
Network Pharmacology and Molecular Docking Based Prediction of Mechanism of Pharmacological Attributes of Glutinol

 , ,
, ,  , , ,
, , , 
Abstract
:1. Introduction
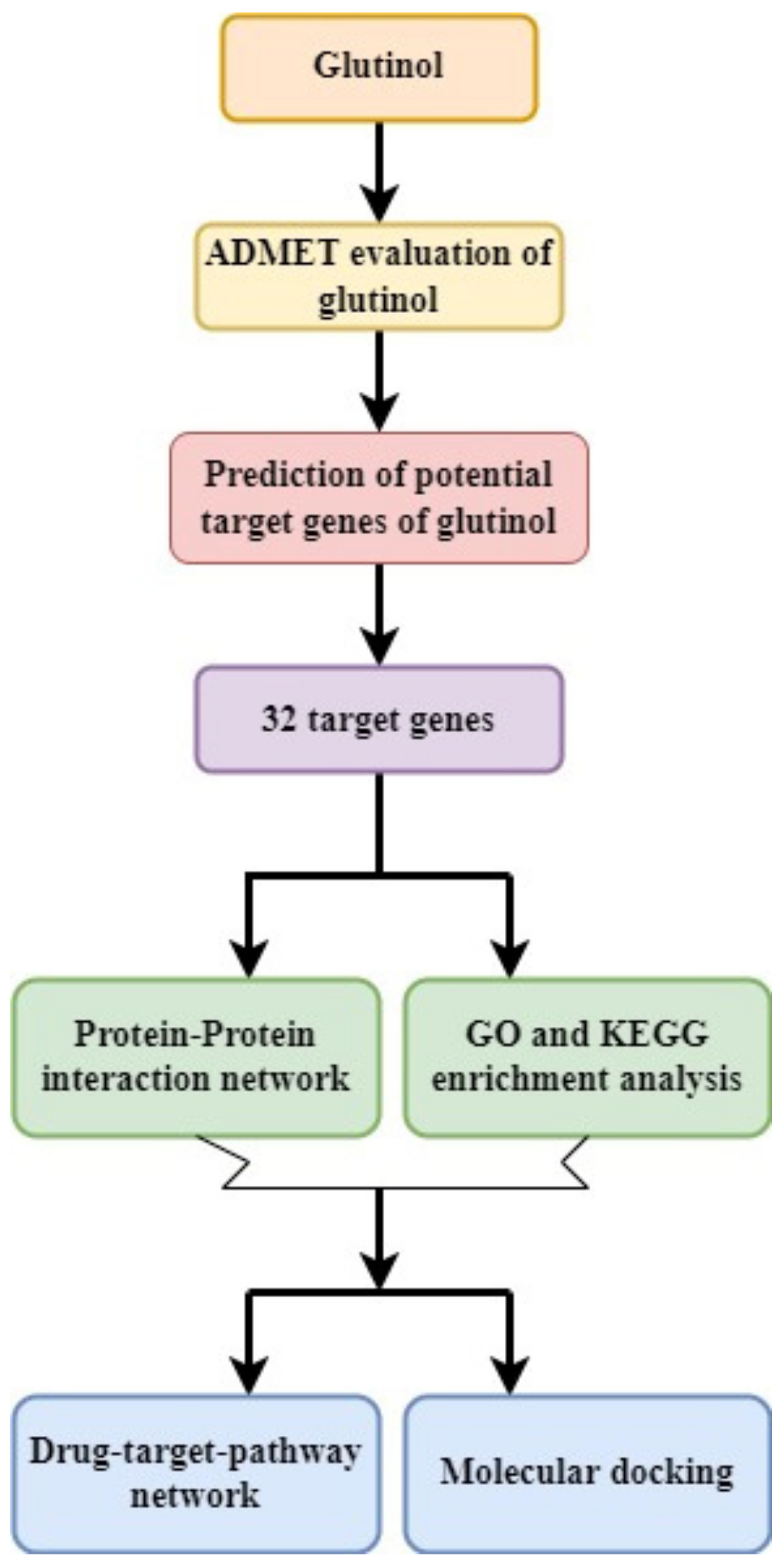
2. Methodology
2.1. PubChem Database-Based Screening of Chemical Structure and ADMET Analysis
2.2. Target Gene Screening by Using Binding DB Database
2.3. Protein–Protein Interaction Network Construction and Analysis
2.4. Analysis of Gene Function and Pathway Enrichment
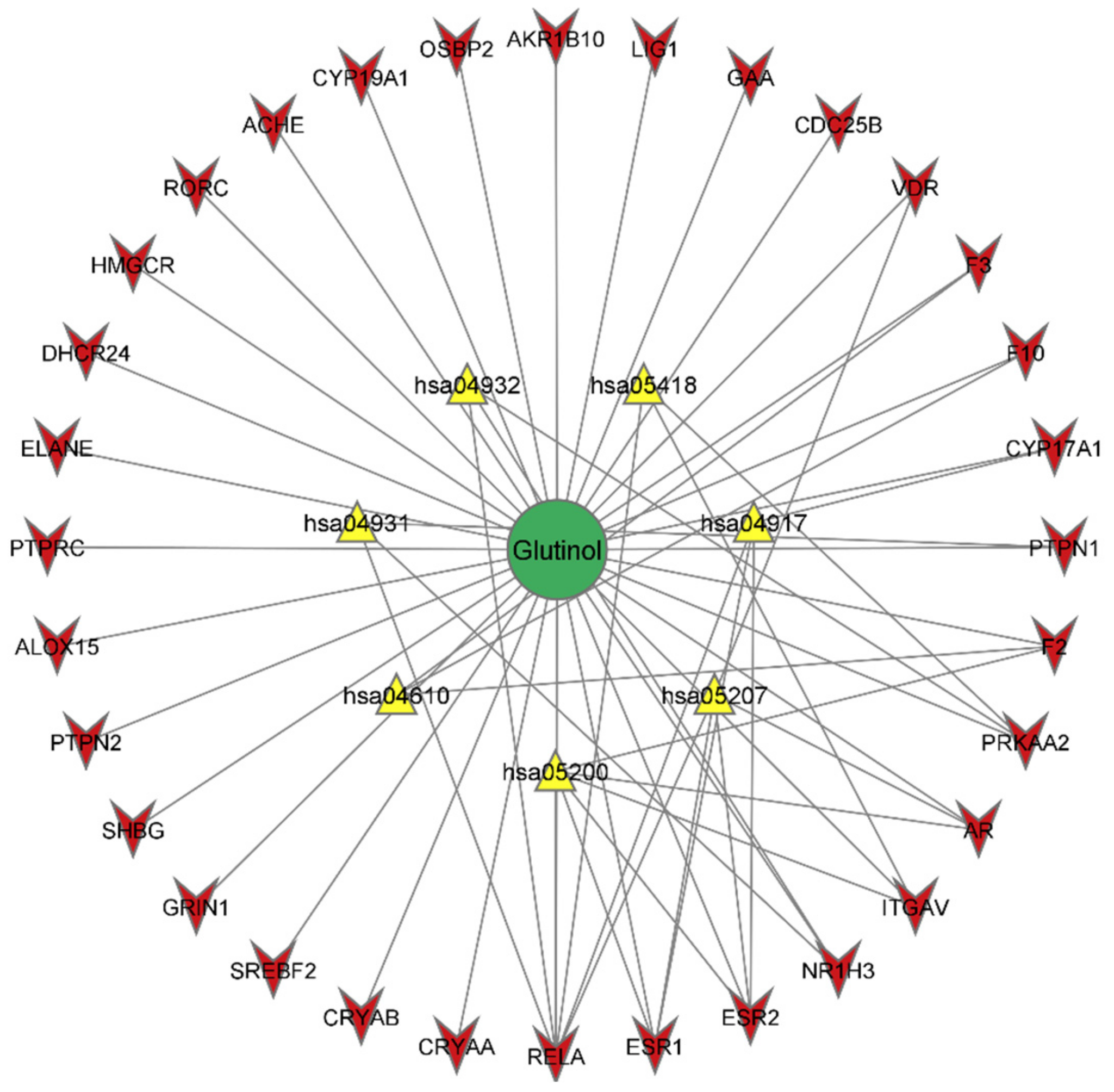
2.5. Construction of Glutinol-Target-Pathway Network
2.6. Molecular Docking
3. Results
3.1. Molecular Formula and ADMET Attributes of Glutinol
3.2. Prediction of Glutinol’s Target Genes
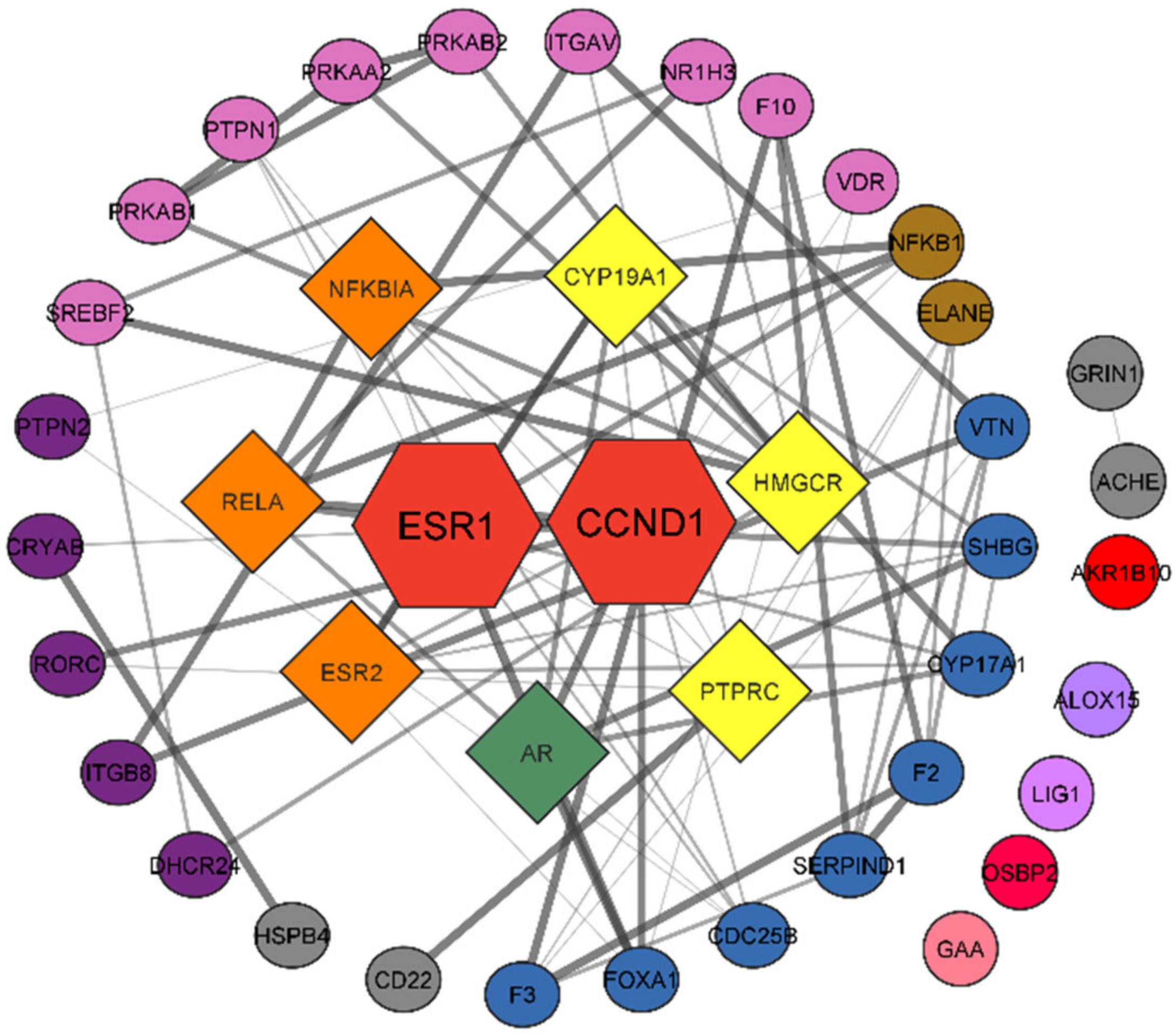
3.3. Protein–Protein Interaction Network
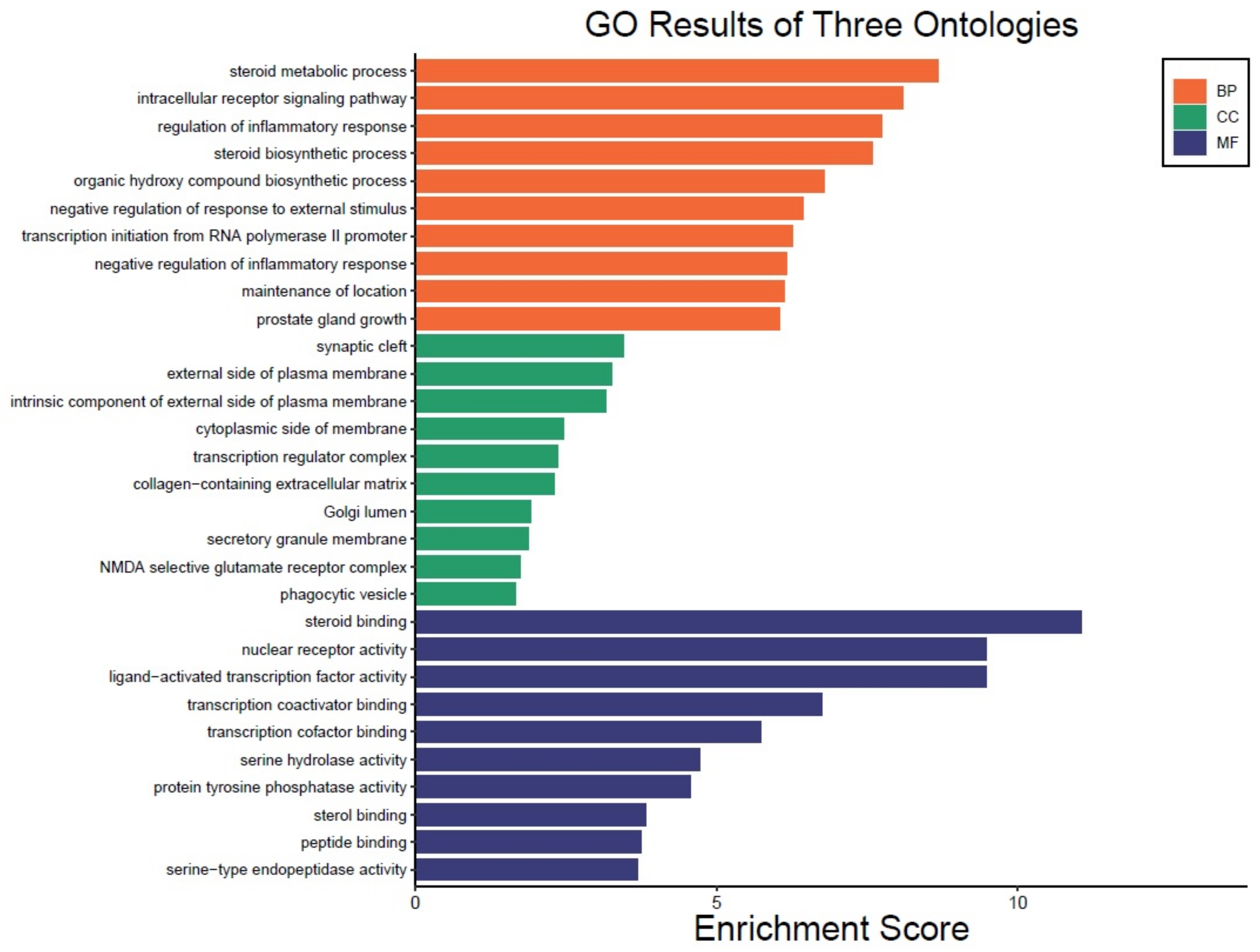
3.4. GO Enrichment Analysis
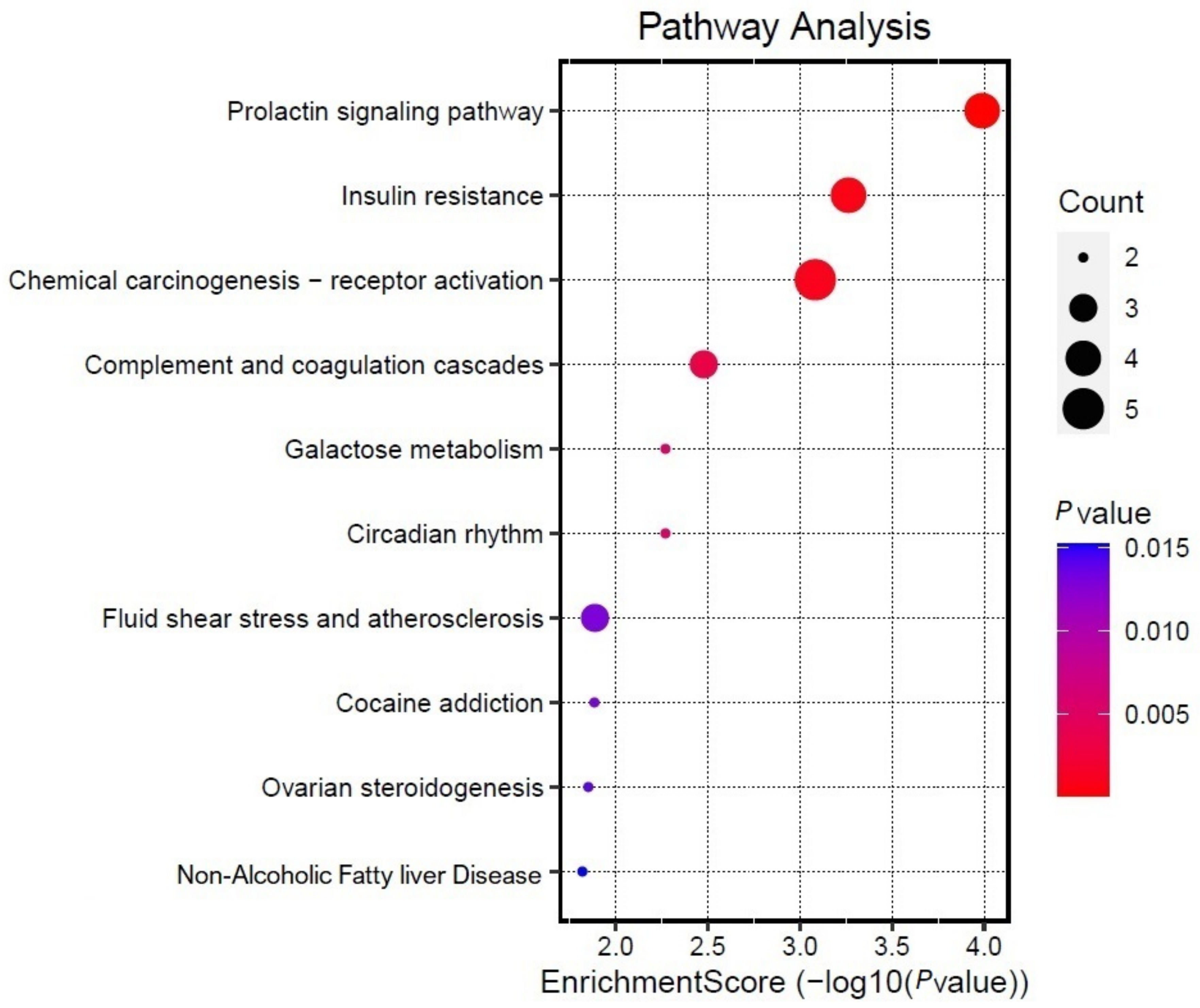
3.5. KEGG Enrichment Analysis
3.6. Network Analysis
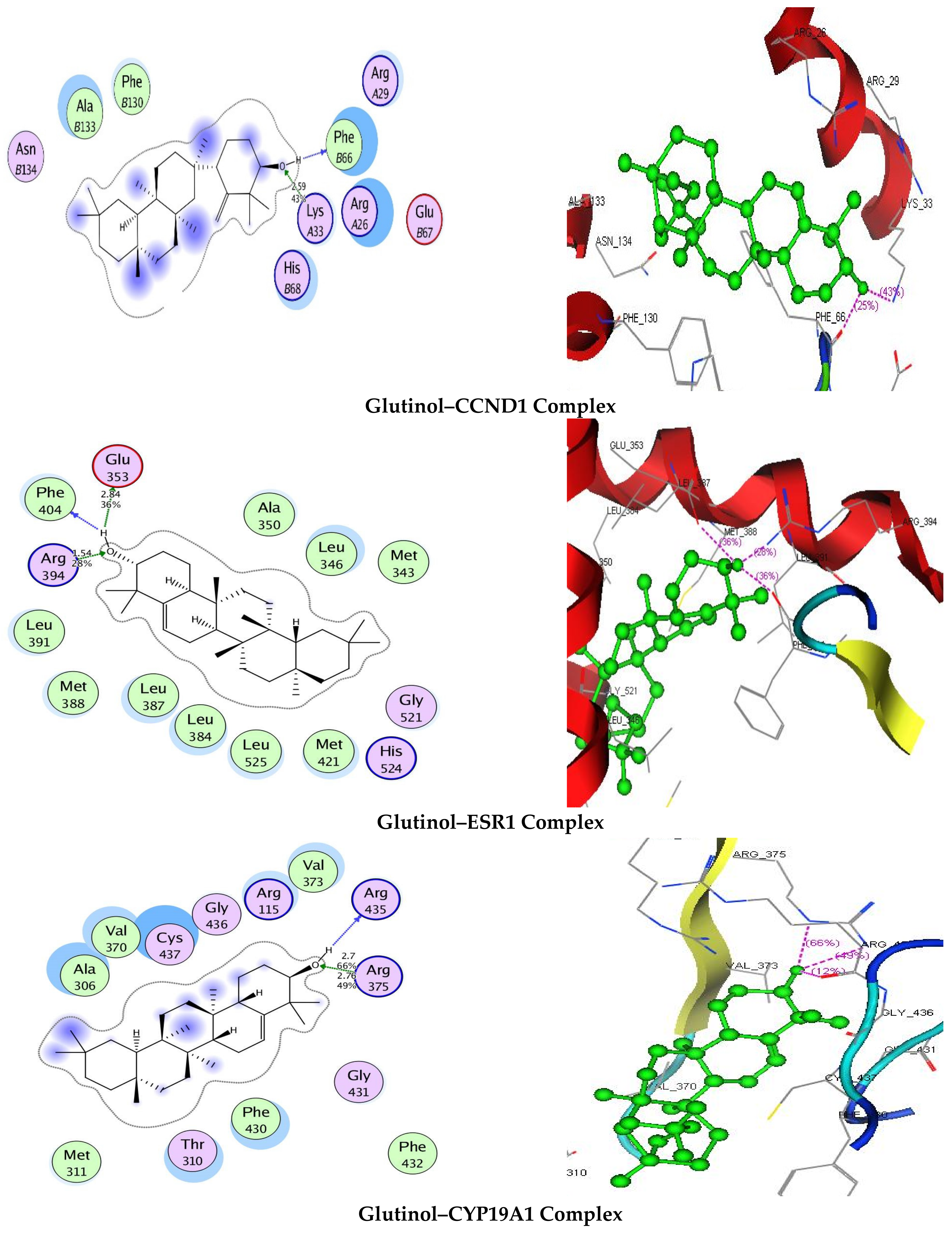
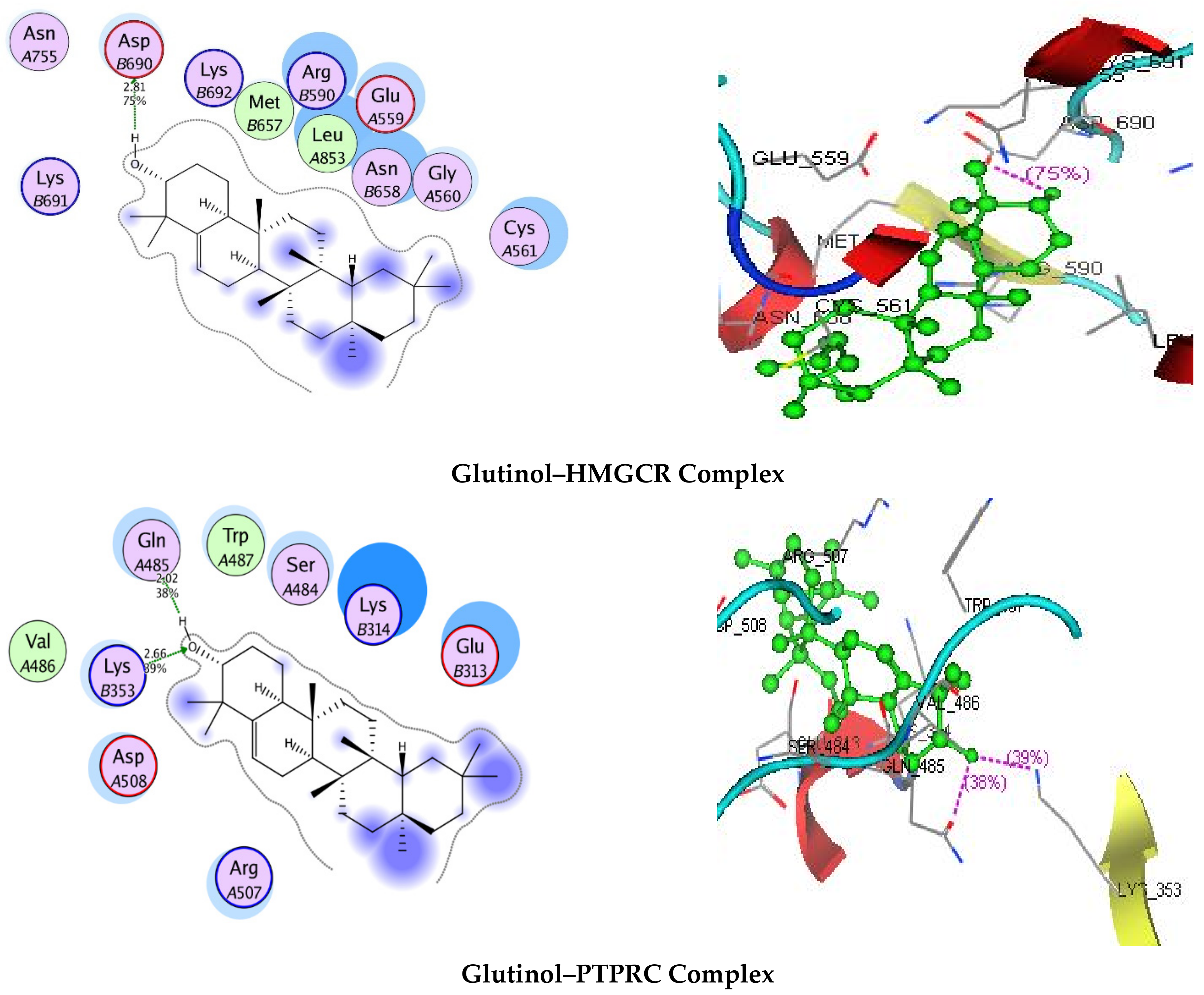
3.7. Molecular Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dar, R.A.; Shahnawaz, M.; Qazi, P.H. General overview of medicinal plants: A review. J. Phytopharm. 2017, 6, 349–351. [Google Scholar] [CrossRef]
- Siddiqui, A.J.; Danciu, C.; Ashraf, S.A.; Moin, A.; Singh, R.; Alreshidi, M.; Patel, M.; Jahan, S.; Kumar, S.; Alkhinjar, M.I.M.; et al. Plants-derived biomolecules as potent antiviral phytomedicines: New insights on ethnobotanical evidences against coronaviruses. Plants 2020, 9, 1244. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S. Biosynthesis of structurally diverse triterpenes in plants: The role of oxidosqualene cyclases. Proc. Indian Natl. Sci. Acad. 2016, 82, 1189–1210. [Google Scholar] [CrossRef]
- Hassan, M.; Azhar, M.; Abbas, Q.; Raza, H.; Moustafa, A.A.; Shahzadi, S.; Ashraf, Z.; Seo, S.Y. Finding novel anti-carcinomas compounds by targeting SFRP4 through molecular modeling, docking and dynamic simulation studies. Curr. Comput. Aided Drug Des. 2018, 14, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Sartori, S.B.; Singewald, N. Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol. Ther. 2019, 204, 107402. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Jeon, E.S.; Lee, J.; Han, S.Y.; Chae, H. Pharmacognostic outlooks on medical herbs of Sasang typology. Integr. Med. Res. 2017, 6, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Pamunuwa, G.; Karunaratne, D.; Waisundara, V.Y. Antidiabetic properties, bioactive constituents, and other therapeutic effects of Scoparia dulcis. Evid. Based Complement. Altern. Med. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebayo, S.A.; Shai, L.J.; Eloff, J.N. First isolation of glutinol and a bioactive fraction with good anti-inflammatory activity from n-hexane fraction of Peltophorum africanum leaf. Asian Pac. J. Trop. Med. 2017, 10, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, J. Glutinol inhibits the proliferation of human ovarian cancer cells via PI3K/AKT signaling pathway. Trop. J. Pharm. Res. 2021, 20, 1331–1335. [Google Scholar] [CrossRef]
- Sharma, K.R.; Adhikari, A.; Hafizur, R.M.; Hameed, A.; Raza, S.A.; Kalauni, S.K.; Miyazaki, J.I.; Choudhary, M.I. Potent insulin secretagogue from Scoparia dulcis Linn of Nepalese origin. Phytother. Res. 2015, 29, 1672–16755. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, U.; Patwardhan, B. Network ethnopharmacological evaluation of the immunomodulatory activity of Withania somnifera. J. Ethnopharmacol. 2017, 197, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhong, M.; Long, F.; Yang, R.; Zhang, Y.; Liu, T. Network pharmacology-based prediction of active ingredients and mechanisms of Lamiophlomis rotata (Benth.) Kudo against rheumatoid arthritis. Front. Pharmacol. 2019, 10, 1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, S.; Kalsoom, S.; Bibi, R.; Asghar, A.; Hameed, A.; Ahmed, W.; Hassan, A. Computational and biological studies of novel thiazolyl coumarin derivatives synthesized through Suzuki coupling. Turk. J. Chem. 2020, 44, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-X.; Zhang, Y.-R.; Luo, S.-Y.; Zhang, Y.-S.; Zhang, Y.; Tang, C. Chlorogenic acid, a natural product as potential inhibitor of COVID-19: Virtual screening experiment based on network pharmacology and molecular docking. Nat. Prod. Res. 2022, 36, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wanga, L.; Schwaigerb, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Weight | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WS | IS | SP | BBB | CNSP | CYP3A4 Substrate | CYP2C19 inhibitor | TC | MTD | ORAT | HT | SS | AMES | |
| 426.72 | −6.49 | 94.41 | −2.816 | 0.665 | −1.905 | Yes | No | −0.037 | −0.603 | 2.298 | No | No | No |
| S. No. | Gene | UniProt ID | Description |
|---|---|---|---|
| 1 | DHCR24 | Q15392 | Delta(24)-sterol reductase |
| 2 | HMGCR | P04035 | 3-hydroxy-3-methylglutaryl-coenzyme reductase |
| 3 | ACHE | P22303 | Acetylcholinesterase |
| 4 | AKR1B10 | O60218 | Aldo-keto reductase family 1 member B10 |
| 5 | GAA | P10253 | Lysosomal alpha-glucosidase |
| 6 | CRYAA | P02489 | Alpha-crystallin A chain |
| 7 | CRYAB | P02511 | Alpha-crystallin B chain |
| 8 | PRKAA2 | P54646 | 5′-AMP-activated protein kinase catalytic subunit alpha-2 |
| 9 | AR | P10275 | Androgen receptor |
| 10 | ALOX15 | P16050 | Polyunsaturated fatty acid lipoxygenase ALOX15 |
| 11 | F3 | P13726 | Tissue factor |
| 12 | F10 | P00742 | Coagulation factor X |
| 13 | CYP17A1 | P05093 | Steroid 17-alpha-hydroxylase/17,20 lyase |
| 14 | CYP19A1 | P11511 | Aromatase |
| 15 | LIG1 | P18858 | DNA ligase 1 |
| 16 | CDC25B | P30305 | M-phase inducer phosphatase 2 |
| 17 | ESR2 | Q92731 | Estrogen receptor beta |
| 18 | ESR1 | P03372 | Estrogen receptor |
| 19 | GRIN1 | Q05586 | Glutamate receptor ionotropic, NMDA 1 |
| 20 | ITGAV | P06756 | Integrin alpha-V |
| 21 | PTPRC | P08575 | Receptor-type tyrosine-protein phosphatase C |
| 22 | ELANE | P08246 | Neutrophil elastase |
| 23 | RELA | Q04206 | Transcription factor p65 |
| 24 | RORC | P51449 | Nuclear receptor ROR-gamma |
| 25 | OSBP2 | Q969R2 | Oxysterol-binding protein 2 |
| 26 | NR1H3 | Q13133 | Oxysterols receptor LXR-alpha |
| 27 | PTPN1 | P18031 | Tyrosine-protein phosphatase non-receptor type 1 |
| 28 | F2 | P00734 | Prothrombin |
| 39 | SREBF2 | Q12772 | Sterol regulatory element-binding protein 2 |
| 30 | SHBG | P04278 | Sex hormone-binding globulin |
| 31 | PTPN2 | P17706 | Tyrosine-protein phosphatase non-receptor type 2 |
| 32 | VDR | P11473 | Vitamin D3 receptor |
| Name | Degree | Betweenness Centrality | Closeness Centrality |
|---|---|---|---|
| CCND1 | 13 | 0.356617 | 0.507463 |
| ESR1 | 13 | 0.243091 | 0.5 |
| CYP19A1 | 7 | 0.253281 | 0.43038 |
| HMGCR | 7 | 0.242254 | 0.343434 |
| PTPRC | 7 | 0.420766 | 0.447368 |
| RELA | 6 | 0.076522 | 0.404762 |
| ELANE | 4 | 0.192513 | 0.34 |
| ITGAV | 3 | 0.096702 | 0.336634 |
| Targets | Binding Energy (kJ/mol) | Interaction | ||
|---|---|---|---|---|
| CCND1 | −8.3554 | 2.59 | 43 | LysA33 |
| ESR1 | −5.3991 | 2.84 1.54 | 36 28 | Glu353 Arg394 |
| CYP19A1 | −10.1795 | 2.7 2.76 | 66 50 | Arg375 Arg375 |
| HMGCR | −5.9682 | 2.81 | 75 | AspB690 |
| PTPRC | −8.8985 | 2.02 2.66 | 38 39 | GlnA485 LysB353 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzarea, S.I.; Qasim, S.; Uttra, A.M.; Khan, Y.H.; Aljoufi, F.A.; Ahmed, S.R.; Alanazi, M.; Malhi, T.H. Network Pharmacology and Molecular Docking Based Prediction of Mechanism of Pharmacological Attributes of Glutinol. Processes 2022, 10, 1492. https://doi.org/10.3390/pr10081492
Alzarea SI, Qasim S, Uttra AM, Khan YH, Aljoufi FA, Ahmed SR, Alanazi M, Malhi TH. Network Pharmacology and Molecular Docking Based Prediction of Mechanism of Pharmacological Attributes of Glutinol. Processes. 2022; 10(8):1492. https://doi.org/10.3390/pr10081492
Chicago/Turabian StyleAlzarea, Sami I., Sumera Qasim, Ambreen Malik Uttra, Yusra Habib Khan, Fakhria A. Aljoufi, Shaimaa Rashad Ahmed, Madhawi Alanazi, and Tauqeer Hussain Malhi. 2022. "Network Pharmacology and Molecular Docking Based Prediction of Mechanism of Pharmacological Attributes of Glutinol" Processes 10, no. 8: 1492. https://doi.org/10.3390/pr10081492