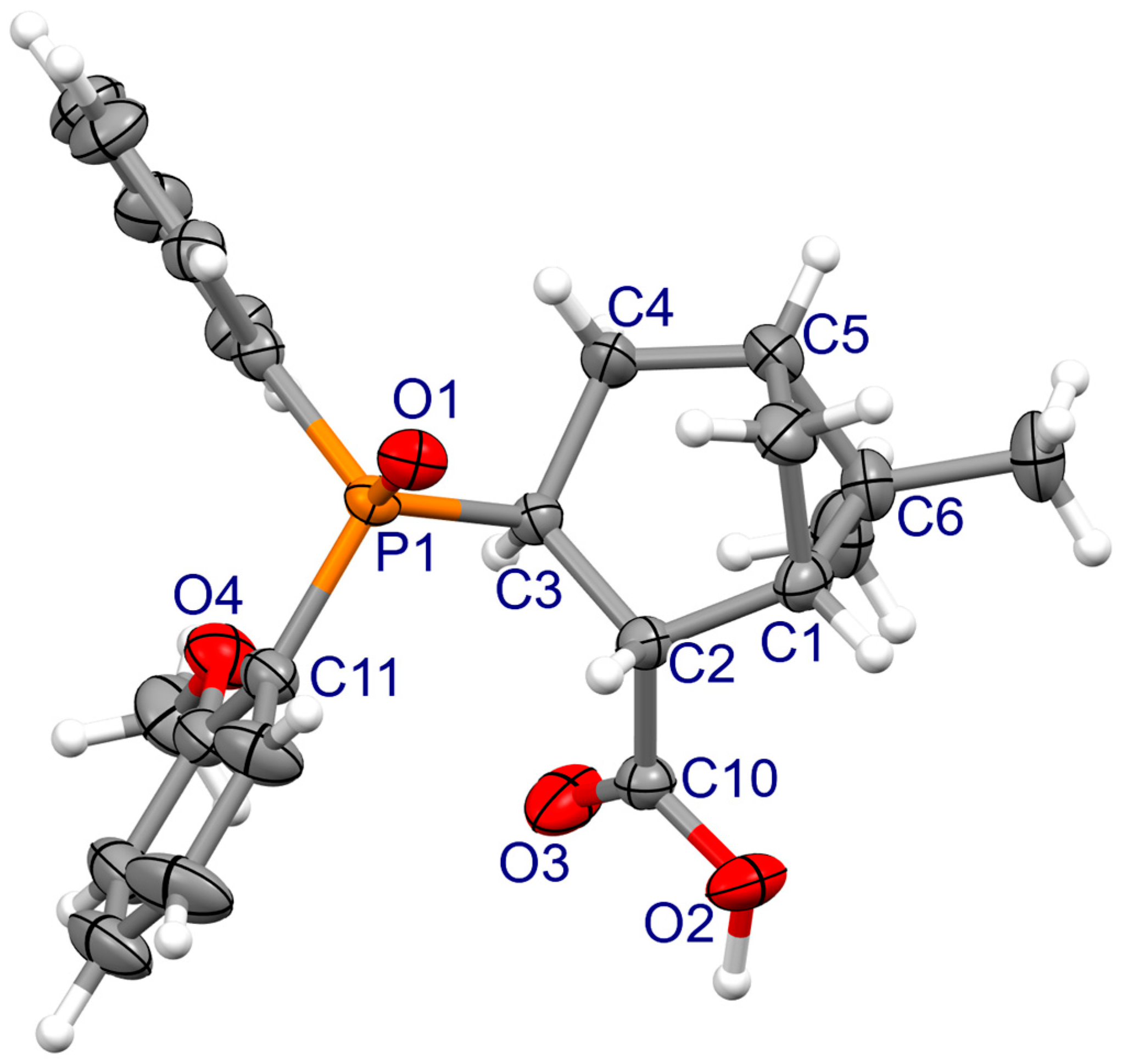
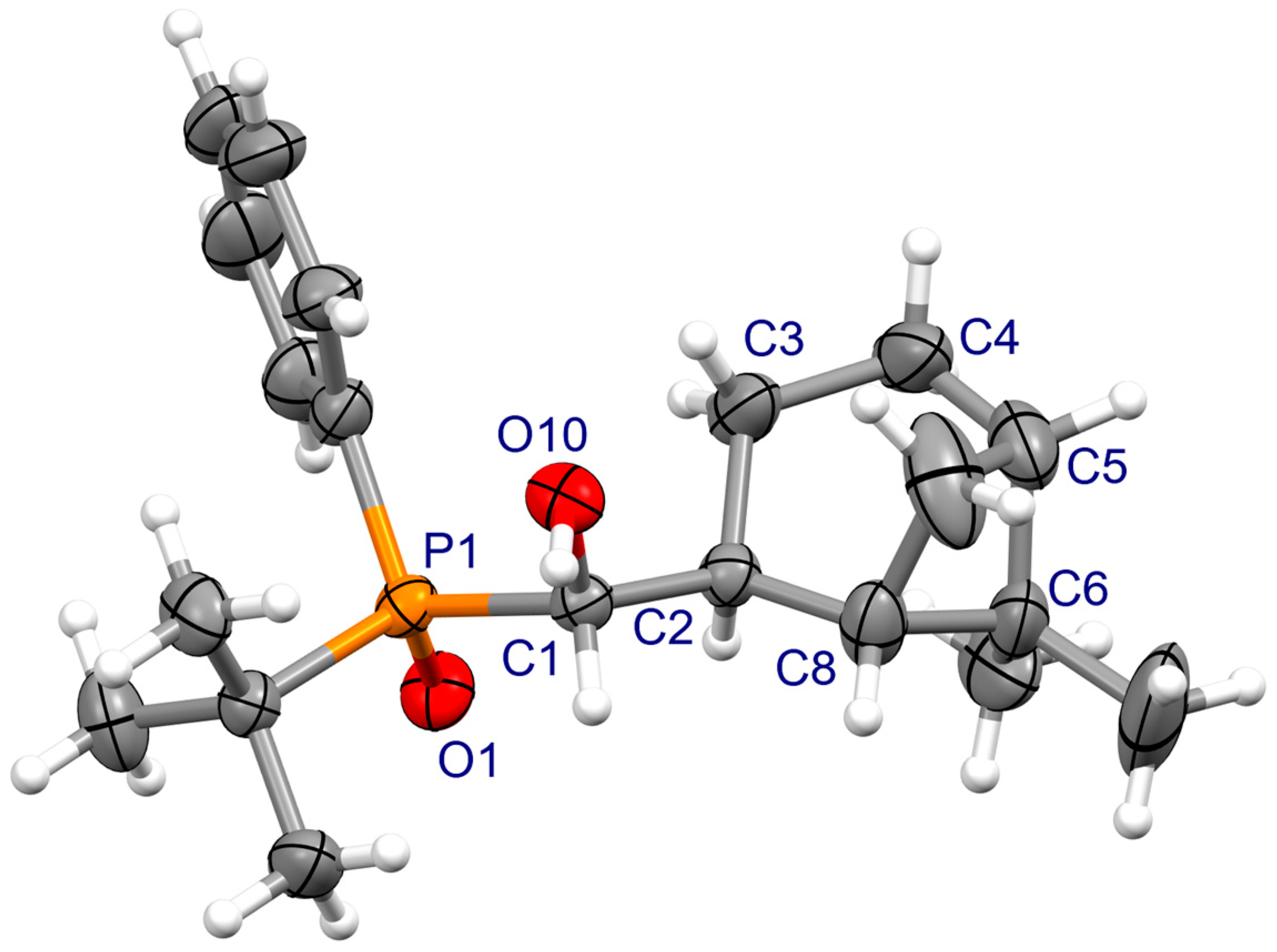
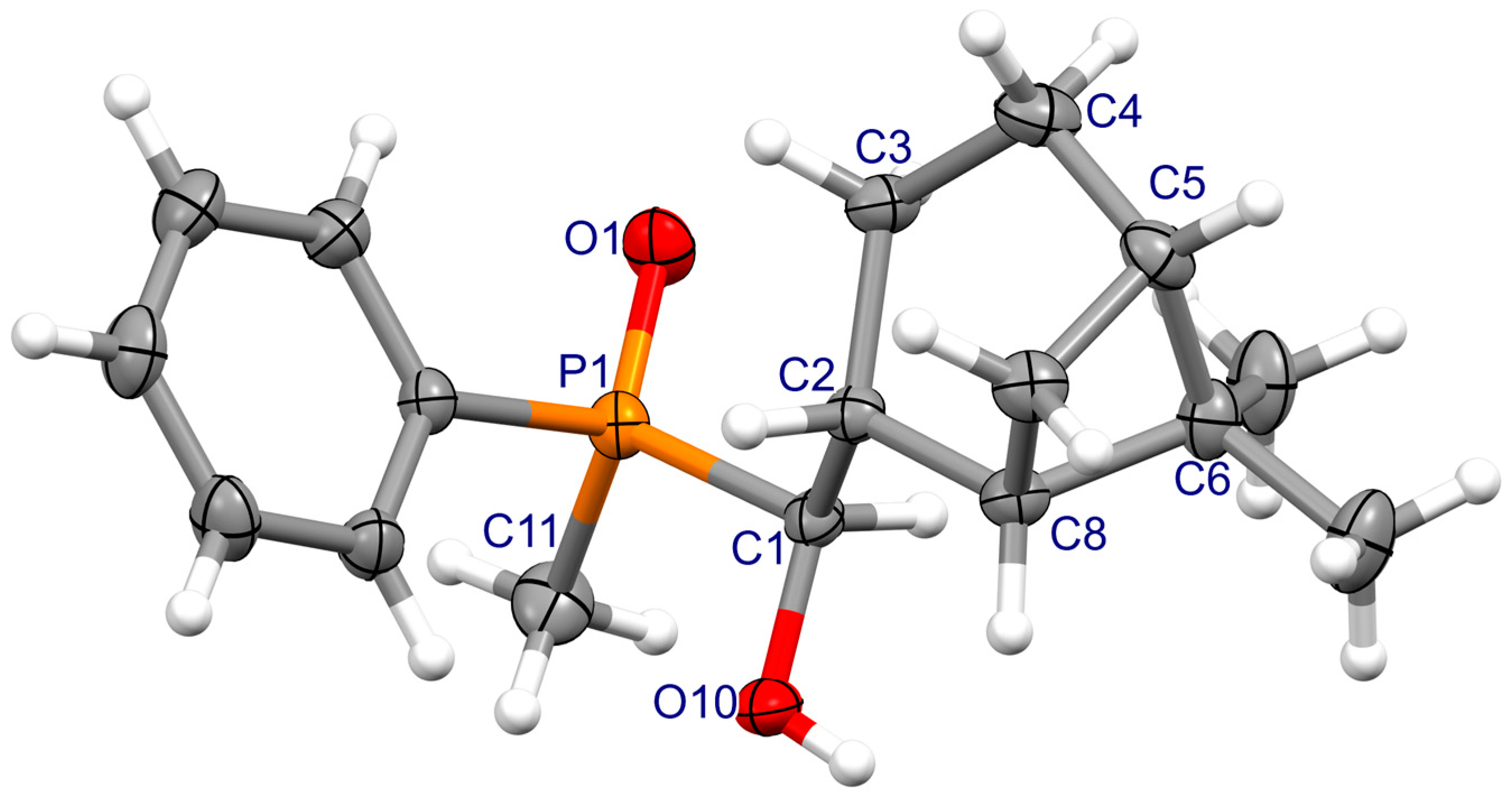
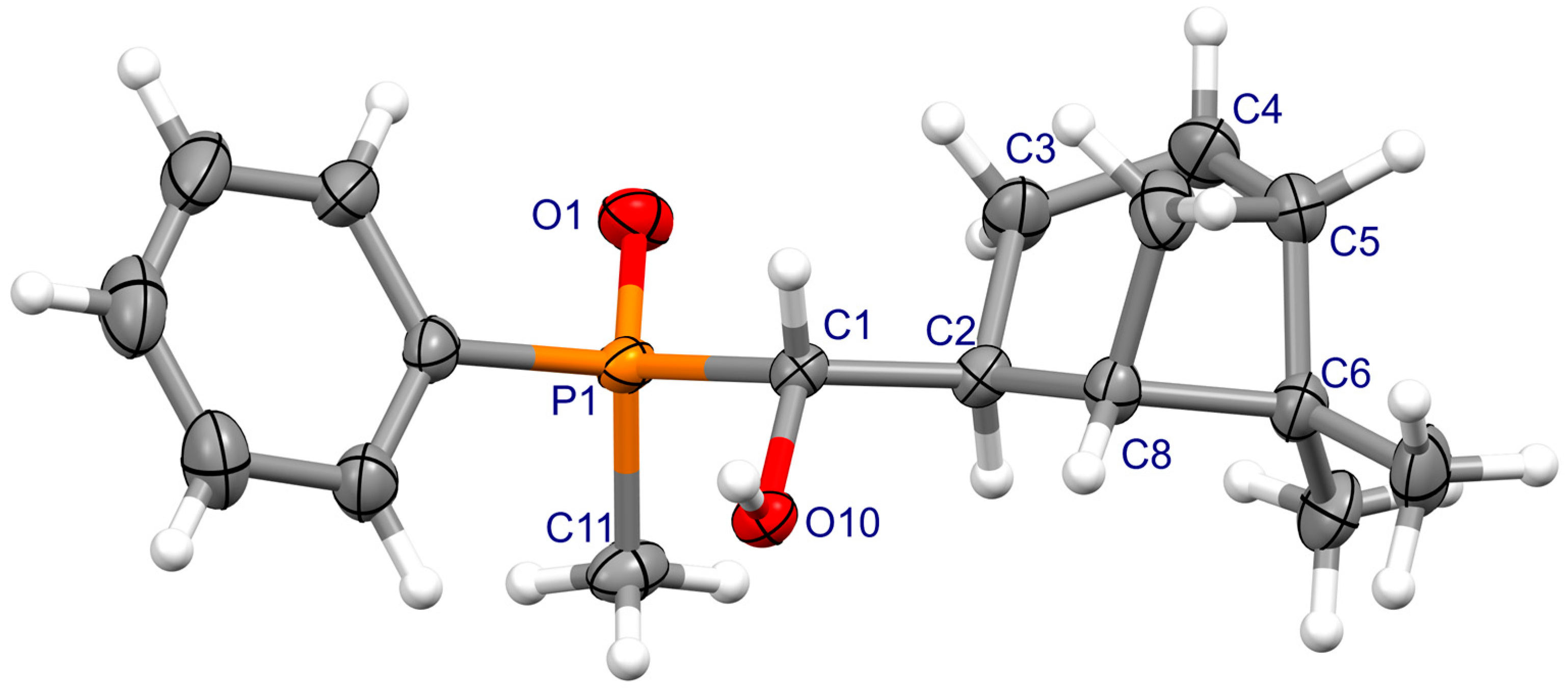
3.2. X-ray Crystallography
4. Experimental
4.1. General Procedure of the Reaction of Phosphine Oxides with (R)-Myrtenal
4.2. Procedure of Reduction of (R)-Myrtenal (2) to Myrtanal (6) [25]
4.3. Procedure of the Synthesis of [6,6-dimethylbicyclo[3.1.1]heptan-2-yl)(hydroxy)methyl](t-butyl)(phenyl)phosphine Oxide (7a) Using n-BuLi
4.4. Procedure of the Synthesis of 6,6-dimethylbicyclo[3.1.1]heptan-2-yl)(hydroxy)methyl)(t-butyl)(phenyl)phosphine Oxide (7a) from (RP)-1a and Myrtanal (6) Using DBU as a Base
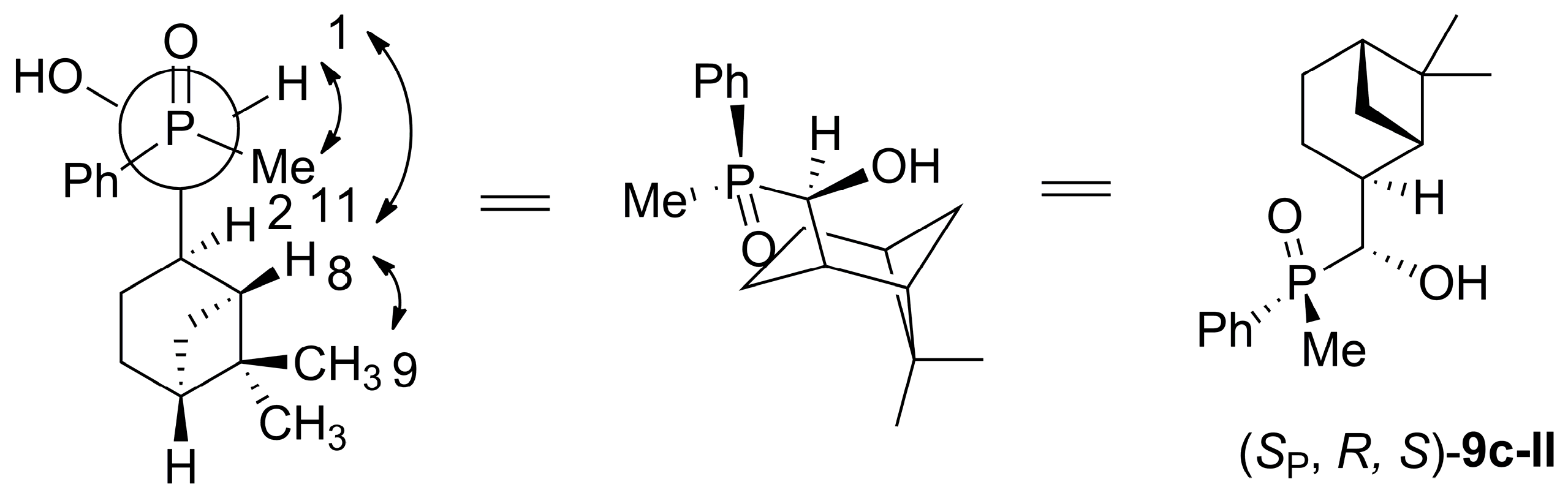
4.5. General Procedure of the Synthesis of [6,6-dimethylbicyclo[3.1.1]heptan-2-yl)(hydroxy)methyl](methyl)(phenyl)phosphine Oxide (9c) from rac-1c and Myrtanal (6) Using DBU as a Base
4.6. General Procedure for the Reaction of α-Hydroxyphosphine Oxides with BH3-THF
4.7. Procedure of the Reduction of (3a-I) Using BH3-SMe2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kagan, H.B.; Sasaki, M. Optically Active Phosphines: Preparation, Uses and Chiroptical Properites. In Chemistry of Organophosphorus Compounds; Hartley, F.R., Ed.; Wiley & Sons: New York, NY, USA, 1990; Volume 1, pp. 51–102. [Google Scholar]
- Imamoto, T. Optically Active Phosphorus Compounds. In Handbook of Organophosphorus Chemistry; Engel, R., Ed.; Marcel Dekker: New York, NY, USA, 1992; pp. 1–53. [Google Scholar]
- Sasaki, M. Chirality in Agrochemicals; Kurihara, N., Miyamoto, J., Eds.; Wiley & Sons: Chichester, NH, USA, 1998; pp. 85–139. [Google Scholar]
- Zabłocka, M.; Pietrusiewicz, K.M. Preparation of scalemic P-chiral phosphines and their derivatives. Chem. Rev. 1994, 94, 1375–1411. [Google Scholar]
- Dubrovina, N.V.; Börner, A. Enantioselective catalysis with chiral phosphine oxide preligands. Angew. Chem. Int. Ed. 2004, 43, 5883–5886. [Google Scholar] [CrossRef]
- Dutartre, M.; Bayardon, J.; Juge, S. Applications and stereoselective syntheses of P-chirogenic phosphorus compounds. Chem. Soc. Rev. 2016, 45, 5771–5794. [Google Scholar] [CrossRef]
- Andrushko, V.; Börner, A. Chiral hydroxyl Phosphines. In Phosphorous Ligands in Asymmetric Catalysis. Synthesis and Applications; Börner, A., Ed.; Wiley-VCH: Weinheim, Germany, 2008; Volume 2, pp. 633–714. [Google Scholar]
- Berger, O.; Montchamp, J.-L. A General strategy for the synthesis of P-stereogenic compounds. Angew. Chem. 2013, 125, 11587–11590. [Google Scholar] [CrossRef]
- Lemouzy, S.; Jean, M.; Giordano, L.; Hérault, D.; Buono, G. The hydroxyalkyl moiety as a protecting group for the stereospecific alkylation of masked secondary phosphine-boranes. Org. Lett. 2016, 18, 140–143. [Google Scholar] [CrossRef]
- Lemouzy, S.; Giordano, L.; Hérault, D.; Buono, G. Introducing chirality at phosphorus atoms: An update on the recent synthetic strategies for the preparation of optically pure P-stereogenic molecules. Eur. J. Org. Chem. 2020, 23, 3351–3366. [Google Scholar] [CrossRef]
- Haynes, R.K.; Lain, W.W.-L.; Yeung, L.-L. Stereoselective preparation of functionalized tertiary P-chiral phosphine oxides by nucleophilic addition of lithiated tert-butylphenylphosphine oxide to carbonyl compounds. Tetrahedron Lett. 1996, 37, 4729–4732. [Google Scholar] [CrossRef]
- Drabowicz, J.; Łyżwa, P.; Omelańczuk, J.; Pietrusiewicz, K.M.; Mikołajczyk, M. New procedures for the resolution of chiral tert-butylphenylphosphine oxide and some of its reactions. Tetrahedron Asymmetry 1999, 10, 2757–2763. [Google Scholar] [CrossRef]
- Leyris, A.; Bigeault, J.; Nuel, D.; Giordano, L.; Buono, G. Enantioselective synthesis of secondary phosphine oxides from (RP)-(-)-Menthyl Hydrogenophenylphosphinate. Tetrahedron Lett. 2007, 48, 5247–5250. [Google Scholar] [CrossRef]
- Kortmann, F.A.; Chang, M.-C.; Otten, E.; Couzijn, E.P.A.; Lutz, M.; Minnaard, A.J. Consecutive dynamic resolutions of phosphine oxides. Chem. Sci. 2014, 5, 1322–1327. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, C.-Q.; Han, L.-B. Stereospecific nucleophilic substitution of optically pure H-phosphinates: A general way for the preparation of chiral P-stereogenic phosphine oxides. J. Am. Chem. Soc. 2008, 130, 12648–12655. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.; Maj, A.M.; Schudde, E.P.; Pietrusiewicz, K.M.; Sieron, L.; Wieczorek, W.; Jerphagnon, T.; Arends, I.W.C.E.; Hanefeld, U.; Minnaard, A.J. On the resolution of secondary phosphine oxides via diastereomeric complex formation: The case of tert-butylphenylphosphine oxide. Synthesis 2009, 2009, 2061–2065. [Google Scholar] [CrossRef]
- Gatineau, D.; Nguyen, D.H.; Hérault, D.; Vanthuyne, N.; Leclaire, J.; Giordano, L.; Buono, G. H-Adamantylphosphinates as universal precursors of P-stereogenic compounds. J. Org. Chem. 2015, 80, 4132–4141. [Google Scholar] [CrossRef]
- Varga, B.; Szemesi, P.; Nagy, P.; Herbay, R.; Holczbauer, T.; Fogassy, E.; Keglevich, G.; Bagi, P. Enantioseparation of P-stereogenic secondary phosphine oxides and their stereospecific transformation to various tertiary phosphine oxides and a thiophosphinate. J. Org. Chem. 2021, 86, 14493–14507. [Google Scholar]
- Zhang, H.; Sun, Y.-M.; Zhao, Y.; Zhou, Z.-Y.; Wang, J.-P.; Xin, N.; Nie, S.-Z.; Zhao, C.-Q.; Han, L.-B. One-pot process that efficiently generates single stereoisomers of 1,3-bisphosphinylpropanes having five chiral centers. Org. Lett. 2015, 17, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Valentine, D., Jr.; Blount, J.F.; Toth, K. Synthesis of phosphines having chiral organic groups ligated to chiral phosphorus. J. Org. Chem. 1980, 45, 3691–3698. [Google Scholar] [CrossRef]
- Bodalski, R.; Rutkowska-Olma, E.; Pietrusiewicz, K.M. Optically active phosphine oxides. Synthesis and absolute configuration of enantiomeric phenylvinylcarbomenthoxymethylphosphine oxide. Tetrahedron 1980, 36, 2353–2355. [Google Scholar] [CrossRef]
- Bogdanović, B.; Henc, B.; Loesler, A.; Meister, B.; Pauling, H.; Wilke, G. Asymmetrische Synthesen mit homogenen Übergangsmetallkatalysatoren. Angew. Chem. 1973, 23, 1013–1023. [Google Scholar] [CrossRef]
- Corey, E.J.; Chen, Z.; Tanoury, G.J. A new and highly enantioselective synthetic route to P-chiral phosphines and diphosphines. J. Am. Chem. Soc. 1993, 115, 11000–11001. [Google Scholar] [CrossRef]
- Martinez-Ramos, F.; Vargas-Diaz, M.E.; Chacon-Garcia, L.; Tamariz, J.; Joseph-Nathan, P. Zepeda Highly diastereoselective nucleophilic additions using a novel myrtenal-derived oxathiane as a chiral auxiliary. Tetrahedron Asymmetry 2001, 12, 3095–3103. [Google Scholar] [CrossRef]
- el Gaied, M.M.; Bessiere-Chretien, Y. Réduction par le lithium dessous dans l’ammoniac liquide de cétones cyclopropaniques dérivées de terpènes. Bull. Soc. Chim. Fr. 1973, 4, 1351–1356. [Google Scholar]
- Sowa, S.; Stankevič, M.; Szmigielska, A.; Małuszyńska, H.; Kozioł, A.E.; Pietrusiewicz, K.M. Reduction of functionalized tertiary phosphine oxides with BH3. J. Org. Chem. 2015, 80, 1672–1688. [Google Scholar] [CrossRef] [PubMed]
- Lemouzy, S.; Nguyen, D.H.; Camy, V.; Jean, M.; Gatineau, D.; Giordano, L.; Naubron, J.-V.; Vanthuyne, N.; Hérault, D.; Buono, G. Stereospecific synthesis of α- and β-hydroxyalkyl P-stereogenic phosphine–boranes and functionalized derivatives: Evidence of the P=O activation in the BH3-mediated reduction. Chem. Eur. J. 2015, 21, 15607–15621. [Google Scholar] [CrossRef] [PubMed]
- Sowa, S.; Stankevič, M.; Flis, A.; Pietrusiewicz, K.M. Reduction of tertiary phosphine oxides by BH3 assisted by neighboring activating groups. Synthesis 2018, 50, 2106–2118. [Google Scholar]
- Sowa, S.; Pietrusiewicz, K.M. Chemoselective reduction of the P=O bond in the presence of P–O and P–N bonds in phosphonate and phosphinate derivatives. Eur. J. Org. Chem. 2019, 2019, 923–928. [Google Scholar] [CrossRef]
- Stankevič, M.; Pietrusiewicz, K.M. Resolution and stereochemistry of t-butylphenylphosphinous acid−borane. J. Org. Chem. 2007, 72, 816–822. [Google Scholar] [CrossRef]
- Stankevič, M.; Pietrusiewicz, K.M. An expedient reduction of sec-phosphine oxides to sec-phosphine-boranes by BH3-SMe2. Synlett 2003, 7, 1012–1016. [Google Scholar]
- Pietrusiewicz, K.M.; Stankevič, M. Phosphinous acid-boranes. Curr. Org. Chem. 2005, 9, 1883–1897. [Google Scholar] [CrossRef]
- CrysAlisPro, Version 1.171.36.20 (Release 27–06–2012); Agilent Technologies Poland: Warsaw, Poland, 2012.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Wife, R.L.; van Oort, A.B.; van Doorn, J.A.; van Leeuwen, P.W.N.M. Phosphine oxide anions in the synthesis of phosphine ligands. Synthesis 1983, 1983, 71–73. [Google Scholar] [CrossRef]
- Stankevic, M.; Wlodarczyk, A. Efficient copper(I)-catalyzed coupling of secondary phosphine oxides with aryl halides. Tetrahedron 2013, 69, 73–81. [Google Scholar] [CrossRef]
- Pommerening, P.; Oestreich, M. Chiral modification of the tetrakis(pentafluorophenyl)borate anion with myrtanyl groups. Eur. J. Org. Chem. 2019, 2019, 7240–7246. [Google Scholar] [CrossRef]
- Hoover, J.M.; Stahl, S.S. Highly practical Copper(I)/TEMPO catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Kobori, T.; Fujisawa, T. Prostaglandin synthesis from a fulvene with the ω-side chain equivalent. Tetrahedron Lett. 1981, 22, 115–118. [Google Scholar] [CrossRef]
- Lee, A.S.-Y.; Cheng, C.-L. A Novel and selective method for hydrolysis of acetals and ketals. Tetrahedron 1997, 53, 14255–14262. [Google Scholar] [CrossRef]























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrusiewicz, K.M.; Kozioł, A.E.; Małuszyńska, H.; Sowa, S. Myrtenal and Myrtanal as Auxiliaries in the Synthesis of Some C,P-Stereogenic Hydroxyphosphine Oxides and Hydroxyphosphine-Boranes Possessing up to Four Contiguous Centers of Chirality. Symmetry 2023, 15, 1172. https://doi.org/10.3390/sym15061172
Pietrusiewicz KM, Kozioł AE, Małuszyńska H, Sowa S. Myrtenal and Myrtanal as Auxiliaries in the Synthesis of Some C,P-Stereogenic Hydroxyphosphine Oxides and Hydroxyphosphine-Boranes Possessing up to Four Contiguous Centers of Chirality. Symmetry. 2023; 15(6):1172. https://doi.org/10.3390/sym15061172
Chicago/Turabian StylePietrusiewicz, K. Michał, Anna E. Kozioł, Hanna Małuszyńska, and Sylwia Sowa. 2023. "Myrtenal and Myrtanal as Auxiliaries in the Synthesis of Some C,P-Stereogenic Hydroxyphosphine Oxides and Hydroxyphosphine-Boranes Possessing up to Four Contiguous Centers of Chirality" Symmetry 15, no. 6: 1172. https://doi.org/10.3390/sym15061172