
Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials


1
Department of Basic Science, Graduate School of Arts and Sciences, The University of Tokyo, 3-8-1, Komaba, Meguro-ku, Tokyo 153-8902, Japan
2
PRESTO, Japan Science and Technology Agency, 4-1-8, Honcho, Kawaguchi, Saitama 332-0012, Japan
3
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan
*
Authors to whom correspondence should be addressed.
Polymers 2023, 15(10), 2389; https://doi.org/10.3390/polym15102389
Submission received: 25 April 2023
/
Revised: 16 May 2023
/
Accepted: 17 May 2023
/
Published: 20 May 2023
(This article belongs to the Section Polymer Physics and Theory)
Abstract
:Polyacrylate-based network materials are widely used in various products owing to their facile synthesis via radical polymerization reactions. In this study, the effects of alkyl ester chains on the toughness of polyacrylate-based network materials were investigated. Polymer networks were fabricated via the radical polymerization of methyl acrylate (MA), ethyl acrylate (EA), and butyl acrylate (BA) in the presence of 1,4-butanediol diacrylate as a crosslinker. Differential scanning calorimetry and rheological measurements revealed that the toughness of MA-based networks drastically increased compared with that of EA- and BA-based networks; the fracture energy of the MA-based network was approximately 10 and 100 times greater than that of EA and BA, respectively. The high fracture energy was attributed to the glass transition temperature of the MA-based network (close to room temperature), resulting in large energy dissipation via viscosity. Our results set a new basis for expanding the applications of polyacrylate-based networks as functional materials.
1. Introduction
Polymer network materials are widely used in the industry because of their high toughness and elasticity [1,2,3,4,5,6]. Their broad range of applications is attributed to the diverse chemical and physical properties of their various polymer chains. Poly(alkyl acrylate)s, such as poly(ethyl acrylate)s and poly(butyl acrylate)s, are easily accessible polymers exhibiting high applicability to various polymerization reactions, including free-radical and living-radical polymerizations [7,8,9,10,11,12,13]. However, poly(alkyl acrylate)-based materials exhibit relatively weaker toughness compared with polymethacrylate, polybutadiene, and polystyrene-based materials [14,15,16,17] and have been seldom used as tough materials; Poly(ethyl acrylate)s and poly(butyl acrylate)s are used as adhesives [18,19,20]. Recently, the toughness of polyacrylate-based network materials has been enhanced by block or random copolymerization with other polymer chains and interpenetrations with polymer networks, such as double-network materials [21,22,23,24,25,26,27,28,29,30,31,32,33,34]. Hence, despite the significant potential of poly(alkyl acrylate)-based network materials, simple methodologies for enhancing their toughness are limited. Therefore, the development of a simple and efficient method for increasing the fracture energy of poly(alkyl acrylate)-based polymer networks could expand their use in strong structural and functional materials.
Herein, the systematic investigation of alkyl ester groups in polyacrylate-based network materials revealed a specific toughening effect in poly(methyl acrylate) compared with other alkyl esters. To date, systematic evaluations of the effects of alkyl groups of polyacrylate-based elastic network materials on their toughness have been rarely reported, while the effects of alkyl groups of linear poly(alkyl acrylate)s [35,36,37,38] and rigid networks [39] have been examined. Our investigation revealed that poly(methyl acrylate) exhibited the highest fracture energy among the polyacrylate-based network materials composed of methyl, ethyl, and butyl esters, which were prepared by the radical copolymerization of alkyl acrylates and 1,4-butanediol diacrylate as a crosslinking agent. The enhancement of toughness was attributed to the higher glass transition temperature (near room temperature) of the poly(methyl acrylate) network material compared with the others.
2. Materials and Methods
2.1. Materials
Methyl acrylate (MA) and 2,2′-azobis(2,4-dimethylvaleronitrile) (ADVN) were purchased from FUJIFILM Wako Pure Chemical. Ethyl acrylate (EA), n-butyl acrylate (BA), and 1,4-butanediol diacrylate were purchased from Tokyo Chemical Industry. Dimethylformamide (DMF) was purchased from KANTO CHEMICAL. All reagents and solvents were commercially obtained and used as received.
2.2. Preparation of Polymer Networks
The polymer networks were prepared via a general free-radical polymerization process according to a previously published procedure [13]. As shown in Table 1, the reaction solution composed of the monomer (MA, EA, or BA, 11.2 mmol), 1,4-butanediol diacrylate (0.0005, 0.0020, or 0.010 eq.), and ADVN (0.011 mmol) in DMF (200 μL) was degassed in triplicate using the freeze-thaw technique. The solution was injected into a polytetrafluoroethylene (PTFE)-coated reaction mold (40 × 40 × 0.5 mm3). The mold was composed of two PTFE-coated glass slides with a 0.5 mm thick spacer, and the glass slides and spacer were held with binder clips (Figure S1). The reaction solution was then placed in an oven (60 °C, 18 h) for polymerization, affording films as network materials containing DMF. After polymerization, the obtained network materials were placed in a vacuum oven at 120 °C for 12 h to remove the remaining solvents and monomers, thus providing elastomeric films.
2.3. Characterization
Differential scanning calorimetry (DSC) measurements were performed using Shimadzu DSC-60A Plus under nitrogen flow. Approximately 10 mg of polymer network materials were loaded into aluminum pans. For the measurement of the glass transition temperature, the samples were first heated to 120 °C at 10 °C/min and equilibrated for 20 min to remove thermal history. A subsequent cool/heat cycle was carried out at 5 °C/min. The Tg data presented in the main text was taken from the second heating curves.
Tensile tests were performed with a Shimadzu EZ-SX tester equipped with a 50 N load-cell. The dumbbell-shaped sample (ISO 37-4 shrunk by 2/3, the initial length of the parallel section was 8 mm) with a thickness of 0.3−0.4 mm was cut from the polymer network materials. The test piece was stretched at a constant crosshead speed at 5 mm/min, 50 mm/min, and 500 mm/min until the test piece failure. At least three test pieces were tested at room temperature, and their mean and standard error were calculated.
The linear viscoelastic properties of the network materials were investigated using a TA Instruments HR30 rheometer equipped with a convection oven and a liquid nitrogen cooling system. The round sample (a radius of 9 mm) with a thickness of 0.3−0.4 mm was cut from the polymer network materials and placed in a parallel plate geometry with a radius of 8 mm. The storage modulus (G′) and loss modulus (G″) were measured with sinusoidal oscillatory shear at a constant strain amplitude (0.2% or 0.5%) and varying frequency.
The analytical size-exclusion chromatography of linear polymers was performed with a GL-Science GL-7400 HPLC System equipped with Shodex KF-802, -803, -804 columns, a GL-7410 HPLC pump, a GL-7400 UV detector, and a GL-7454 RI detector using THF as the eluent at a flow rate of 0.6 mL/min.
3. Results and Discussion
3.1. Preparation of Network Materials
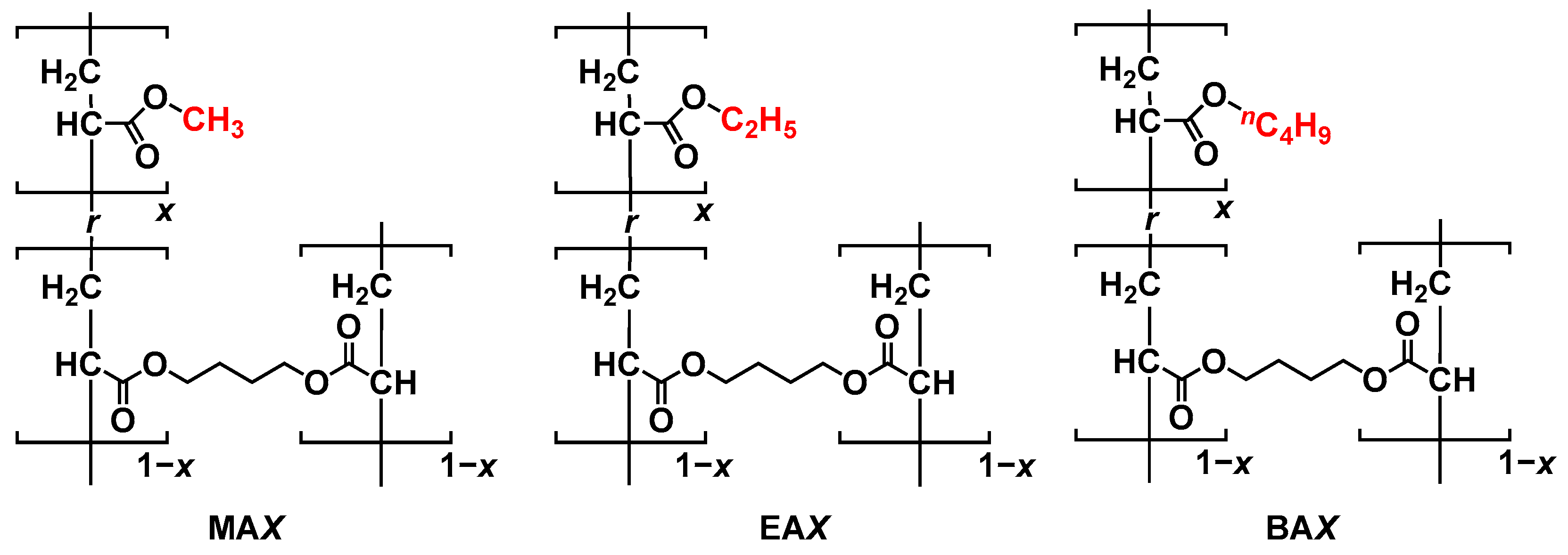
As shown in Figure 1, three types of poly(alkyl acrylate) network materials were prepared via free-radical polymerization. We polymerized 11 mmol of MA, EA, and BA monomers with 1,4-butanediol diacrylate as a crosslinker in the presence of a radical initiator (ADVN, 0.1 mol%) in DMF at 60 °C for 18 h [40]. Three types of crosslinking densities (0.0005, 0.0020, and 0.010 eq.) were introduced to each poly(alkyl acrylate) network. Consequently, nine types of network materials were prepared with different alkyl chains and crosslinking densities, named MAX, EAX, and BAX, where X is 005, 020, and 100 (Table 1). Polymerization proceeded in a polytetrafluoroethylene (PTFE)-coated reaction mold (40 × 40 × 0.5 mm3) affording films as network materials containing DMF.
After polymerization, the obtained films were vacuum-dried to remove the residual monomers and solvents because they possibly influence the mechanical properties of the network materials during the heating processes. The vacuum-drying of the films was conducted at 120 °C for 12–18 h, resulting in a 17–19% decrease in material mass. The complete removal of the solvent and monomer was confirmed from the constancy of the material mass. After vacuum drying, transparent elastic films of a 0.3–0.4 mm thickness were obtained as network materials (Figure S2). The films were cut into specimens for characterization.
3.2. Thermal Properties of Polymer Materials
The thermal properties of the poly(alkyl acrylate)-based network materials were investigated. The degradation temperatures of MA020, EA020, and BA020 were determined by thermogravimetry (TG) analysis. The 5% weight reduction temperature was greater than 250 °C for all the network materials, indicating their high thermal resistance (Figure S5). Differential scanning calorimetry (DSC) measurements were conducted on nine network samples (MAX, EAX, and BAX; X = 005, 020, and 100). The results clearly revealed the reversible base shifts due to glass transitions in all the samples (Figure S4). For example, the glass transition temperature (Tg) of MA020 was at 16.1 °C, which was remarkably higher than that of EA020 and BA020, −14.7 and −50.2 °C, respectively (Table 2). The high Tg of MA0 was derived from the small mobility of its side chains with methyl groups, which decreased the activation barrier for the segmental motion of the polymer chains [40]. However, the increase of crosslinking density slightly affected the Tg of the network materials; the Tg of MAX, EAX, and BAX (X = 005, 020, and 100) were in the range of 16 to 18 °C, −15 to −12 °C, and −46 to −50 °C, respectively. These results indicate that the Tg of the poly(alkyl acrylate)-based network materials is significantly influenced by the type of alkyl ester chains rather than by the crosslinking density. Hence, as the number of carbon atoms on the ester chains decreased, the Tg of the network materials increased near room temperature, particularly in the case of MA (16–18 °C).
The dependence of Tg on the alkyl chain length was consistent with the behavior of linear homopolymers without crosslinkers, namely poly(methyl acrylate) (MA0), poly(ethyl acrylate) (EA0), and poly(butyl acrylate) (BA0); the Tg of the linear polymers increased in the order of BA0 (−54 °C), EA0 (−24 °C), and MA0 (10 °C) as previously reported [40,41]. Notably, the Tg of the network materials was 3–10 °C higher than that of the respective linear homopolymers, because the presence of crosslinks causes an increase in Tg [3,42,43]. As a result, the Tg of MAX (X = 005, 020, and 100) approached room temperatures because of the crosslinking and the potential high Tg of poly(methyl acrylate).
3.3. Viscoelastic Properties of Polymer Materials
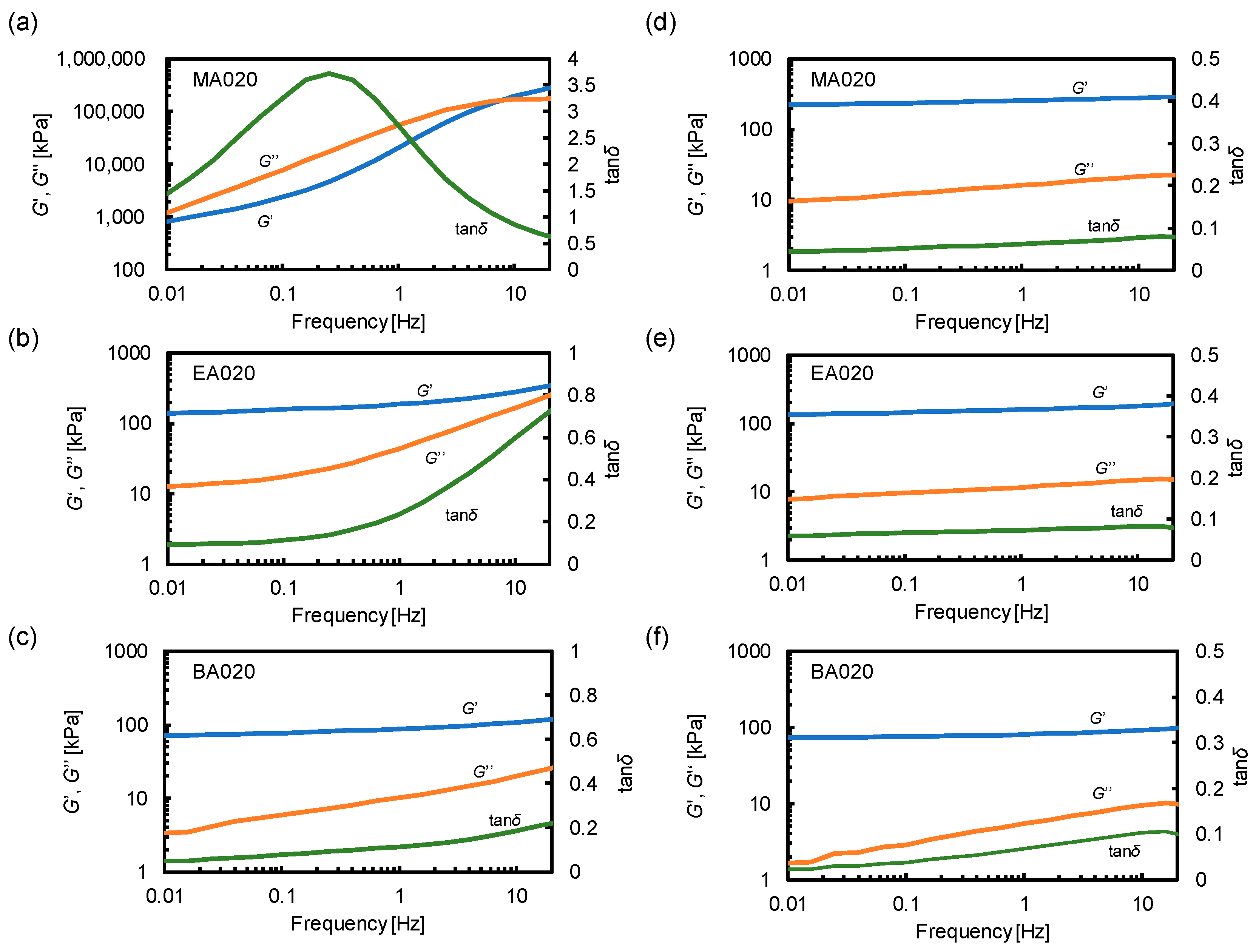
The dynamic behavior of the poly(alkyl acrylate)-based network materials was investigated by conducting rheological measurements. The frequency-dependent characteristics of MA020, EA020, and BA020 were examined at room temperature as shown in Figure 2a–c. Remarkably, the G′ and G″ of MA020 at 1 Hz were 100 and 1000 times higher, respectively, than those of EA020 and BA020. Notably, below 10 Hz, only MA020 exhibited a G″ larger than G′ and the intersection between G′ and G″ was observed. Consequently, only MA020 exhibited a relaxation as a peak in tanδ at 0.3 Hz, indicating segmental motions that resulted in energy dissipation. The viscoelastic differences among MA020, EA020, and BA020 were attributed to the Tg of MA020 at approximately room temperature.
Additionally, the frequency-dependent moduli of MA020, EA020, and BA020 were measured at temperatures 100–120 °C above the corresponding Tg. This condition enables the removal of the effects of segmental relaxation and highlights the elasticity of the network. G′ was independent of the frequency and was much higher than G″ for MA020, EA020, and BA020 (Figure 2d–f). This confirms that these materials consist of permanent covalent networks. The rubber elasticity theory states that the shear elastic modulus G is determined by the number density of the elastically effective chains ν, and is described by Equation (1),
where R and T are the gas constant and temperature, respectively, assuming an affine network model. Equation (1) was used to calculate ν from the observed G values. The ν of MA020, EA020, and BA020 was obtained to be 7.64 × 10−5, 5.29 × 10−5, and 2.94 × 10−5 mol/cm3, respectively, showing a decreasing trend upon increase in the length of the alkyl side chain.
G = νRT,
To rationalize the difference in the obtained ν, we estimated the ideal number density of elastically effective chains (νideal) from the monomer and crosslinker feeds. For considering ideal networks, we assumed that all crosslinkers (1,4-butanediol diacrylate) serve as a 4-branch point and the network is free of any defects such as dangling chains and loops. This led to Equation (2),
where ρ is the physical density of the polymer, x is the crosslinker-to-monomer ratio, and M is the molecular weight of the repeat unit. Detailed calculation procedures can be found in the Supporting Information. The νideal was determined to be 5.7 × 10−5, 4.5 × 10−5, and 3.3 × 10−5 mol/cm3 for MA020, EA020, and BA020, respectively. The order and the trend of the experimental values (ν) were generally consistent with those of the ideal values (νideal). From Equation (2), the difference in νideal among the above three samples comes from the difference in M/ρ, which can be recognized as the apparent molar volume per repeat unit. Therefore, the different crosslinking densities and plateau modulus can be explained based on the bulkiness of the repeat units. At the same degree of polymerization, less bulky monomers result in less bulky polymer chains. Therefore, the use of less bulky monomers leads to higher number density of chains and hence higher ν in the bulk network material.
νideal = 2ρx/M,
3.4. Tensile Testing to Reveal the Toughness of the Network Materials
The mechanical characteristics of the network materials were assessed via uniaxial tensile testing, conducted at an elongation speed of 50 mm/min, which approximately corresponded to the nominal strain rate of 0.10 s−1. As shown in Figure 3a and Figure S6, MAX exhibited the highest toughness compared with the corresponding EAX and BAX. The fracture strains of MAX (X = 020 and 100) were 1.5 times and 2.5 times larger than those of EAX and BAX, respectively (Figure S7). The fracture strain of MA005 was similar to that of EA005 but 2.5 times larger than that of BA005. In contrast, the fracture stresses of MAX significantly surpassed those of EAX and BAX (Figure 3b). Similarly, MAX exhibited a four- and eight-fold higher Young’s modulus compared with EAX and BAX, respectively, for X = 005, 020, and 100 (Figure 3c).
Consequently, as shown in Figure 3d, the fracture energy of MA020 was approximately 10 and 100 times greater than those of EAX and BAX, respectively, demonstrating the large resistance to mechanical strain. Comparisons of the fracture energies of MA005, MA020, and MA100 revealed that a decrease in the crosslink density increased the fracture energy of the material [44]; MA005 exhibited the highest fracture energy (83 MJ/m3). Regardless of the amount of crosslinker, MAX exhibited remarkably high fracture energies, which were improved by one or two orders of magnitude compared with the corresponding EAX and BAX, suggesting the superior mechanical properties of the poly(methyl acrylate)-based network materials.
The superior mechanical performance of MAX can be attributed to the unique glass transition temperature of MA-based polymer network materials (close to room temperature); regarding MA020, such a performance was attributed to the methyl ester groups and crosslinking. As mentioned in Section 3.3, a large relaxation via segmental motion was observed in MAX at room temperature. The relaxation took place in the time scale ranging from 0.1 to 100 s (corresponding to the frequency of 0.01–10 Hz), which should cover the time scale of the uniaxial tensile tests (~0.1 s−1 or ~10 s). Therefore, a considerable amount of energy is absorbed and dissipated by the segmental motion of the poly(methyl acrylate) chains during stretching of the network material, probably owing to the small activation barrier for the segmental motion of the polymer chains derived from the small side-chain methyl groups in MAX [40]. However, EA- and BA-based network materials lack this energy dissipation mechanism due to their Tg, which is much lower than room temperature. The energy dissipation contributes to the high Young’s modulus of the MA-based networks and is also responsible for the large strain at break, leading to the high stress at break and toughness of MA-based networks. In general, the fracture of a polymer network begins with a tiny crack that propagates through the material, eventually leading to catastrophic failure. The extent of the energy concentration at the crack tip is considered to play an important role in the fracture process [45,46,47,48,49]. In MAX, the energy dissipation via segmental motions should help dissipate the strain energy at the crack tip, delaying crack propagation and hence the fracture of the material. Consequently, MAX exhibited significantly superior strain at break, stress at break, and fracture toughness compared with EAX and BAX.
To verify the toughening mechanism, the strain rate dependence of the tensile behavior was investigated. MA020, EA020, and BA020 were tested at the additional elongation speed of 5 and 500 mm/min, which approximately corresponded to the nominal strain rate of 0.01 and 1.0 s−1, respectively. Figure S8 compares the fracture energies at different elongation speeds derived from the stress-strain curves. The mechanical properties of MA020 and BA020 were independent of the elongation speed, whereas increasing the elongation speed improved the toughness of EA020; the fracture energy of EA020 at 50 mm/min was approximately 3 times higher compared with that at 5 mm/min. The enhancement of the toughness of EA020 can be attributed to the segmental relaxation. In Figure 2b, EA020 showed an upturn in tanδ at the high-frequency limit (>1 Hz), indicating that the relaxation via segmental motion becomes more prominent at a shorter time scale. Thus, more energy dissipation takes place at a higher elongation speed, leading to an increased toughness. BA020 did not show an increase in tanδ within the frequency range tested (Figure 2c). There was almost no energy dissipation by the segmental motion in BA020 in the accessible time scale, resulting in the independence of the mechanical properties on the elongation speed. In MA020, dynamic moduli and tanδ considerably varied with the frequency (Figure 2a). Therefore, it was rather surprising that MA020 did not show the elongation speed dependence. We speculate that the time scale of the tensile tests corresponded to a relatively high-frequency region in the rheological spectra. In Figure 2a, the storage and loss moduli almost become independent of the frequency at >1 Hz. Thus, MA020 had a similar degree of energy dissipation at all elongation speeds, leading to the toughness independent of the elongation speed. These results support the proposed mechanism, i.e., the toughness of poly(alkyl acrylate)-based network materials was drastically enhanced through the energy dissipation from the segmental relaxation.
4. Conclusions
In this study, the effects of the alkyl groups of polyacrylate-based network materials on their thermal and rheological properties were systematically evaluated. We synthesized nine types of polymer network materials with different crosslinking densities and different alkyl ester lengths on the polymer side chains: methyl, ethyl, and n-butyl esters. The MA network materials exhibited elevated glass transition temperature (Tg), around room temperature, which was attributed to the potentially high Tg of poly(methyl acrylate) and the crosslinking of the network. Rheological measurements indicated that the loss modulus of the MA-based network materials exceeds their storage modulus at room temperature. Consequently, the materials exhibited high toughness owing to their large fracture energy, which was induced by energy dissipation from the viscosity, compared with other poly(alkyl acrylate)-based counterparts, poly(ethyl acrylate), and poly(butyl acrylate).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/polym15102389/s1, Figure S1. Schematic representation of reaction mold for the synthesis of network materials: Figure S2. The photograph of network materials (MA020, EA020, and BA020) cut out in a round shape: Figure S3. SEC analyses of the polymerization of methyl acrylate, ethyl acrylate, and butyl acrylate without a crosslinker (1,4-butanediol diacrylate) (Detector: RI): Detailed discussion for calculation of ideal number density of elastically effective chains (νideal): Figure S4. DSC thermograms of MA020 during the 2nd and 3rd heating processes: Figure S5. Thermogravimetric analyses of MA020, EA020, and BA020: Figure S6. Representative stress-strain curves for (a) MA005, EA005, and BA005 and (b) MA100, EA100, and BA100: Figure S7. The comparison of fracture strain for MAX, EAX, and BAX (X = 005, 020, and 100): Figure S8. Fracture energies of MA020, EA020, and BA020, that were measured with tensile tests at the elongation speed of 5, 50, and 500 mm/min: Figure S9–S23. Stress-strain curves of MA005, EA005, BA005, MA020, EA020, BA020, MA100, EA100, and BA100 [50].
Author Contributions
Conceptualization, H.M. and S.N.; methodology, Y.K., H.M. and S.N.; validation, Y.K., H.M. and S.N.; formal analysis, Y.K.; investigation, Y.K.; data curation, Y.K., H.M. and S.N.; writing—original draft preparation, Y.K., H.M. and S.N.; writing—review and editing, H.M., S.N., N.Y. and J.T.; supervision, N.Y. and J.T.; project administration, H.M. and J.T.; funding acquisition, H.M., N.Y. and J.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by JST PRESTO Grant Number JPMJPR21N8; JST CREST Grant Number JPMJCR19I2; JSPS KAKENHI Grant Numbers 21K05181, 21K18714, 21K18948, and 22H02060; Ogasawara Foundation, “Innovation inspired by Nature” Research Support Program, SEKISUI CHEMICAL CO., LTD, and The Murata Science Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Acknowledgments
The authors thank Sayaka Uchida and Naoki Ogiwara for providing instrumental access and advice on thermogravimetric analyses.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zheng, K.; Zhang, Y.; Li, B.; Granick, S. Phosphorescent Extensophores Expose Elastic Nonuniformity in Polymer Networks. Nat. Commun. 2023, 14, 537. [Google Scholar] [CrossRef] [PubMed]
- Kannurpatti, A.R.; Anseth, J.W.; Bowman, C.N. A Study of the Evolution of Mechanical Properties and Structural Heterogeneity of Polymer Networks Formed by Photopolymerizations of Multifunctional (Meth)Acrylates. Polymer 1998, 39, 2507–2513. [Google Scholar] [CrossRef]
- Bandzierz, K.; Reuvekamp, L.; Dryzek, J.; Dierkes, W.; Blume, A.; Bielinski, D. Influence of Network Structure on Glass Transition Temperature of Elastomers. Materials 2016, 9, 607. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, W.; Liu, Z.; Xue, Y.; Lei, X.; Gong, G.; Zhang, Q. Towards a Tough Reprocessable and Self-Healable Acrylonitrile-Butadiene Rubber Based on Strong Hydrogen Bonding Interactions. J. Mater. Chem. C 2021, 9, 6241–6250. [Google Scholar] [CrossRef]
- Ferry, J.D.; Mancke, R.G.; Maekawa, E.; Oyanagi, Y.; Dickie, R.A. Dynamic Mechanical Properties of Cross-Linked Rubbers. I. Effects of Cross-Link Spacing in Natural Rubber. J. Phys. Chem. 1964, 68, 3414–3418. [Google Scholar] [CrossRef]
- Osaka, N.; Kato, M.; Saito, H. Mechanical Properties and Network Structure of Phenol Resin Crosslinked Hydrogenated Acrylonitrile-Butadiene Rubber. J. Appl. Polym. Sci. 2013, 129, 3396–3403. [Google Scholar] [CrossRef]
- Bendre, A.M.; Inamdar, S.R.; Parulekar, S.J. Multi-Equation Bifurcation Analysis of a Free Radical Polymerization in a CSTR. Mater. Today Proc. 2023, 72, 242–249. [Google Scholar] [CrossRef]
- Gao, H.; Li, W.; Matyjaszewski, K. Synthesis of Polyacrylate Networks by ATRP: Parameters Influencing Experimental Gel Points. Macromolecules 2008, 41, 2335–2340. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Tan, M.; Hu, Z.; Dai, Y.; Peng, Y.; Zhou, Y.; Shi, Y.; Li, Y.; Chen, Y. A Simple Mechanochromic Mechanophore Based on Aminothiomaleimide. ACS Macro Lett. 2021, 10, 1423–1428. [Google Scholar] [CrossRef]
- Kury, M.; Ehrmann, K.; Gorsche, C.; Dorfinger, P.; Koch, T.; Stampfl, J.; Liska, R. Regulated Acrylate Networks as Tough Photocurable Materials for Additive Manufacturing. Polym. Int. 2022, 71, 897–905. [Google Scholar] [CrossRef]
- Ide, N.; Fukuda, T. Nitroxide-Controlled Free-Radical Copolymerization of Vinyl and Divinyl Monomers. 2. Gelation. Macromolecules 1999, 32, 95–99. [Google Scholar] [CrossRef]
- Russell, G.M.; Kaneko, T.; Ishino, S.; Masai, H.; Terao, J. Transient Photodegradability of Photostable Gel Induced by Simultaneous Treatment with Acid and UV–Light for Phototuning of Optically Functional Materials. Adv. Funct. Mater. 2022, 32, 2205855. [Google Scholar] [CrossRef]
- Corsaro, C.; Neri, G.; Santoro, A.; Fazio, E. Acrylate and Methacrylate Polymers’ Applications: Second Life with Inexpensive and Sustainable Recycling Approaches. Materials 2022, 15, 282. [Google Scholar] [CrossRef]
- Moretti, G.; Sarina, L.; Agostini, L.; Vertechy, R.; Berselli, G.; Fontana, M. Styrenic-Rubber Dielectric Elastomer Actuator with Inherent Stiffness Compensation. Actuators 2020, 9, 44. [Google Scholar] [CrossRef]
- Chen, Y.; Agostini, L.; Moretti, G.; Fontana, M.; Vertechy, R. Dielectric Elastomer Materials for Large-Strain Actuation and Energy Harvesting: A Comparison between Styrenic Rubber, Natural Rubber and Acrylic Elastomer. Smart Mater. Struct. 2019, 28, 114001. [Google Scholar] [CrossRef]
- Jin, F.L.; Lu, S.L.; Song, Z.B.; Pang, J.X.; Zhang, L.; De Sun, J.; Cai, X.P. Effect of Rubber Contents on Brittle-Tough Transition in Acrylonitrile-Butadiene-Styrene Blends. Mater. Sci. Eng. A 2010, 527, 3438–3441. [Google Scholar] [CrossRef]
- Beharaj, A.; Ekladious, I.; Grinstaff, M.W. Poly(Alkyl Glycidate Carbonate)s as Degradable Pressure-Sensitive Adhesives. Angew. Chem. Int. Ed. 2019, 58, 1407–1411. [Google Scholar] [CrossRef]
- Inui, T.; Sato, E.; Matsumoto, A. High-Molecular-Weight Polar Acrylate Block Copolymers as High-Performance Dismantlable Adhesive Materials in Response to Photoirradiation and Postbaking. RSC Adv. 2014, 4, 24719–24728. [Google Scholar] [CrossRef]
- Sakdapipanich, J.; Thananusont, N.; Pukkate, N. Synthesis of Acrylate Polymers by a Novel Emulsion Polymerization for Adhesive Applications. J. Appl. Polym. Sci. 2006, 100, 413–421. [Google Scholar] [CrossRef]
- Qin, X.; Li, X.; Wang, W.; Li, Q.; Wang, T.; Cao, L.; Piao, J.; Chen, S. Synthesis of Poly Ethyl Acrylate Interpenetrated Poly Methyl Acrylate Elastomer Coating and Its Corrosion Resistance Properties. Mater. Lett. 2022, 314, 131880. [Google Scholar] [CrossRef]
- Ducrot, E.; Creton, C. Characterizing Large Strain Elasticity of Brittle Elastomeric Networks by Embedding Them in a Soft Extensible Matrix. Adv. Funct. Mater. 2016, 26, 2482–2492. [Google Scholar] [CrossRef]
- Smith, K.E.; Parks, S.S.; Hyjek, M.A.; Downey, S.E.; Gall, K. The Effect of the Glass Transition Temperature on the Toughness of Photopolymerizable (Meth)Acrylate Networks under Physiological Conditions. Polymer 2009, 50, 5112–5123. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Mark, J.E.; Jethmalani, J.M.; Ford, W.T. Effects of Dispersion and Aggregation of Silica in the Reinforcement of Poly(Methyl Acrylate) Elastomers. Chem. Mater. 1997, 9, 2442–2447. [Google Scholar] [CrossRef]
- Huang, G.S.; Jiang, L.X.; Li, Q. Molecular Design of Damping Rubber Based on Polyacrylate-Containing Silicone. J. Appl. Polym. Sci. 2002, 85, 746–751. [Google Scholar] [CrossRef]
- Ducrot, E.; Chen, Y.; Bulters, M.; Sijbesma, R.P.; Creton, C. Toughening Elastomers with Sacrificial Bonds and Watching Them Break. Science 2014, 344, 186–189. [Google Scholar] [CrossRef]
- Kawai, Y.; Park, J.; Ishii, Y.; Urakawa, O.; Murayama, S.; Ikura, R.; Osaki, M.; Ikemoto, Y.; Yamaguchi, H.; Harada, A.; et al. Preparation of Dual-Cross Network Polymers by the Knitting Method and Evaluation of Their Mechanical Properties. NPG Asia Mater. 2022, 14, 32. [Google Scholar] [CrossRef]
- Wu, C.L.; Zhang, M.Q.; Rong, M.Z.; Friedrich, K. Tensile Performance Improvement of Low Nanoparticles Filled-Polypropylene Composites. Compos. Sci. Technol. 2002, 62, 1327–1340. [Google Scholar] [CrossRef]
- Zhengcai, P.; James, E.M.; Jagdish, M.J.; Warren, T.F. Mechanical properties of a poly(methyl acrylate) nanocomposite containing regularly-arranged silica particles. Polym. Bull. 1996, 37, 545–551. [Google Scholar]
- Huang, X.; Nakagawa, S.; Houjou, H.; Yoshie, N. Insights into the Role of Hydrogen Bonds on the Mechanical Properties of Polymer Networks. Macromolecules 2021, 54, 4070–4080. [Google Scholar] [CrossRef]
- Kim, C.; Ejima, H.; Yoshie, N. Non-Swellable Self-Healing Polymer with Long-Term Stability under Seawater. RSC Adv. 2017, 7, 19288–19295. [Google Scholar] [CrossRef]
- Hayashi, M.; Noro, A.; Matsushita, Y. Highly Extensible Supramolecular Elastomers with Large Stress Generation Capability Originating from Multiple Hydrogen Bonds on the Long Soft Network Strands. Macromol. Rapid Commun. 2016, 37, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Hayashi, M. Potential of Graft Polymers Bearing Inner Molten Block and Outer Glassy Block at the Graft Chains for Thermoplastic Elastomers with Enhanced Properties. Macromol. Chem. Phys. 2022, 223, 2200073. [Google Scholar] [CrossRef]
- Urban, M.W.; Davydovich, D.; Yang, Y.; Demir, T.; Zhang, Y.; Casabianca, L. Key-and-lock commodity self-healing copolymers. Science 2018, 362, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Fytas, G.; Patkowski, A.; Meier, G.; Dorfmüller, T. A High Pressure Photon Correlation Study of Bulk Poly(Methylacrylate). Comparison with Relaxation Processes in Poly(Ethylacrylate) and Related Polymethacrylates. J. Chem. Phys. 1983, 80, 2214–2220. [Google Scholar] [CrossRef]
- Ribelles, J.L.G.; Duenas, J.M.M.; Pradas, M.M. Dielectric Relaxations in Poly(methyl Acrylate), Poly(ethyl Acrylate), and Poly(butyl Acrylate). J. Appl. Polym. Sci. 1989, 38, 1145–1157. [Google Scholar] [CrossRef]
- Coumoulos, G.D. The Electron Diffraction by Amorphous Polymers. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1943, 182, 166–179. [Google Scholar]
- Dahlquist, C.A.; Hendricks, J.O.; Taylor, N.W. Elasticity of Soft Polymers—Constant-Stress Elongation Tests. Ind. Eng. Chem. 1951, 43, 1404–1410. [Google Scholar] [CrossRef]
- Safranski, D.L.; Gall, K. Effect of Chemical Structure and Crosslinking Density on the Thermo-Mechanical Properties and Toughness of (Meth)Acrylate Shape Memory Polymer Networks. Polymer 2008, 49, 4446–4455. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Wang, H.; Lu, W.; Osa, M.; Wang, Y.; Mays, J.; Hong, K. Chain Flexibility and Glass Transition Temperatures of Poly(n-Alkyl (Meth)Acrylate)s: Implications of Tacticity and Chain Dynamics. Polymer 2021, 213, 123207. [Google Scholar] [CrossRef]
- Jakubowski, W.; Juhari, A.; Best, A.; Koynov, K.; Pakula, T.; Matyjaszewski, K. Comparison of Thermomechanical Properties of Statistical, Gradient and Block Copolymers of Isobornyl Acrylate and n-Butyl Acrylate with Various Acrylate Homopolymers. Polymer 2008, 49, 1567–1578. [Google Scholar] [CrossRef]
- Ueberreiter, K.; Kanig, G. Second-Order Transitions and Mesh Distribution Functions of Cross-Linked Polystyrenes. J. Chem. Phys. 1950, 18, 399–406. [Google Scholar] [CrossRef]
- Loshaek, S. Crosslinked Polymers. II. Glass Temperatures of Copolymers of Methyl Methacrylate and Glycol Dimethacrylates. J. Polym. Sci. 1955, 15, 391–404. [Google Scholar] [CrossRef]
- Qu, L.; Nie, Y.; Huang, G.; Weng, G.; Wu, J. Dynamic Fatigue Behavior of Natural Rubber Reinforced with Nanoclay and Carbon Black. J. Macromol. Sci. Part B Phys. 2011, 50, 1646–1657. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Gong, J.P. Why Are Double Network Hydrogels so Tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar] [CrossRef]
- Nakajima, T.; Ozaki, Y.; Namba, R.; Ota, K.; Maida, Y.; Matsuda, T.; Kurokawa, T.; Gong, J.P. Tough Double-Network Gels and Elastomers from the Nonprestretched First Network. ACS Macro Lett. 2019, 8, 1407–1412. [Google Scholar] [CrossRef]
- Kang, B.; Lang, Q.; Tu, J.; Bu, J.; Ren, J.; Lyu, B.; Gao, D. Preparation and Properties of Double Network Hydrogel with High Compressive Strength. Polymers 2022, 14, 966. [Google Scholar] [CrossRef]
- Lee, H.; Hong, H.J.; Ahn, S.; Kim, D.; Kang, S.H.; Cho, K.; Koh, W.G. One-Pot Synthesis of Double-Network PEG/Collagen Hydrogel for Enhanced Adipogenic Differentiation and Retrieval of Adipose-Derived Stem Cells. Polymers 2023, 15, 1777. [Google Scholar] [CrossRef]
- Mashita, K.; Hirooka, M. Alternating copolymers of isobutylene and acrylic ester by complexed copolymerization. Polymers 1995, 36, 2983–2988. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of MAX, EAX, and BAX. (X = 005, 020, and 100 in the case of x = 0.0005, 0.0020, and 0.010, respectively).
Figure 1.
Chemical structures of MAX, EAX, and BAX. (X = 005, 020, and 100 in the case of x = 0.0005, 0.0020, and 0.010, respectively).

Figure 2.
Storage modulus (G′), loss modulus (G″), and tanδ at room temperature for (a) MA020, (b) EA020, and (c) BA020. The G′, G″, and tanδ for (d) MA020 measured at 130 °C, (e) EA020 measured at 90 °C, and (f) BA020 measured at 60 °C.
Figure 2.
Storage modulus (G′), loss modulus (G″), and tanδ at room temperature for (a) MA020, (b) EA020, and (c) BA020. The G′, G″, and tanδ for (d) MA020 measured at 130 °C, (e) EA020 measured at 90 °C, and (f) BA020 measured at 60 °C.

Figure 3.
Tensile testing for elastomer materials. (a) Representative stress-strain curves for MA020, EA020, and BA020. Comparison of (b) fracture stress, (c) Young’s modulus, and (d) fracture energy between MAX, EAX, and BAX. The tests were conducted in triplicate at room temperature, and their average and standard deviation were calculated.
Figure 3.
Tensile testing for elastomer materials. (a) Representative stress-strain curves for MA020, EA020, and BA020. Comparison of (b) fracture stress, (c) Young’s modulus, and (d) fracture energy between MAX, EAX, and BAX. The tests were conducted in triplicate at room temperature, and their average and standard deviation were calculated.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Composition of the reaction solution for the synthesis of the network materials MAX, EAX, and BAX.
Table 1.
Composition of the reaction solution for the synthesis of the network materials MAX, EAX, and BAX.
| Network Material | Monomer (MA, EA, or BA) | Crosslinker (1,4-Butanediol Diacrylate) | Initiator (ADVN) | Solvent (DMF) | |||
|---|---|---|---|---|---|---|---|
| [mL] | [mmol] | [μL] | [mmol] | [mg] | [mmol] | [mL] | |
| MA005 | 1.00 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| MA020 | 1.00 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| MA100 | 1.00 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
| EA005 | 1.19 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| EA020 | 1.19 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| EA100 | 1.19 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
| BA005 | 1.59 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| BA020 | 1.59 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| BA100 | 1.59 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
Table 2.
Glass transition temperatures of the network materials MAX, EAX, and BAX.
| X | Tg of Network Materials [°C] | ||
|---|---|---|---|
| MAX | EAX | BAX | |
| 0 (homopolymer) | 10.041 | −24.041 | −54.041 |
| 005 | 16.5 | −15.1 | −49.0 |
| 020 | 16.1 | −14.7 | −50.2 |
| 100 | 17.6 | −12.1 | −46.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kawano, Y.; Masai, H.; Nakagawa, S.; Yoshie, N.; Terao, J. Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials. Polymers 2023, 15, 2389. https://doi.org/10.3390/polym15102389
AMA Style
Kawano Y, Masai H, Nakagawa S, Yoshie N, Terao J. Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials. Polymers. 2023; 15(10):2389. https://doi.org/10.3390/polym15102389
Chicago/Turabian StyleKawano, Yutaro, Hiroshi Masai, Shintaro Nakagawa, Naoko Yoshie, and Jun Terao. 2023. "Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials" Polymers 15, no. 10: 2389. https://doi.org/10.3390/polym15102389
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.