
Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling 1FUV Peptide
2.2. Calculation of Properties
3. Results
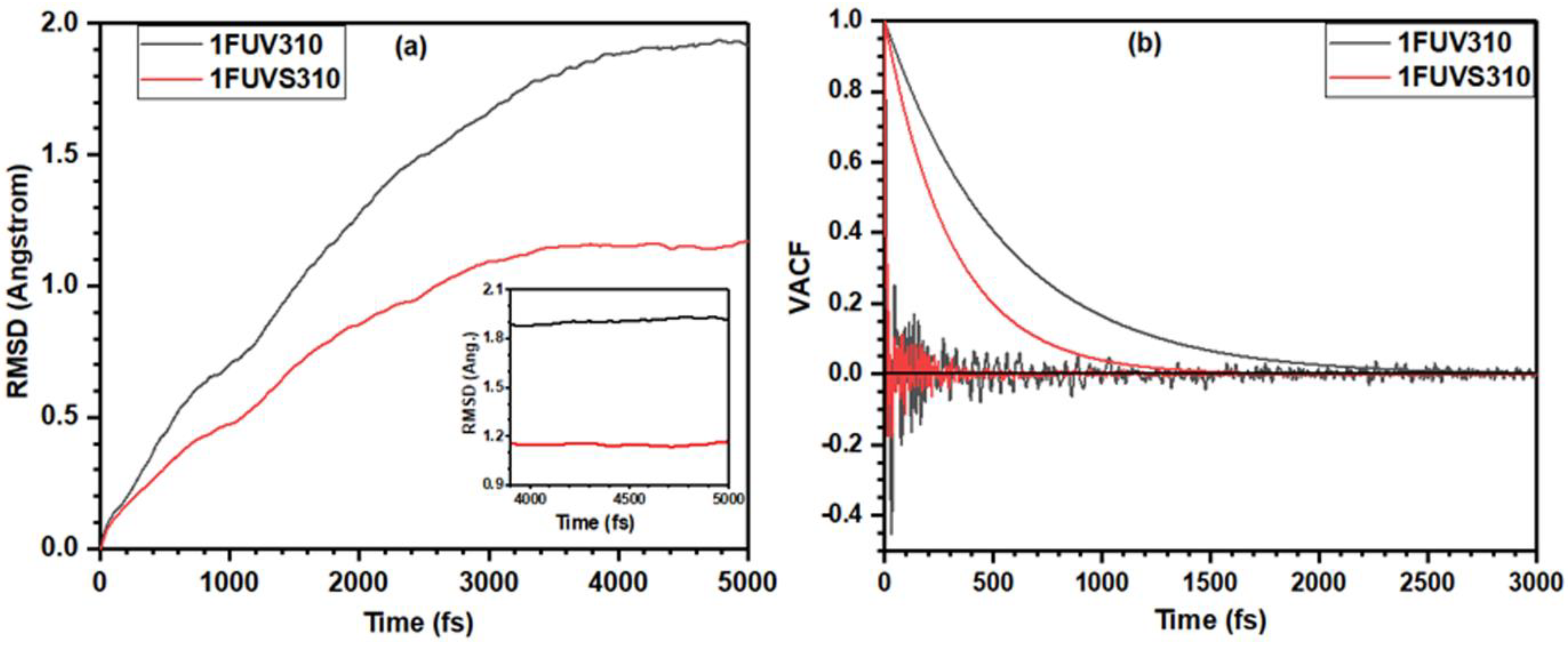
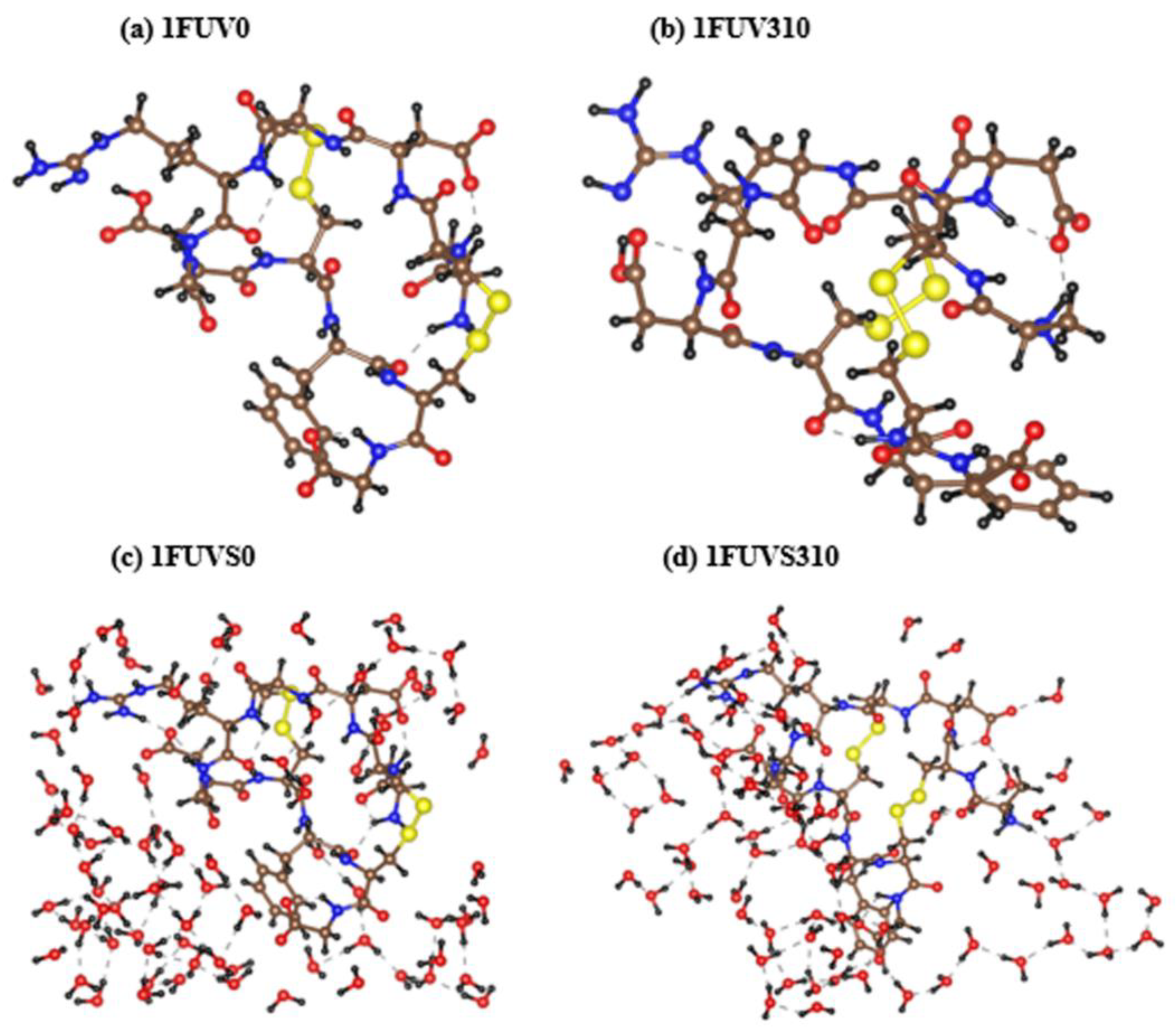
3.1. Analysis of 1FUV Structures
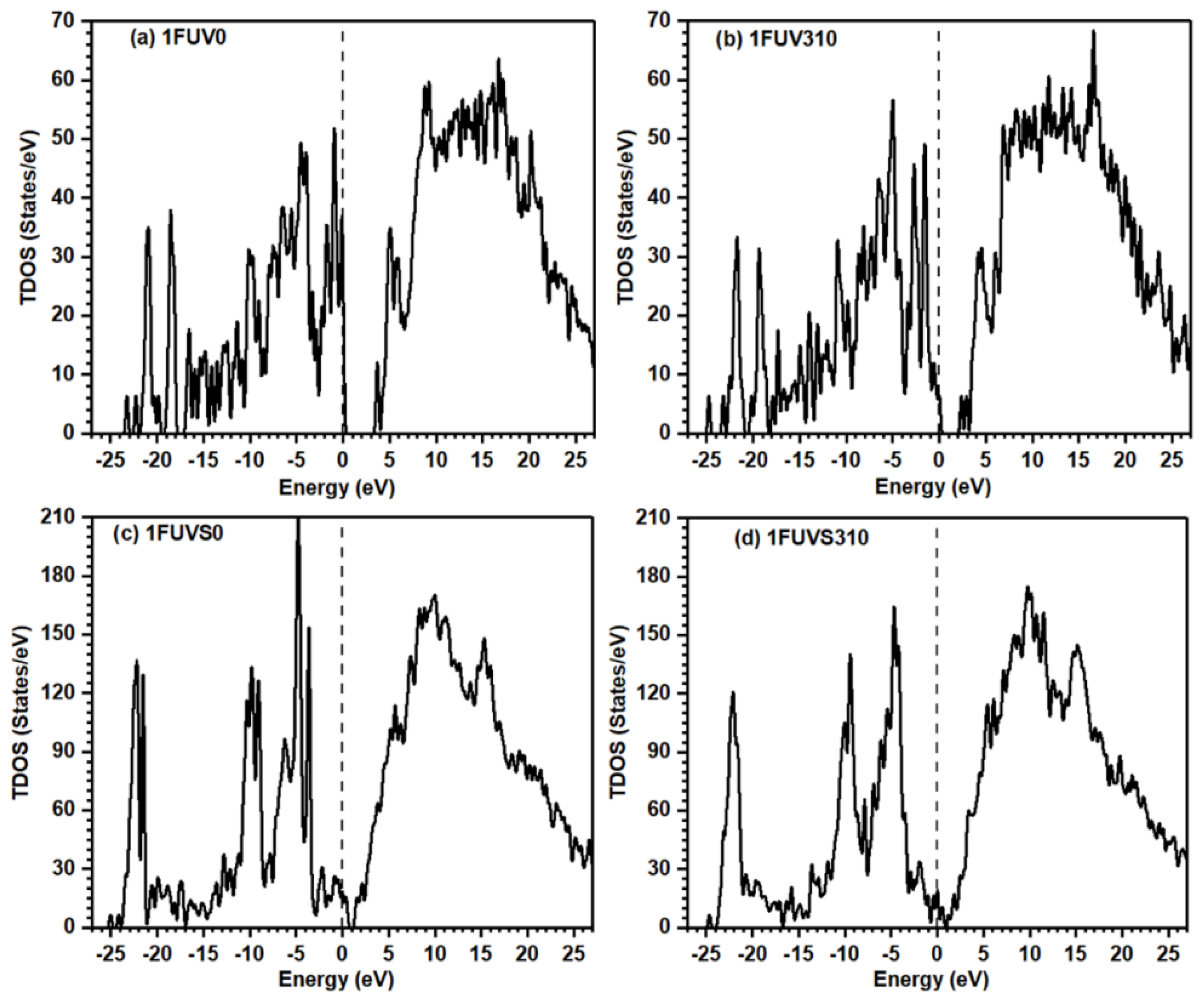
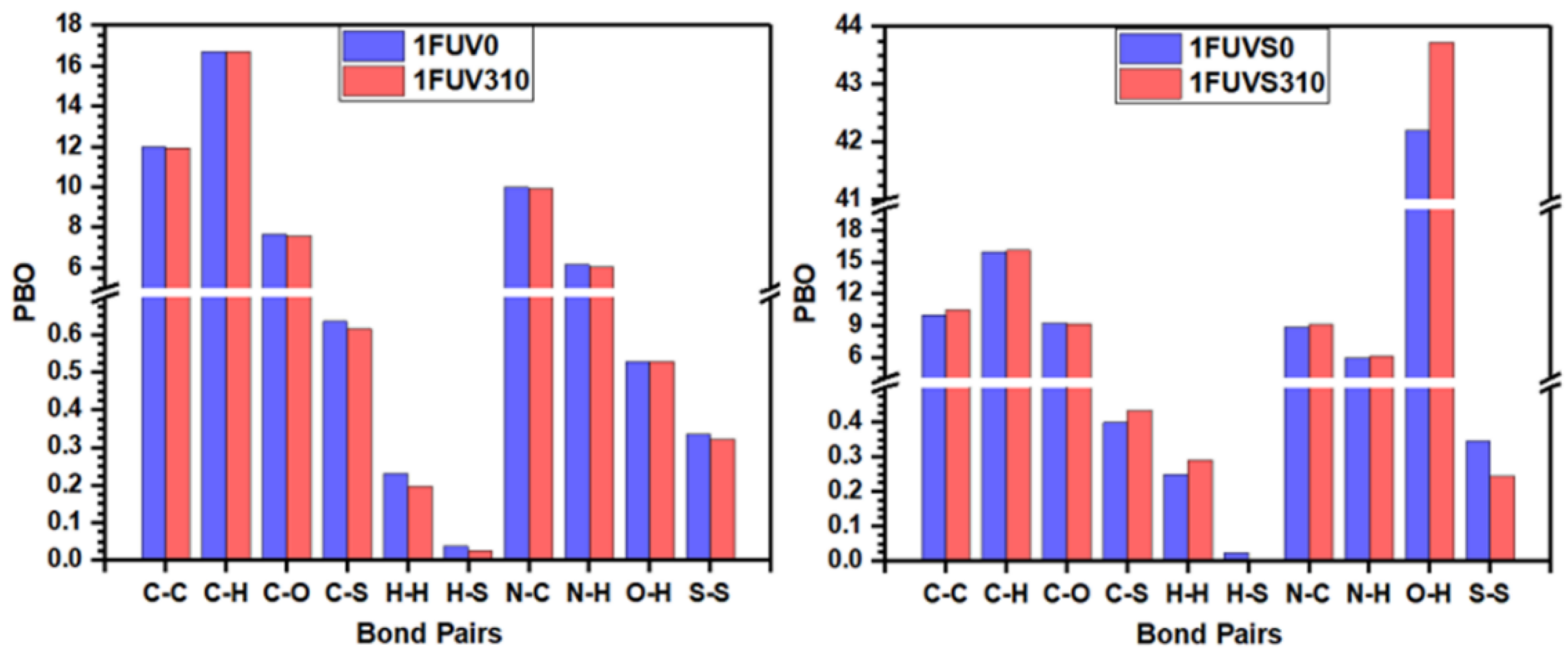
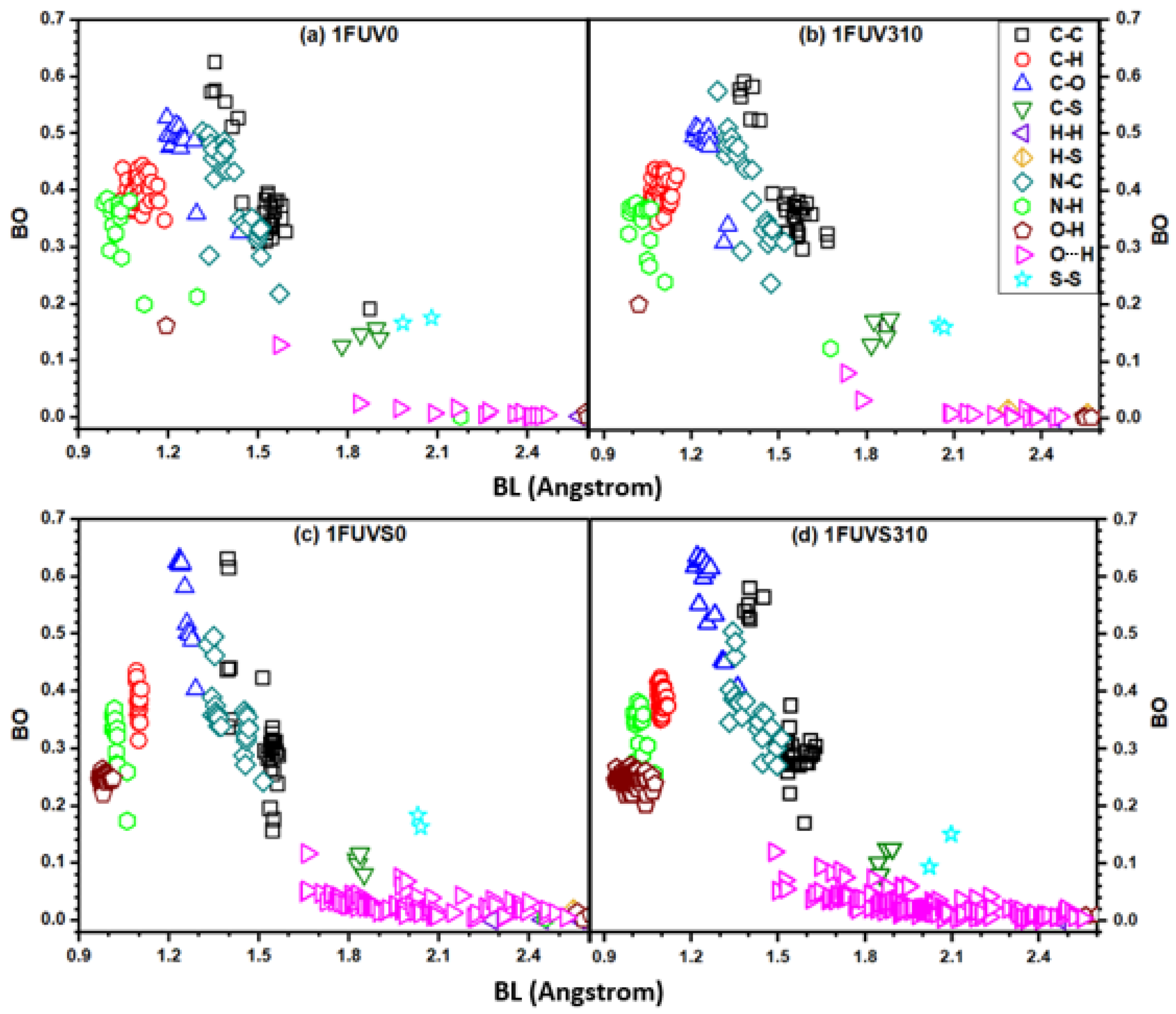
3.2. Electronic Structure and Interatomic Bonding
3.3. Hydrogen Bonding Analysis
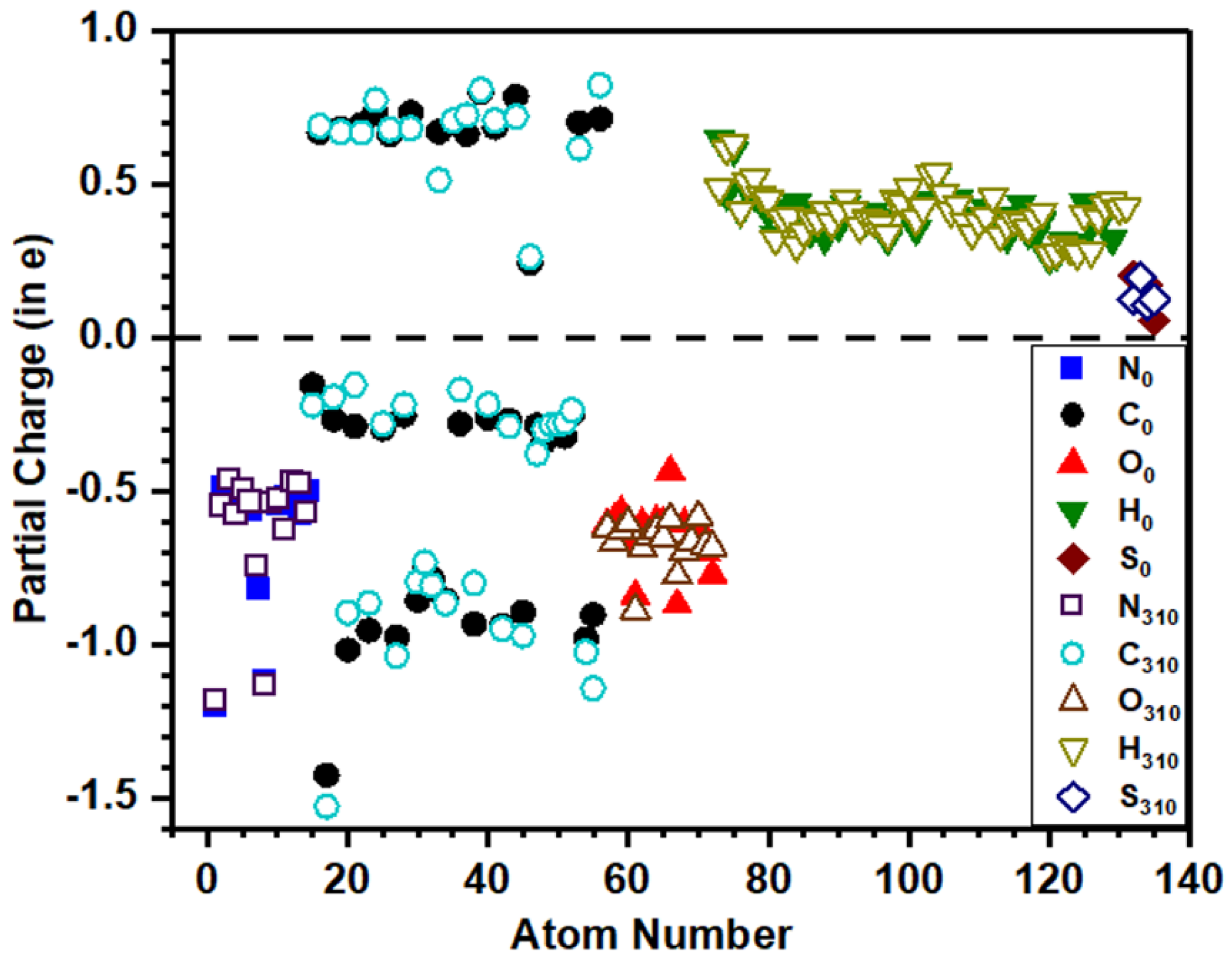
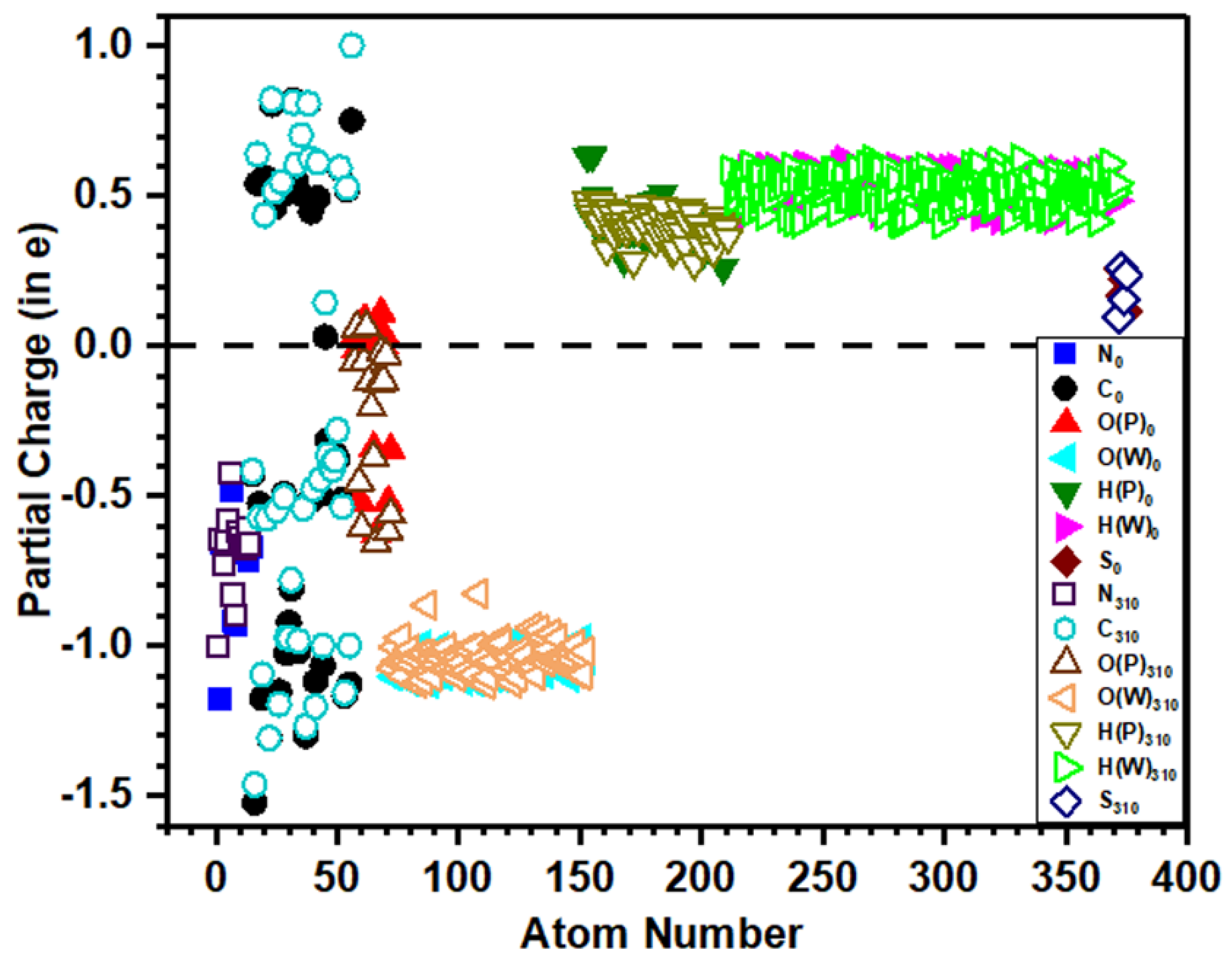
3.4. Partial Charge Distribution
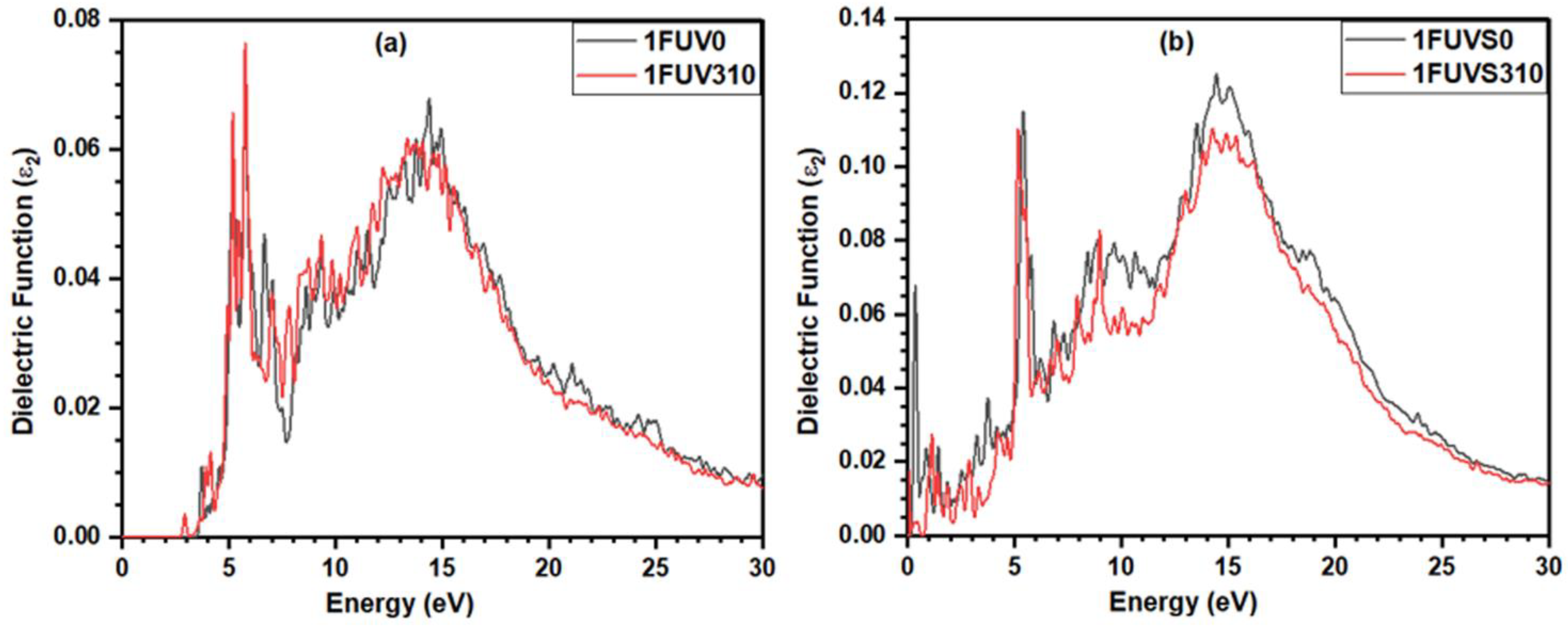
3.5. Dielectric Response
4. Discussions and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alipour, M.; Baneshi, M.; Hosseinkhani, S.; Mahmoudi, R.; Jabari Arabzadeh, A.; Akrami, M.; Mehrzad, J.; Bardania, H. Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review. J. Biomed. Mater. Res. A 2020, 108, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, B.; Meyer, J.; Jonczyk, A.; Kessler, H.; Adamietz, P.; Meenen, N.M.; Kantlehner, M.; Goepfert, C.; Nies, B. RGD-peptides for tissue engineering of articular cartilage. Biomaterials 2002, 23, 3455–3463. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Beer, A.J.; Wester, H.-J. Application of RGD-containing peptides as imaging probes for alphavbeta3 expression. Front. Biosci. 2009, 14, 887–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.Y.; Choi, K.-J.; Lee, C.H.; Kim, S. A multimodal nanoparticle-based cancer imaging probe simultaneously targeting nucleolin, integrin αvβ3 and tenascin-C proteins. Biomaterials 2011, 32, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol. Pharm. 2006, 3, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, S. Radiolabeled cyclic RGD peptide bioconjugates as radiotracers targeting multiple integrins. Biocon. Chem. 2015, 26, 1413–1438. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Le Breton, A.; Préat, V. RGD-based strategies to target Alpha(v) Beta (3) integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Yin, H.-Q.; Bi, F.-L.; Gan, F. Rapid synthesis of cyclic RGD conjugated gold nanoclusters for targeting and fluorescence imaging of Melanoma A375 Cells. Biocon. Chem. 2015, 26, 243–249. [Google Scholar] [CrossRef]
- Fu, S.; Xu, X.; Ma, Y.; Zhang, S.; Zhang, S. RGD peptide-based non-viral gene delivery vectors targeting integrin αvβ3 for cancer therapy. J. Drug Target. 2019, 27, 1–11. [Google Scholar] [CrossRef]
- Sun, Y.; Kang, C.; Liu, F.; Zhou, Y.; Luo, L.; Qiao, H. RGD peptide-based target drug delivery of doxorubicin nanomedicine. Drug Dev. Res. 2017, 78, 283–291. [Google Scholar] [CrossRef]
- Nikfar, Z.; Shariatinia, Z. Tripeptide arginyl-glycyl-aspartic acid (RGD) for delivery of cyclophosphamide anticancer drug: A computational approach. Int. J. Nano Dimen. 2020, 11, 312–336. [Google Scholar]
- Nikfar, Z.; Shariatinia, Z. The RGD tripeptide anticancer drug carrier: DFT computations and molecular dynamics simulations. J. Mol. Liq. 2019, 281, 565–583. [Google Scholar] [CrossRef]
- Jin, H.; Varner, J. Integrins: Roles in cancer development and as treatment targets. Br. J. Cancer 2004, 90, 561–565. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Zeng, X.; Meng, Q.-F.; Zhang, X.-Z.; Cheng, S.-X.; Zhuo, R.-X. The influence of RGD addition on the gene transfer characteristics of disulfide-containing polyethyleneimine/DNA complexes. Biomaterials 2008, 29, 4356–4365. [Google Scholar] [CrossRef] [PubMed]
- Temming, K.; Schiffelers, R.; Molema, G.; Kok, R.J. RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumour vasculature. Drug Resist. Updates 2005, 8, 381–402. [Google Scholar] [CrossRef]
- Assa-Munt, N.; Jia, X.; Laakkonen, P.; Ruoslahti, E. Solution structures and integrin binding activities of an RGD peptide with two isomers. Biochemistry 2001, 40, 2373–2378. [Google Scholar] [CrossRef]
- Ruoslahti, E.; Pierschbacher, M.D. New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef]
- Paradise, R.K.; Lauffenburger, D.A.; Van Vliet, K.J. Acidic extracellular pH promotes activation of integrin αvβ3. PLoS ONE 2011, 6, e15746. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.; Gao, M.; Schulten, K.; Vogel, V. Structural insights into how the MIDAS ion stabilizes integrin binding to an RGD peptide under force. Structure 2004, 12, 2049–2058. [Google Scholar] [CrossRef] [Green Version]
- Ruoslahti, E. Integrins. J. Clin. Investig. 1991, 87, 1–5. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tiss. Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, Y.; Liang, R.; An, X.; Wang, K.; Shen, G.; Tu, Y.; Zhu, J.; Tao, J. Recent advances in targeted nanoparticles drug delivery to melanoma. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 769–794. [Google Scholar] [CrossRef]
- Marinelli, L.; Lavecchia, A.; Gottschalk, K.-E.; Novellino, E.; Kessler, H. Docking studies on αvβ3 integrin ligands: Pharma-cophore refinement and implications for drug design. J. Med. Chem. 2003, 46, 4393–4404. [Google Scholar] [CrossRef]
- Li, N.; Qiu, S.; Fang, Y.; Wu, J.; Li, Q. Comparison of Linear vs. Cyclic RGD Pentapeptide Interactions with Integrin αvβ3 by molecular dynamics simulations. Biology 2021, 10, 688. [Google Scholar] [CrossRef]
- Damjanovic, J.; Miao, J.; Huang, H.; Lin, Y.-S. Elucidating solution structures of cyclic peptides using molecular dynamics simulations. Chem. Rev. 2021, 121, 2292–2324. [Google Scholar] [CrossRef] [PubMed]
- Muir, J.; Costa, D.; Idriss, H. DFT computational study of the RGD peptide interaction with the rutile TiO2 (110) surface. Surf. Sci. 2014, 624, 8–14. [Google Scholar] [CrossRef]
- Xu, M.; Zhu, T.; Zhang, J.Z.H. A force balanced fragmentation method for ab initio molecular dynamic simulation of protein. Front. Chem. 2018, 6, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isborn, C.M.; Mar, B.D.; Curchod, B.F.E.; Tavernelli, I.; Martínez, T.J. The charge transfer problem in density functional theory calculations of aqueously solvated molecules. J. Phys. Chem. B 2013, 117, 12189–12201. [Google Scholar] [CrossRef]
- Ufimtsev, I.S.; Luehr, N.; Martinez, T.J. Charge transfer and polarization in solvated proteins from ab initio molecular dynamics. J. Phys. Chem. Let. 2011, 2, 1789–1793. [Google Scholar] [CrossRef]
- Wei, D.; Guo, H.; Salahub, D. Conformational dynamics of an alanine dipeptide analog: An ab initio molecular dynamics study. Phys. Rev. E 2001, 64, 011907. [Google Scholar] [CrossRef]
- Jois, S.D.; Tambunan, U.S.; Chakrabarti, S.; Siahaan, T.J. Solution structure of a cyclic RGD peptide that inhibits platelet ag-gregation. J. Biomol. Struct. Dyn. 1996, 14, 1–11. [Google Scholar] [CrossRef]
- Portela, A.; Yano, T.-A.; Santschi, C.; Martin, O.J.F.; Tabata, H.; Hara, M. Highly sensitive SERS analysis of the cyclic Arg-Gly-Asp peptide ligands of cells using nanogap antennas. J. Biophotonics 2016, 10, 294–302. [Google Scholar] [CrossRef]
- Stevens, J.S.; de Luca, A.C.; Pelendritis, M.; Terenghi, G.; Downes, S.; Schroeder, S.L.M. Quantitative analysis of complex amino acids and RGD peptides by X-ray photoelectron spectroscopy (XPS). Surf. Interf. Anal. 2013, 45, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Huang, G.; Chen, L.; Wu, W. Studies on the conformations and Hydrogen-bonding interactions of RGD tri-peptide in aqueous solutions by molecular dynamics simulations and 2D-NOESY spectroscopy. J. Solut. Chem. 2015, 44, 1281–1291. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.E.; Wuest, W.M.; Voelz, V.A. Molecular simulation of conformational pre-organization in cyclic RGD peptides. J. Chem. Inf. Model. 2015, 55, 806–813. [Google Scholar] [CrossRef]
- Adhikari, P.; Wen, A.M.; French, R.H.; Parsegian, V.A.; Steinmetz, N.F.; Podgornik, R.; Ching, W.-Y. Electronic structure, dielectric response and surface charge distribution of RGD (1FUV) peptide. Sci. Rep. 2014, 4, 5605. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Strömblad, S.; Klemke, R.; Visscher, D.; Sarkar, F.H.; Cheresh, D.A. Antiintegrin alpha v beta 3 blocks human breast cancer growth and angiogenesis in human skin. J. Clin. Investig. 1995, 96, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.-P.; Brownlee, M.; Jonczyk, A.; Sutter, A.; Preissner, K.T. Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor–type integrins inhibits retinal neovascularization. Nat. Med. 1996, 2, 529–533. [Google Scholar] [CrossRef]
- Yamamoto, M.; Fisher, J.E.; Gentile, M.; Seedor, J.G.; Leu, C.-T.; Rodan, S.B.; Rodan, G.A. The integrin ligand echistatin prevents bone loss in ovariectomized mice and rats. Endocrinology 1998, 139, 1411–1419. [Google Scholar] [CrossRef]
- Arap, W.; Pasqualini, R.; Ruoslahti, E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Sciences 1998, 279, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Solution Structure of an RGD Peptide Isomer-A. Available online: https://www.rcsb.org/structure/1FUV (accessed on 7 June 2021).
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 169–186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Vienna Ab-Initio Simulation Package (VASP); University of Vienna: Vienna, Austria, 2001. [Google Scholar]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Eifler, J.; Podgornik, R.; Steinmetz, N.F.; French, R.H.; Parsegian, V.A.; Ching, W. Charge distribution and hydrogen bonding of a collagen α2-chain in vacuum, hydrated, neutral, and charged structural models. Int. J. Quantum Chem. 2016, 116, 681–691. [Google Scholar] [CrossRef]
- Poudel, L.; Steinmetz, N.F.; French, R.; Parsegian, V.A.; Podgornik, R.; Ching, W.-Y. Implication of the solvent effect, metal ions and topology in the electronic structure and hydrogen bonding of human telomeric G-quadruplex DNA. Phys. Chem. Chem. Phys. 2016, 18, 21573–21585. [Google Scholar] [CrossRef] [PubMed]
- Poudel, L.; Tamerler, C.; Misra, A.; Ching, W.-Y. Atomic-scale quantification of interfacial binding between peptides and in-organic crystals: The case of calcium carbonate binding peptide on aragonite. J. Phys. Chem. C 2017, 121, 28354–28363. [Google Scholar] [CrossRef]
- Jawad, B.; Poudel, L.; Podgornik, R.; Steinmetz, N.F.; Ching, W.-Y. Molecular mechanism and binding free energy of doxoru-bicin intercalation in DNA. Phys. Chem. Chem. Phys. 2019, 21, 3877–3893. [Google Scholar] [CrossRef]
- Adhikari, P.; Ching, W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020, 10, 39831–39841. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Shin, M.; Steinmetz, N.F.; Twarock, R.; Podgornik, R.; Ching, W.-Y. Intra-and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: Implication for ACE2 receptor binding. Phys. Chem. Chem. Phys. 2020, 22, 18272–18283. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Ultra-large-scale ab initio quantum chemical computation of bio-molecular systems: The case of spike protein of SARS-CoV-2 virus. Comput. Struct. Biotech. J. 2021, 19, 1288–1301. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: Combination of molecular dynamic simulation and density functional calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Poudel, L.; Podgornik, R.; Ching, W.-Y. Thermodynamic dissection of the intercalation binding process of doxoru-bicin to dsDNA with implications of ionic and solvent effects. J. Phys. Chem. B 2020, 124, 7803–7818. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. II. overlap populations, bond orders, and covalent bond energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Dharmawardhana, C.C.; Misra, A.; Ching, W.-Y. Quantum mechanical metric for internal cohesion in cement crystals. Sci. Rep. 2014, 4, 7332. [Google Scholar] [CrossRef]
- Wang, Y.; Goh, S.-Y.; Kuczera, K. Molecular dynamics study of disulfide bond influence on properties of an RGD peptide. J. Pept. Res. 1999, 53, 188–200. [Google Scholar] [CrossRef]
- Löffler, G.; Schreiber, H.; Steinhauser, O. Calculation of the dielectric properties of a protein and its solvent: Theory and a case study. J. Mol. Biol. 1997, 270, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Schutz, C.N.; Warshel, A. What are the dielectric?constants? of proteins and how to validate electrostatic models? Proteins Struct. Funct. Bioinform. 2001, 44, 400–417. [Google Scholar] [CrossRef]
- King, G.; Lee, F.S.; Warshel, A. Microscopic simulations of macroscopic dielectric constants of solvated proteins. J. Chem. Phys. 1991, 95, 4366–4377. [Google Scholar] [CrossRef]
- Gilson, M.K.; Honig, B.H. Calculation of electrostatic potentials in an enzyme active site. Nature 1987, 330, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Rulis, P.; Ouyang, L.; Ching, W.Y. Structure and properties of the low-density phase ι-Al2O3 from first principles. Phys. Rev. B 2011, 84, 174123. [Google Scholar] [CrossRef] [Green Version]
- Baral, K.; Adhikari, P.; Ching, W.Y. Ab initio modeling of the electronic structures and physical properties of a-Si1− xGexO2 Glass (x = 0 to 1). J. Am. Ceram. Soc. 2016, 99, 3677–3684. [Google Scholar] [CrossRef]
- Hasan, S.; Baral, K.; Li, N.; Ching, W.-Y. Structural and physical properties of 99 complex bulk chalcogenides crystals using first-principles calculations. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Hussain, A.; Mehmood, S.; Rasool, M.; Aryal, S.; Rulis, P.; Ching, W. Electronic structure, mechanical, and optical properties of CaO Al2O3 system: A first principles approach. Ind. J. Phys. 2016, 90, 917–929. [Google Scholar] [CrossRef]
- Rajter, R.F.; French, R.; Ching, W.Y.; Carter, W.C.; Chiang, Y.M. Calculating van der Waals-London dispersion spectra and Hamaker coefficients of carbon nanotubes in water from ab initio optical properties. J. Appl. Phys. 2007, 101, 054303. [Google Scholar] [CrossRef]
- Baral, K.; Li, A.; Ching, W.-Y. Ab initio modeling of structure and properties of single and mixed alkali silicate glasses. J. Phys. Chem. A 2017, 121, 7697–7708. [Google Scholar] [CrossRef] [PubMed]
- Baral, K.; Li, A.; Ching, W.-Y. Ab initio molecular dynamics simulation of Na-doped aluminosilicate glasses and glass-water interaction. AIP Adv. 2019, 9, 075218. [Google Scholar] [CrossRef] [Green Version]
- Baral, K.; Li, A.; Ching, W.-Y. Ab initio study of hydrolysis effects in single and ion-exchanged alkali aluminosilicate glasses. J. Phys. Chem. B 2020, 124, 8418–8433. [Google Scholar] [CrossRef] [PubMed]
- Baral, K.; San, S.; Sakidja, R.; Couet, A.; Sridharan, K.; Ching, W.-Y. Temperature-dependent properties of molten Li2BeF4 Salt using Ab initio molecular dynamics. ACS Omega 2021, 6, 19822–19835. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | TBO | PBO | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C‒C | C‒H | C‒O | C‒S | H‒H | H‒S | H‒O | N‒C | N‒H | N‒O | S‒S | ||
| 1FUV0 | 54.272 | 11.991 | 16.680 | 7.669 | 0.637 | 0.232 | 0.037 | 0.529 | 9.979 | 6.181 | 0.001 | 0.336 |
| 1FUV310 | 53.887 | 11.944 | 16.705 | 7.581 | 0.617 | 0.198 | 0.027 | 0.530 | 9.926 | 6.034 | 0.000 | 0.323 |
| 1FUVS0 | 93.166 | 9.990 | 15.947 | 9.234 | 0.400 | 0.251 | 0.025 | 42.207 | 8.859 | 5.905 | 0.003 | 0.346 |
| 1FUVS310 | 95.663 | 10.479 | 16.119 | 9.125 | 0.434 | 0.292 | 0.005 | 43.724 | 9.111 | 6.119 | 0.000 | 0.246 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baral, K.; Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics. Polymers 2021, 13, 3434. https://doi.org/10.3390/polym13193434
Baral K, Adhikari P, Jawad B, Podgornik R, Ching W-Y. Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics. Polymers. 2021; 13(19):3434. https://doi.org/10.3390/polym13193434
Chicago/Turabian StyleBaral, Khagendra, Puja Adhikari, Bahaa Jawad, Rudolf Podgornik, and Wai-Yim Ching. 2021. "Solvent Effect on the Structure and Properties of RGD Peptide (1FUV) at Body Temperature (310 K) Using Ab Initio Molecular Dynamics" Polymers 13, no. 19: 3434. https://doi.org/10.3390/polym13193434