
Determination of 19 Psychoactive Substances in Premortem and Postmortem Whole Blood Samples Using Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. Stock Standard Solutions
2.3. Working Standard Solutions and Calibration Whole Blank Samples
2.4. Sample Preparation Procedure
2.5. UHPLC-ESI-MS/MS
2.6. Method Validation
2.7. Analysis of Real Samples
3. Results and Discussion
3.1. Method Development
3.1.1. Mass Spectrometry
3.1.2. Chromatography
3.1.3. Optimization of the Sample Preparation Procedure
3.2. Statistical Analysis of Data
3.2.1. Limits of Detection (LODs) and Limits of Quantitation (LOQs)
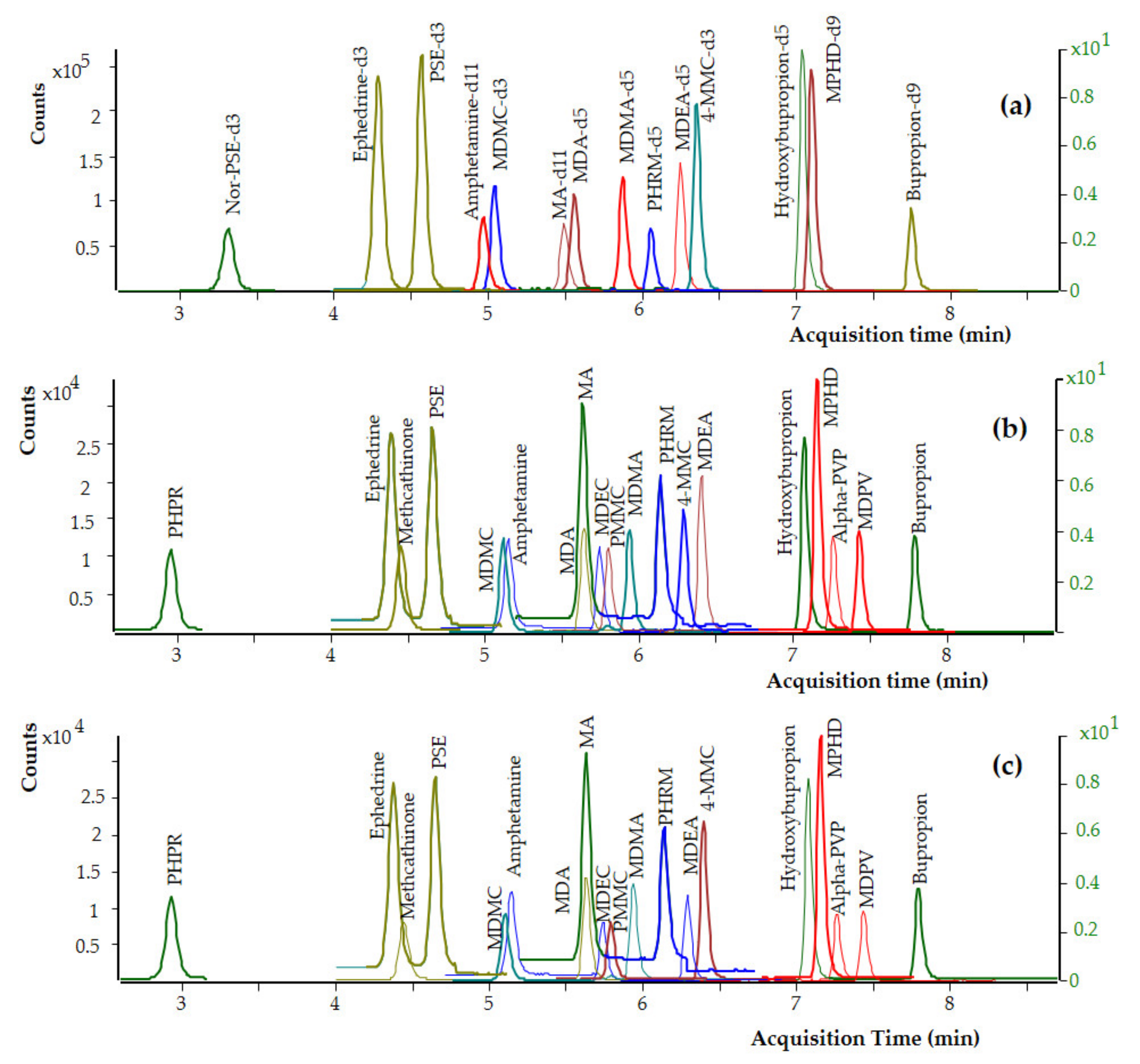
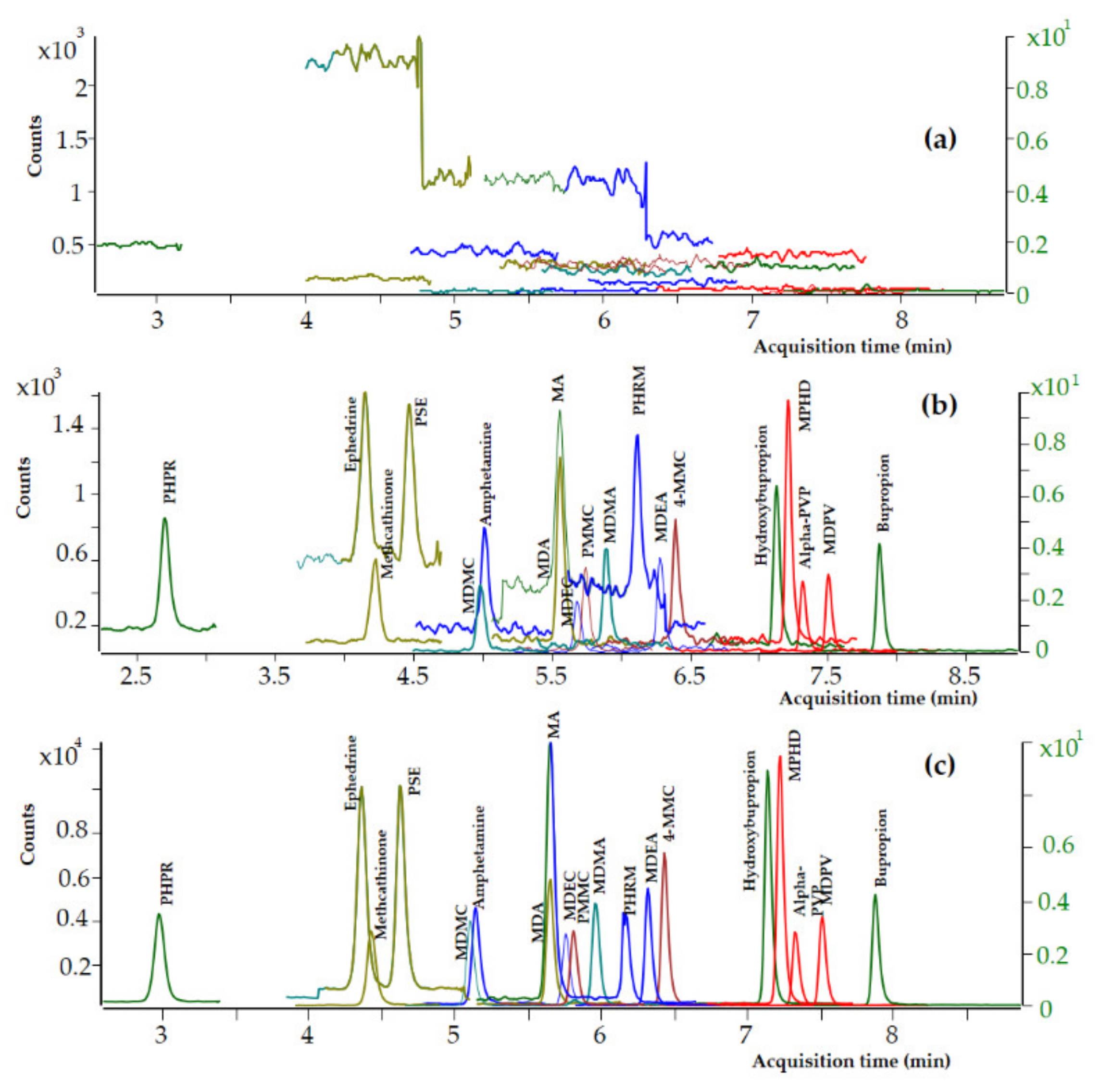
3.2.2. Selectivity and Specificity
3.2.3. Calibration Model, Precision, and Accuracy
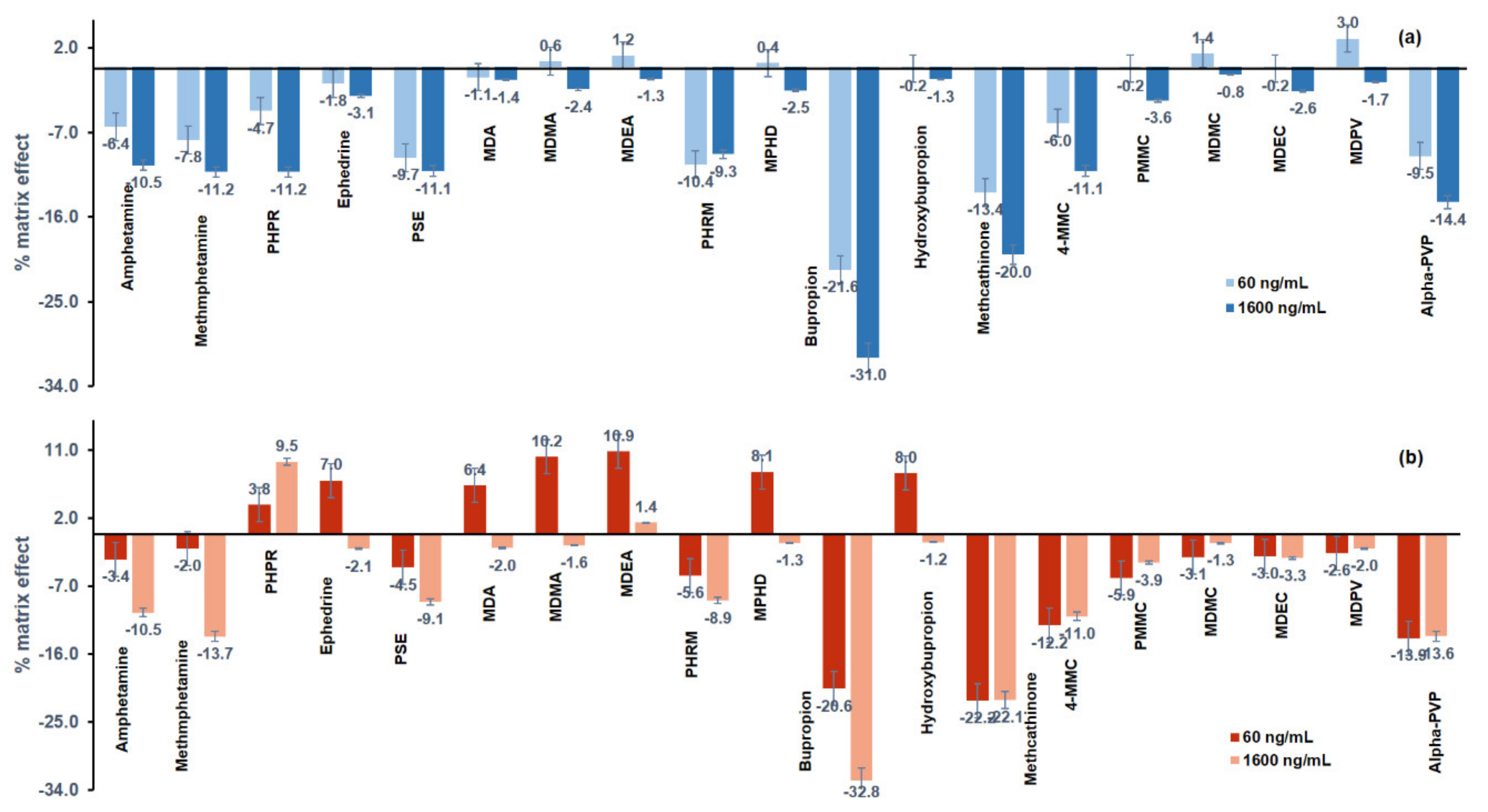
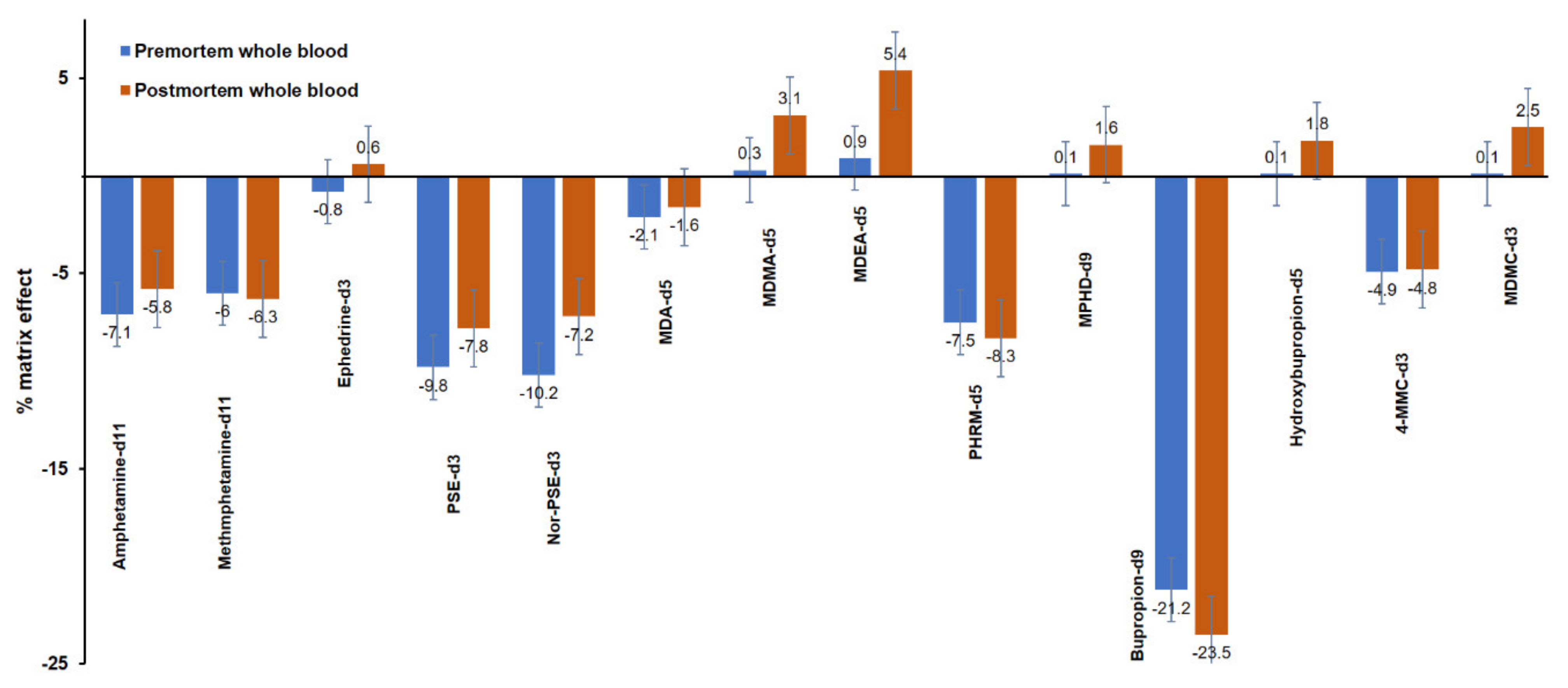
3.2.4. Matrix Effect and Extraction Efficiency
3.3. Applicability of the Method to the Analysis of Real Cases
3.4. Comparison with Other LC-MS/MS Analytical Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vandrey, R.; Johnson, M.V.; Johnson, P.S.; Khali, M.A. Novel Drugs of Abuse: A Snapshot of an Evolving Marketplace. Adolesc. Psychiatry 2013, 3, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Calpe-López, C.; García-Pardo, M.P.; Aguilar, M.A. Cannabidiol Treatment Might Promote Resilience to Cocaine and Methamphetamine Use Disorders: A Review of Possible Mechanisms. Molecules 2019, 24, 2583. [Google Scholar] [CrossRef] [Green Version]
- Drasch, G.; Dahlmann, F.; von Meyer, L.; Roider, G.; Eisenmenger, W. Frequency of different anti-depressants associated with suicides and drug deaths. Int. J. Legal Med. 2008, 122, 115–121. [Google Scholar] [CrossRef]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2019: Trends and Developments; Publications Office of the European Union: Luxemburg, 2020; Available online: https://www.emcdda.europa.eu/ (accessed on 7 June 2020).
- Langman, L.J.; Snozek, C.L.H. Introduction to Drugs of Abuse. In Critical Issues in Alcohol and Drugs of Abuse Testing, 2nd ed.; Mercolini, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 6, pp. 71–78. [Google Scholar]
- Ellefsen, K.N.; Concheiro, M.; Huestis, M.A. Synthetic cathinone pharmacokinetics, analytical methods, and toxicological findings from human performance and postmortem cases. Drug Metab. Rev. 2016, 48, 237–265. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. World Drug Report 2015; United Nations Publications: New York, NY, USA, 2015. [Google Scholar]
- Fineschi, M.; Masti, A. Fatal poisoning by MDMA (ecstasy) and MDEA: A case report. Int. J. Legal Med. 1996, 108, 272–275. [Google Scholar] [CrossRef]
- Milroy, C.M. “Ecstasy” associated deaths: What is a fatal concentration? Analysis of a case series. Forensic Sci. Med. Pathol. 2011, 7, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Wyman, J.F.; Lavins, E.S.; Engelhart, D.; Armstrong, E.J.; Snell, K.D.; Boggs, P.D.; Taylor, S.M.; Norris, R.N.; Miller, F.P. Postmortem Tissue Distribution of MDPV Following Lethal Intoxication by “Bath Salts”. J. Anal. Toxicol. 2013, 37, 182–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Chronister, C.W.; Hoyer, J.; Goldberger, B.A. Ethylone-Related Deaths: Toxicological Findings. J. Anal. Toxicol. 2015, 39, 567–571. [Google Scholar] [CrossRef] [Green Version]
- National Institute on Drug Abuse. Advancing Addiction Science, Center for Health Statistics, Overdose Death Rates. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 25 May 2021).
- Kraemer, T.; Paul, L. Bioanalytical procedures for determination of drugs of abuse in blood. Anal. Bioanal. Chem. 2004, 388, 1415–1435. [Google Scholar] [CrossRef]
- Meyer, M.R. New psychoactive substances: An overview on recent publications on their toxicodynamics and toxicokinetics. Arch. Toxicol. 2016, 90, 2421–2444. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.T.; Wissenbach, D.K.; Busardo, F.P.; Marchei, E.; Pichini, S. Method development in forensic toxicology. Curr. Pharm. Des. 2018, 23, 5455–5467. [Google Scholar] [CrossRef]
- Nieddu, M.; Burrai, L.; Baralla, E.; Pasciu, V.; Varoni, M.V.; Briguglio, I.; Demontis, M.P.; Boatto, G. ELISA detection of 30 new amphetamine designer drugs in whole blood, urine and oral fluid using Neogen® “amphetamine” and “methamphetamine/MDMA” kits. J. Anal. Toxicol. 2016, 40, 492–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roda, E.; Buscaglia, E.; Papa, P.; Rocchi, L.; Locatelli, C.A.; Coccini, T. Evaluation of two different screening ELISA assays for synthetic cathinones (mephedrone/methcathinone and MDPV) with LC-MS method in intoxicated patients. J. Clin. Toxicol. 2016, 6. [Google Scholar] [CrossRef]
- Marquet, P.; Lacassie, E.; Battu, C.; Faubert, H.; Lachâtre, G. Simultaneous determination of amphetamine and its analogs in human whole blood by gas chromatography–mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1997, 24, 77–82. [Google Scholar] [CrossRef]
- Pelição, F.S.; Peres, M.D.; Pissinate, J.F.; Martinis, B.S. A one-step extraction procedure for the screening of cocaine, amphetamines and cannabinoids in postmortem blood samples. J. Anal. Toxicol. 2014, 38, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercieca, G.; Odoardi, S.; Cassar, M.; Rossi, S.S. Rapid and simple procedure for the determination of cathinones, amphetamine-like stimulants and other new psychoactive substances in blood and urine by GC-MS. J. Pharm. Biomed. Anal. 2018, 149, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.K.; Banaszkiewicz, L.; Wiergowski, M.; Tomczak, E.; Kata, M.; Szpiech, B.; Namieśnik, J.; Biziuk, M. Development and validation of a GC-MS/MS method for the determination of 11 amphetamines and 34 synthetic cathinones in whole blood. Forensic Toxicol. 2020, 38, 42–58. [Google Scholar] [CrossRef] [Green Version]
- Alexandridou, A.; Mouskeftara, T.; Raikos, N.; Gika, H.G. GC-MS analysis of underivatised new psychoactive substances in whole blood and urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020. [Google Scholar] [CrossRef]
- Maurer, H.H. Advances in analytical toxicology: The current role of liquid chromatography–mass spectrometry in drug quantification in blood and oral fluid. Anal. Bioanal. Chem. 2005, 381, 110–118. [Google Scholar] [CrossRef]
- Moretti, M.; Freni, F.; Valentini, B.; Vignali, C.; Groppi, A.; Visonà, S.D.; Osculati, A.M.M.; Morini, L. Determination of Antidepressants and Antipsychotics in Dried Blood Spots (DBSs) Collected from Post-Mortem Samples and Evaluation of the Stability over a Three-Month Period. Molecules 2019, 24, 3636. [Google Scholar] [CrossRef] [Green Version]
- Øiestad, E.L.; Johansen, U.; Øiestad, A.M.L.; Christophersen, A.S. Drug screening of whole blood by ultra-performance liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2011, 35, 280–293. [Google Scholar] [CrossRef]
- Wang, C.C.; Hartmann-Fischbach, P.; Krueger, T.R.; Lester, A.; Simonson, A.; Wells, T.L.; Wolk, M.O.; Hidlay, N.J. Fast and Sensitive Chiral Analysis of Amphetamines and Cathinones in Equine Urine and Plasma Using Liquid Chromatography Tandem Mass Spectrometry. Am. J. Anal. Chem. 2015, 6, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Bjørk, M.K.; Nielsen, M.K.K.; Markussen, L.Ø.; Klinke, H.B.; Linnet, K. Determination of 19 drugs of abuse and metabolites in whole blood by high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 2393–2401. [Google Scholar] [CrossRef]
- Adamowicz, P.; Tokarczyk, B. Simple and rapid screening procedure for 143 new psychoactive substances by liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2016, 8, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Castaneto, M.; Kronstrand, R.; Huestis, M.A. Simultaneous determination of 40 novel psychoactive stimulants in urine by liquid chromatography-high resolution mass spectrometry and library matching. J. Chromatogr. A 2015, 1397, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, L.K. Determination of cathinones and related ephedrines in forensic whole-blood samples by liquid-chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Middleberg, R.A.; Homan, J. Quantitation of amphetaminetype stimulants by LC-MS/MS. Methods Mol. Biol. 2012, 902, 105–114. [Google Scholar]
- Montesano, C.; Vannutelli, G.; Gregori, A.; Ripani, L.; Compagnone, D.; Curini, R.; Sergi, M. Broad screening and identification of novel psychoactive substances in plasma by high-performance liquid chromatography-high-resolution mass spectrometry and post-run library matching. J. Anal. Toxicol. 2016, 40, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Montesano, C.; Vannutelli, G.; Massa, M.; Simeoni, M.C.; Gregori, A.; Ripani, L.; Compagnone, D.; Curini, R.; Sergi, M. Multi-class analysis of new psychoactive substances and metabolites in hair by pressurized liquid extraction coupled to HPLC-HRMS. Drug Test. Anal. 2017, 9, 798–807. [Google Scholar] [CrossRef]
- Stephanson, N.N.; Signell, P.; Helander, A.; Beck, O. Use of LC-HRMS in full scan-XIC mode for multi-analyte urine drug testing—A step towards a ‘black-box’ solution? J. Mass Spectrom. 2017, 52, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Maurer, H.H. Review: LC coupled to low- and high-resolution mass spectrometry for new psychoactive substance screening in biological matrices—where do we stand today? Anal. Chim. Acta 2016, 927, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Anizan, S.; Ellefsen, K.; Huestis, M.A. Simultaneous quantification of 28 synthetic cathinones and metabolites in urine by liquid chromatography-high resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 9437–9448. [Google Scholar] [CrossRef]
- Vaiano, F.; Busardo, F.P.; Palumbo, D.; Kyriakou, C.; Fioravanti, A.; Catalani, V.; Mari, F.; Bertol, E. A novel screening method for 64 new psychoactive substances and 5 amphetamines in blood by LC-MS/MS and application to real cases. J. Pharm. Biomed. Anal. 2016, 129, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Al-Saffar, Y.; Stephanson, N.N.; Beck, O. Multicomponent LC-MS/MS screening method for detection of new psychoactive drugs, legal highs, in urine-experience from the Swedish population. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 930, 112–120. [Google Scholar] [CrossRef]
- Olesti, E.; Pascual, J.A.; Ventura, M.; Papaseit, E.; Farré, M.; Torre, R.; Pozo, O.J. LC-MS/MS method for the quantification of new psychoactive substances and evaluation of their urinary detection in humans for doping control analysis. Drug Test. Anal. 2020, 12, 785–797. [Google Scholar] [CrossRef]
- Maurer, H.H.; Brandt, S.D. New Psychoactive Substances, Pharmacology, Clinical, Forensic and Analytical Toxicology in Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 252, pp. 413–439. [Google Scholar] [CrossRef]
- Levine, B. Principles of Forensic Toxicology, 4th ed.; AACC: Washington, DC, USA, 2013; p. 5. [Google Scholar]
- The International Association of Forensic Toxicologists Committee of Systematic Toxicological Analysis. Recommendations on Sample Collection. TIAFT-Bulletin XXIX, Number 1. Available online: http://www.tiaft.org/data/uploads/documents/tiaft-sta-recommendations-on-sample-collection.pdf (accessed on 27 May 2021).
- The Delaware Code Online. Available online: https://delcode.delaware.gov/title21/c041/sc09/ (accessed on 27 May 2021).
- Ondra, P.; Válka, I.; Knob, R.; Ginterová, G.; Maier, V. Analysis of Amphetamine-Derived Designer Drugs by Gas Chromatography with Mass Spectrometry. J. Anal. Toxicol. 2016, 40, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.H.; Ching, C.K.; Lee, C.Y.; Lam, Y.H.; Mak, T.W. Simultaneous detection of 93 conventional and emerging drugs of abuse and their metabolites in urine by UHPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 969, 272–284. [Google Scholar] [CrossRef]
- Kalovidouris, M.; Michalea, S.; Robola, N.; Koutsopoulou, M.; Panderi, I. Ultra-performance liquid chromatography/tandem mass spectrometry method for the determination of lercanidipine in human plasma. Rapid Commun. Mass Spectrom. 2006, 20, 2939–2946. [Google Scholar] [CrossRef]
- Rathod, R.H.; Chaudhari, S.R.; Patil, A.S.; Shirkhedkar, A.A. Ultra-high-performance liquid chromatography-MS/MS (UHPLC-MS/MS) in practice: Analysis of drugs and pharmaceutical formulations. Future J. Pharm. Sci. 2019, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Scientific Working Group for Forensic Toxicology. Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J. Anal. Toxicol. 2013, 37, 452–474. [Google Scholar] [CrossRef]
- Wille, S.M.R.; Coucke, W.; De Baere, T.; Peters, F.T. Update of Standard Practices for New Method Validation in Forensic Toxicology. Curr. Pharm. Des. 2017, 23, 5442–5454. [Google Scholar] [CrossRef] [PubMed]
- European Comission. Commission Decision No. 2002/657/EC of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretations of Results; EUROPEAN Comission: Brussel, Belgium, 2002; Volume 221, pp. 8–36. [Google Scholar]
- Organizational Scientific Area Committee. Standard for Mass Spectral Data Acceptance in Forensic Toxicology. Available online: https://www.nist.gov/system/files/documents/2019/03/20/standard_for_mass_spec_spectral_data_acceptance_-_asb.pdf (accessed on 27 May 2021).
- Cunha, R.L.; Olivieta, G.S.L.; Oliviera, A.L.; Maldaner, A.O.; Pereira, P.A.P. Fast determination of amphetamine-type stimulants and synthetic cathinones in whole blood samples using protein precipitation and LC-MS/MS. Microchem. J. 2021, 163, 105895. [Google Scholar] [CrossRef]
- Lau, T.; Concheiro, M.; Cooper, G. Determination of 30 synthetic cathinones in postmortem blood using LC–MS-MS. J. Anal. Toxicol. 2020, 44, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Ong, R.S.; Kappatos, D.C.; Russell, S.G.G.; Poulsen, H.A.; Banister, S.D.; Gerona, R.R.; Glass, M.; Johnson, C.S.; McCarthy, M.J. Simultaneous analysis of 29 synthetic cannabinoids and metabolites, amphetamines, and cannabinoids in human whole blood by liquid chromatography-tandem mass spectrometry—A New Zealand perspective of use in 2018. Drug Test. Anal. 2020, 12, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.H.; Li, H.; Liu, Y.; Zhao, M.; Jiang, Y.; Zhao, W.S. Simultaneous determination of 12 illicit drugs in whole blood and urine by solid phase extraction and UPLC-MS/MS. J. Chromatogr. B Biomed. Appl. 2014, 995–996, 10–19. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | tR (min) | Precursor Ion (Q1, m/z) | Product Ion (Q3, m/z) | Collision Energy (V) | Fragmentor (V) |
|---|---|---|---|---|---|
| Amphetamine-d11 | 4.59 | 147.2 | 98.1 | 18 | 65 |
| Methamphetamine-d11 | 5.25 | 161.2 | 97.1 | 18 | 75 |
| Ephedrine-d3 | 3.90 | 169.1 | 151.1 | 10 | 70 |
| Pseudoephedrine-d3 (PSE-d3) | 4.23 | 169.1 | 151.1 | 10 | 75 |
| Norpseudoephedrine-d3 | 3.22 | 155.1 | 137.1 | 6 | 60 |
| 3,4-methylenedioxyamphetamine-d5 (MDA-d5) | 5.32 | 185.1 | 168.1 | 6 | 65 |
| 3,4-methylenedioxy methamphetamine-d5 (MDMA-d5) | 5.67 | 199.2 | 165.1 | 10 | 85 |
| 3,4-methylenedioxy-N-ethylamphetamine-d5 (MDEA-d5) | 6.2 | 213.2 | 163.1 | 10 | 80 |
| Phentermine-d5 (PHRM-d5) | 5.89 | 155.2 | 96.1 | 22 | 65 |
| Methylphenidate-d9 (MPHD-d9) | 7.03 | 243.2 | 93.2 | 20 | 90 |
| Bupropion-d9 | 7.68 | 249.2 | 185 | 10 | 70 |
| Hydroxybupropion-d6 | 6.94 | 262.2 | 244.1 | 6 | 80 |
| Mephedrone-d3 (4-MMC-d3) | 6.09 | 181.1 | 163.1 | 10 | 75 |
| Methylone-d3 (MDMC-d3) | 4.78 | 211.1 | 163.1 | 14 | 75 |
| Compound | tR (min) | Precursor ion (Q1, m/z) | Product ions (Q3, m/z) 1 | Collision Energy (V) | Fragmentor (V) |
|---|---|---|---|---|---|
| Amphetamine (A) | 4.81 | 136.1 | 91.1 | 17 | 70 |
| 119.1 | 6 | ||||
| Methamphetamine (MA) | 5.35 | 150.1 | 91.1 119.1 | 21 6 | 80 |
| Phenylpropanolamine (PHPR) | 2.81 | 152.1 | 134.1 91.1 | 6 34 | 60 |
| Ephedrine | 3.9 | 166.1 | 148.1 91.1 | 10 38 | 75 |
| Pseudoephedrine (PSE) | 4.25 | 166.1 | 148.1 91.1 | 10 34 | 70 |
| 3,4-methylenedioxyamphetamine (MDA) | 5.35 | 180.1 | 163.1 105.1 | 6 22 | 70 |
| 3,4-methylenedioxymethamphetamine (MDMA) | 5.69 | 194.1 | 163.1 105.1 | 10 26 | 80 |
| 3,4-methylenedioxy-N-ethylamphetamine (MDEA) | 6.21 | 208.1 | 163 135 | 10 18 | 90 |
| Phentermine (PHRM) | 5.94 | 150.1 | 91.1 65.1 | 22 46 | 65 |
| Methylphenidate (MPHD) | 7.05 | 234.2 | 84.1 56.1 | 18 54 | 90 |
| Bupropion | 7.71 | 240.1 | 184.1 139 | 10 26 | 80 |
| Hydroxybupropion | 6.96 | 256.1 | 238.1 139 | 6 30 | 70 |
| Methcathinone (ephedrone) | 3.99 | 164.1 | 146.1 51.1 | 10 73 | 90 |
| Mephedrone (4-MMC) | 6.09 | 178.1 | 160.1 145.1 | 10 22 | 85 |
| Methedrone (PMMC) | 5.54 | 194.1 | 176.1 161.1 | 10 22 | 80 |
| Methylone (MDMC) | 4.78 | 208.1 | 160.1 132.1 | 14 30 | 90 |
| Ethylone (MDEC) | 5.48 | 222.1 | 174.1 91.1 | 18 46 | 80 |
| 3,4-methylenedioxypyrovalerone (MDPV) | 7.33 | 276.2 | 126.1 135 | 30 30 | 95 |
| Alpha-pyrrolidinopentiophenone (alpha-PVP) | 7.15 | 232.2 | 91.1 126.1 | 22 26 | 115 |
| Compound | Intra-assay Precision 1 (% CV, n = 15) | Total Precision 1 (% CV, n = 15) | Total Accuracy 2 (% Relative Recovery) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration (ng mL−1) | |||||||||
| Added Concentration | 60 | 800 | 1600 | 60 | 800 | 1600 | 60 | 800 | 1600 |
| Amphetamine (A) | 4.6 | 1.8 | 2.2 | 4.3 | 3.1 | 7.3 | 98.9 | 97.8 | 106.8 |
| Methamphetamine (MA) | 5.5 | 3.0 | 1.7 | 4.9 | 4.6 | 5.5 | 100.6 | 96.2 | 98.7 |
| Phenylpropanolamine (PHPR) | 4.3 | 2.3 | 2.0 | 5.3 | 5.6 | 6.1 | 101.2 | 95.8 | 100.4 |
| Ephedrine | 4.1 | 2.3 | 1.8 | 4.2 | 3.9 | 4.7 | 97.6 | 99.4 | 102.6 |
| Pseudoephedrine (PSE) | 4.2 | 2.8 | 1.7 | 4.9 | 5.1 | 4.3 | 98.5 | 99.3 | 100.4 |
| 3,4-methylene dioxyamphetamine (MDA) | 4.3 | 2.8 | 2.1 | 4.6 | 3.9 | 5.1 | 100.3 | 101.2 | 103.5 |
| 3,4-methylenedioxymethamphetamine (MDMA) | 3.8 | 2.5 | 1.8 | 4.0 | 3.3 | 4.6 | 98.5 | 99.8 | 102.2 |
| 3,4-methylenedioxy-N-ethylamphetamine (MDEA) | 3.3 | 2.9 | 1.4 | 3.3 | 3.2 | 4.5 | 102.5 | 104.1 | 101.9 |
| Phentermine (PHRM) | 2.0 | 2.0 | 4.3 | 4.9 | 1.8 | 5.3 | 103.4 | 99.5 | 101.1 |
| Methylphenidate (MPHD) | 3.0 | 2.7 | 2.0 | 4.2 | 4.4 | 2.4 | 98.9 | 105.3 | 104.4 |
| Bupropion | 4.1 | 2.3 | 1.9 | 5.1 | 3.8 | 4.8 | 100.4 | 102.7 | 103.7 |
| Hydroxybupropion | 3.8 | 7.1 | 1.5 | 3.8 | 7.5 | 2.2 | 98.2 | 107.2 | 103.8 |
| Compound | Intra-assay Precision 1 (% CV, n = 15) | Total Precision 1 (% CV, n = 15) | Total Accuracy 2 (% Relative Recovery) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Concentration (ng mL−1) | |||||||||
| Added Concentration | 60 | 800 | 1600 | 60 | 800 | 1600 | 60 | 800 | 1600 |
| Amphetamine (A) | 3.3 | 2.1 | 2.8 | 4.8 | 3.7 | 6.9 | 96.7 | 100.5 | 106.8 |
| Methamphetamine (MA) | 3.9 | 2.6 | 2.7 | 6.6 | 3.4 | 6.0 | 97.4 | 99.1 | 99.2 |
| Phenylpropanolamine (PHPR) | 4.2 | 2.6 | 2.7 | 6.4 | 4.3 | 5.1 | 101.1 | 97.8 | 100.7 |
| Ephedrine | 3.5 | 2.7 | 1.8 | 5.6 | 3.8 | 1.9 | 96.2 | 101.8 | 102.4 |
| Pseudoephedrine (PSE) | 3.0 | 3.3 | 1.9 | 5.2 | 4.4 | 4.6 | |||
| 3,4-methylene dioxyamphetamine (MDA) | 3.4 | 2.3 | 2.9 | 5.2 | 3.6 | 7.3 | 99.7 | 102.4 | 102.6 |
| 3,4-methylenedioxymethamphetamine (MDMA) | 3.0 | 2.5 | 2.1 | 5.0 | 3.5 | 4.4 | 97.5 | 102.5 | 102.3 |
| 3,4-methylenedioxy-N-ethylamphetamine (MDEA) | 4.0 | 2.5 | 2.1 | 6.8 | 3.2 | 5.4 | 99.6 | 105.8 | 102.5 |
| Phentermine (PHRM) | 3.6 | 3.5 | 2.1 | 6.0 | 3.4 | 4.5 | 98.9 | 102.2 | 101.4 |
| Methylphenidate (MPHD) | 3.5 | 3.0 | 3.0 | 4.0 | 4.8 | 3.8 | 95.8 | 102.2 | 100.4 |
| Bupropion | 3.4 | 3.6 | 3.1 | 5.1 | 4.5 | 5.1 | 98.8 | 105.8 | 104.1 |
| Hydroxybupropion | 4.2 | 2.7 | 3.4 | 5.4 | 4.5 | 4.5 | 96.5 | 108.7 | 104.4 |
| Premortem Whole Blood | Postmortem Whole Blood | |||
|---|---|---|---|---|
| Compound | Concentration (ng mL−1) | |||
| Added Concentration | 60 | 1600 | 60 | 1600 |
| Amphetamine (A) | 91 ± 10 | 91 ± 12 | 93.0 ± 8.1 | 90 ± 16 |
| Methamphetamine (MA) | 92 ± 10 | 91 ± 16 | 93.5 ± 9.7 | 91 ± 23 |
| Phenylpropanolamine (PHPR) | 86.1 ± 5.8 | 86.7 ± 1.6 | 85.4 ± 7.6 | 86.2 ± 2.4 |
| Ephedrine | 87.5 ± 6.9 | 90.1 ± 1.6 | 87.2 ± 7.4 | 91.6 ± 2.3 |
| Pseudoephedrine (PSE) | 79.9 ± 7.2 | 87.1 ± 3.6 | 84.9 ± 9.6 | 90.3 ± 2.2 |
| 3,4-methylenedioxyamphetamine (MDA) | 89.4 ± 6.4 | 90.9 ± 1.8 | 87.2 ± 7.8 | 92.6 ± 2.7 |
| 3,4-methylenedioxymethamphetamine (MDMA) | 89.1 ± 6.9 | 89.9 ± 2.3 | 88.2 ± 7.7 | 92.1 ± 1.9 |
| 3,4-methylenedioxy-N-ethylamphetamine (MDEA) | 89.2 ± 7.9 | 90.7 ± 1.9 | 87.4 ± 8.2 | 91.1 ± 1.6 |
| Phentermine (PHRM) | 90.5 ± 9.7 | 90 ± 10 | 93.1 ± 6.5 | 90 ± 16 |
| Methylphenidate (MPHD) | 88.7 ± 7.1 | 93.2 ± 2.4 | 87.9 ± 8.1 | 93.1 ± 4.3 |
| Bupropion | 89 ± 14 | 81 ± 24 | 91.5 ± 8.1 | 87 ± 25 |
| Hydroxybupropion | 88.9 ± 6.9 | 92.6 ± 1.9 | 87.7 ± 8.4 | 93.4 ± 1.3 |
| Methcathinone (ephedrone) | 98 ± 14 | 90 ± 22 | 95.6 ± 16 | 89.7 ± 16 |
| Mephedrone (4-MMC) | 97 ± 12 | 91 ± 15 | 94 ± 15 | 91.6 ± 20 |
| Methedrone (PMMC) | 96.1 ± 9.6 | 91.6 ± 3.7 | 93 ± 14 | 93.9 ± 3.1 |
| Methylone (MDMC) | 97.1 ± 9.0 | 92.9 ± 1.5 | 93 ± 14 | 95.1 ± 1.4 |
| Ethylone (MDEC) | 97.5 ± 10.2 | 92.2 ± 2.6 | 92 ± 13 | 94.3 ± 2.6 |
| 3,4-methylenedioxypyrovalerone (MDPV) | 96.5 ± 9.8 | 90.1 ± 2.1 | 91 ± 14 | 92.7 ± 0.8 |
| Alpha-pyrrolidinopentiophenone (alpha-PVP) | 98 ± 13 | 88 ± 20 | 93 ± 12 | 90 ± 22 |
| Analytes | Analytical Method; Column; Injection Volume; Flow Rate | Run Time (min) | Sample Preparation | Sample Volume | %Extraction Efficiency | Repeatability (%CV) | Linearity Range; Correlation Coefficient (r) | LOQ; LOD | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 9 amphetamines, 10 cathinones | UHPLC-ESI-MS/MS; Poroshell-120 EC-C18 (75 × 2.1 mm, 2.7 μm); 1 μL; 0.4 mL/min | 9 | SPE | 1 mL | 79.9–98.5% (premortem); 86.2–95% (postmortem) | 1.4–7.1% (premortem); 1.4 to 4.2%(postmortem) | 20–2000 ng/mL; r > 0.994 | LOQ: 2 ng/mL; LOD: 0.5 ng/mL | Current method |
| 8 amphetamines, 3 cathinones | LC-ESI-MS/MS; Biphenyl (100 × 2.1 mm, 1.8 μm); 3 μL; 0.4 mL/min | 7.5 | Protein precipitation | 0.25 mL | 60.2–86.2% | 0.7–14.7% (premortem) | 5–500 ng/mL; r > 0.99 | LOQ: 5 ng/mL; LOD: 0.5–1.7 ng/mL | [52] |
| 30 cathinones | LC-ESI-MS/MS; Poroshell-120 EC-C18 (100 × 2.1 mm, 2.7 μm); 10 μL; 0.4 mL/min | 16 | SPE | 0.25 mL | 84.9–91.5% | 1.1–11.2% | 1–500 ng/mL; r2 > 0.99 | LOQ:1 ng/mL; LOD: 1 ng/mL | [53] |
| 3 amphetamines, 31 cannabinoids and metabolites | LC-ESI-QTRAP-MS/MS; Kinetex™ Biphenyl (50 × 2.1 mm, 2.6 μm); 5 μL; 0.5 mL/min | 9 | supported-liquid-extraction | 0.20 mL | - | 2.6–19.9% (40 analytes); >20% | 1–100 ng/mL; r2 > 0.99 | LOC: 1–6 ng/mL LOD: 0.1–6 ng/mL | [54] |
| 5 amphetamines, 19 cathinones, 45 psychoactive substances | LC-ESI-MS/MS screening method; Zorbax Eclipse Plus C18 (2.1 × 50 mm, 1.8 μm; 7 μL; 0.4 mL/min (6 min)- 0.6 mL/min (8 min) | 15 | Protein precipitation | 0.20 mL | 71–110% | 2–17.9% | 1–100 ng/mL; r2 > 0.99 | LOQ: 0.1–5 ng/mL; LOD: 1 ng/mL | [37] |
| 9 amphetamines, 3 cathinones | UHPLC-ESI-MS/MS; ACQUITY UPLC BEH Phenyl (×2.1 mm, 1.7 μm); 0.4 mL/min | 6 | SPE | 0.30 mL | 70.1–107.9% | 2.4–8.3% | LOQ--50 ng/mL r > 0.995 | LOQ: 0.1–0.5 ng/mL; LOD: 0.02–0.1 ng/mL | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karampela, S.; Smith, J.; Panderi, I. Determination of 19 Psychoactive Substances in Premortem and Postmortem Whole Blood Samples Using Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Separations 2021, 8, 78. https://doi.org/10.3390/separations8060078
Karampela S, Smith J, Panderi I. Determination of 19 Psychoactive Substances in Premortem and Postmortem Whole Blood Samples Using Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Separations. 2021; 8(6):78. https://doi.org/10.3390/separations8060078
Chicago/Turabian StyleKarampela, Sevasti, Jessica Smith, and Irene Panderi. 2021. "Determination of 19 Psychoactive Substances in Premortem and Postmortem Whole Blood Samples Using Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry" Separations 8, no. 6: 78. https://doi.org/10.3390/separations8060078