
New Principles of Polymer Composite Preparation. MQ Copolymers as an Active Molecular Filler for Polydimethylsiloxane Rubbers

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
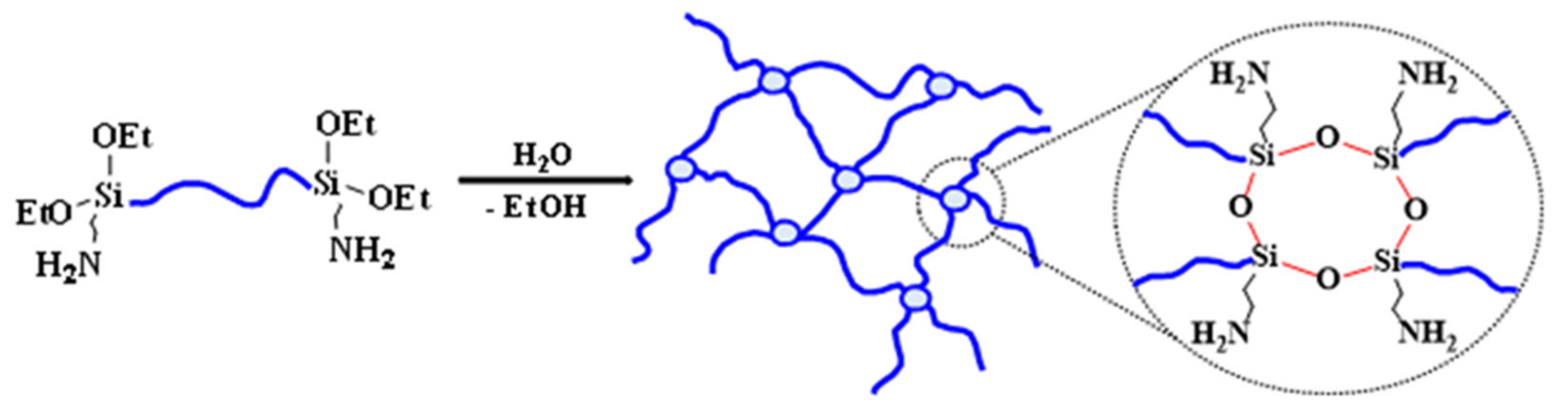
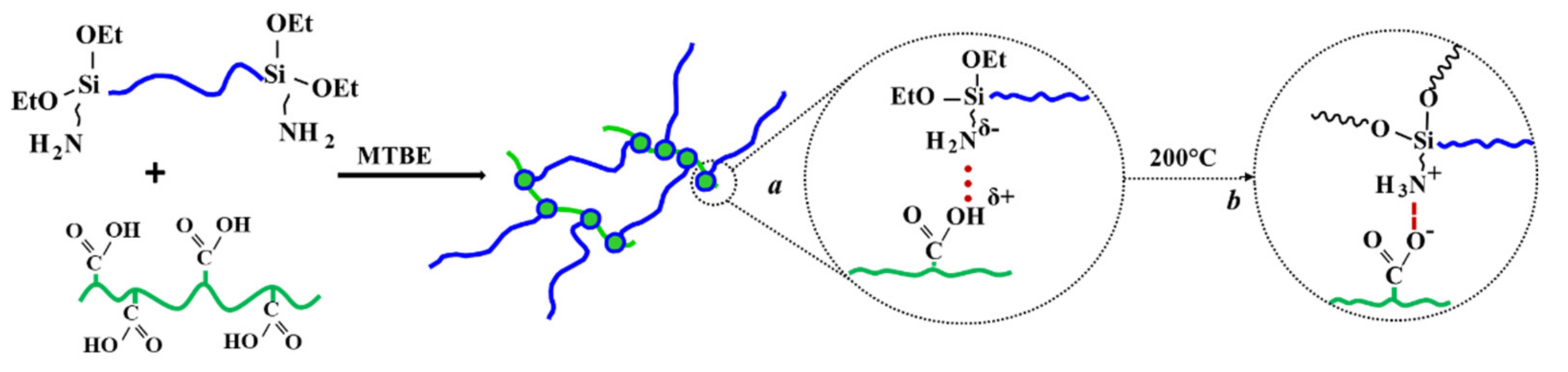
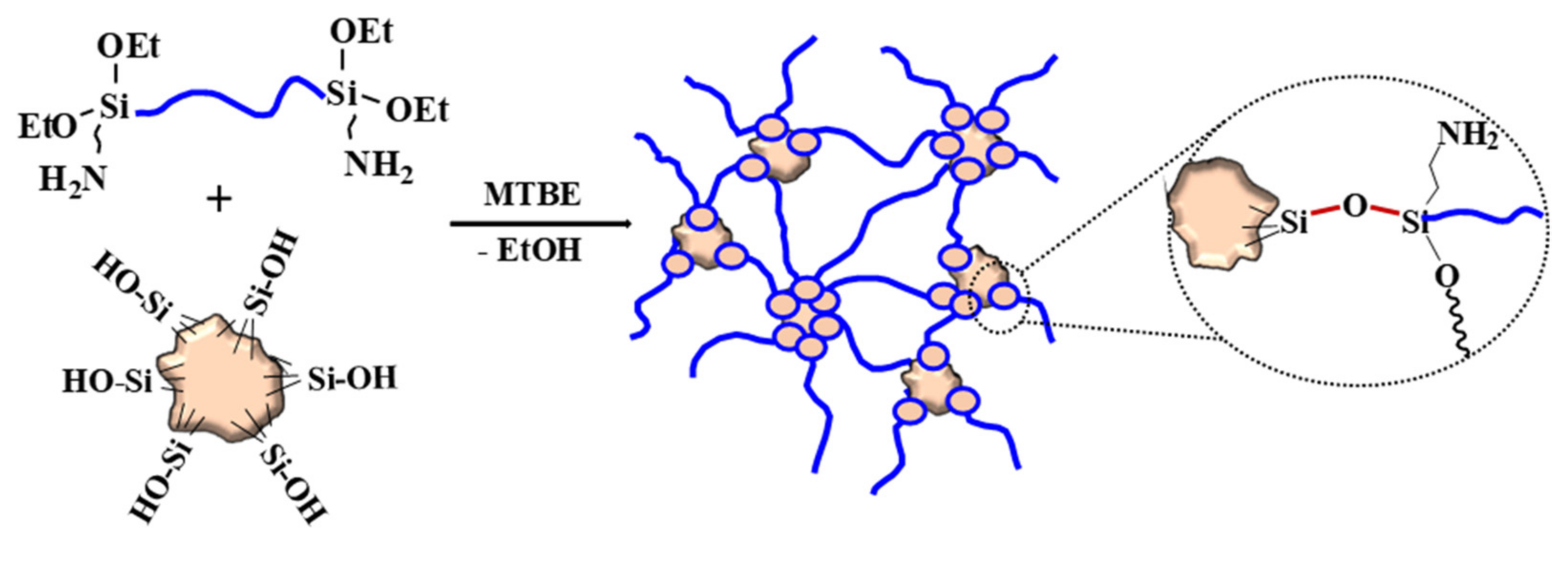
3.1. Preparation of Elastomeric Composites
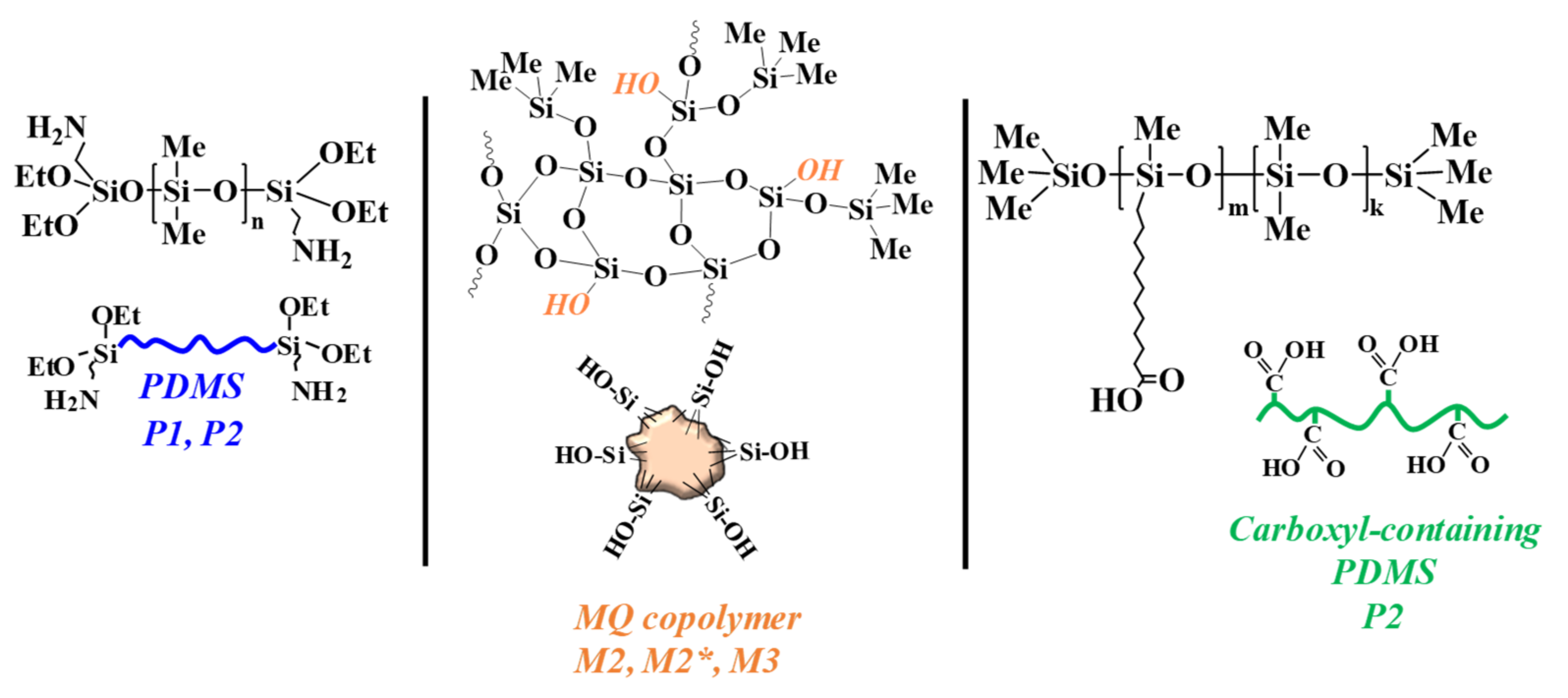
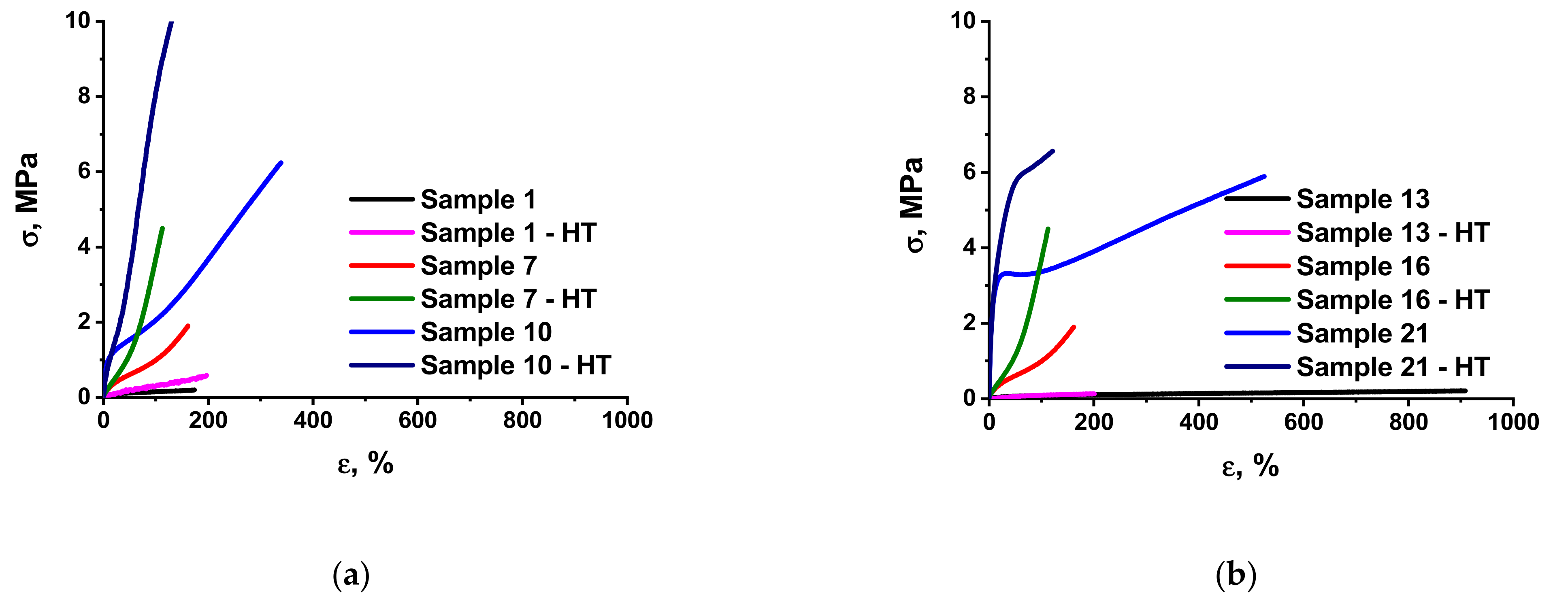
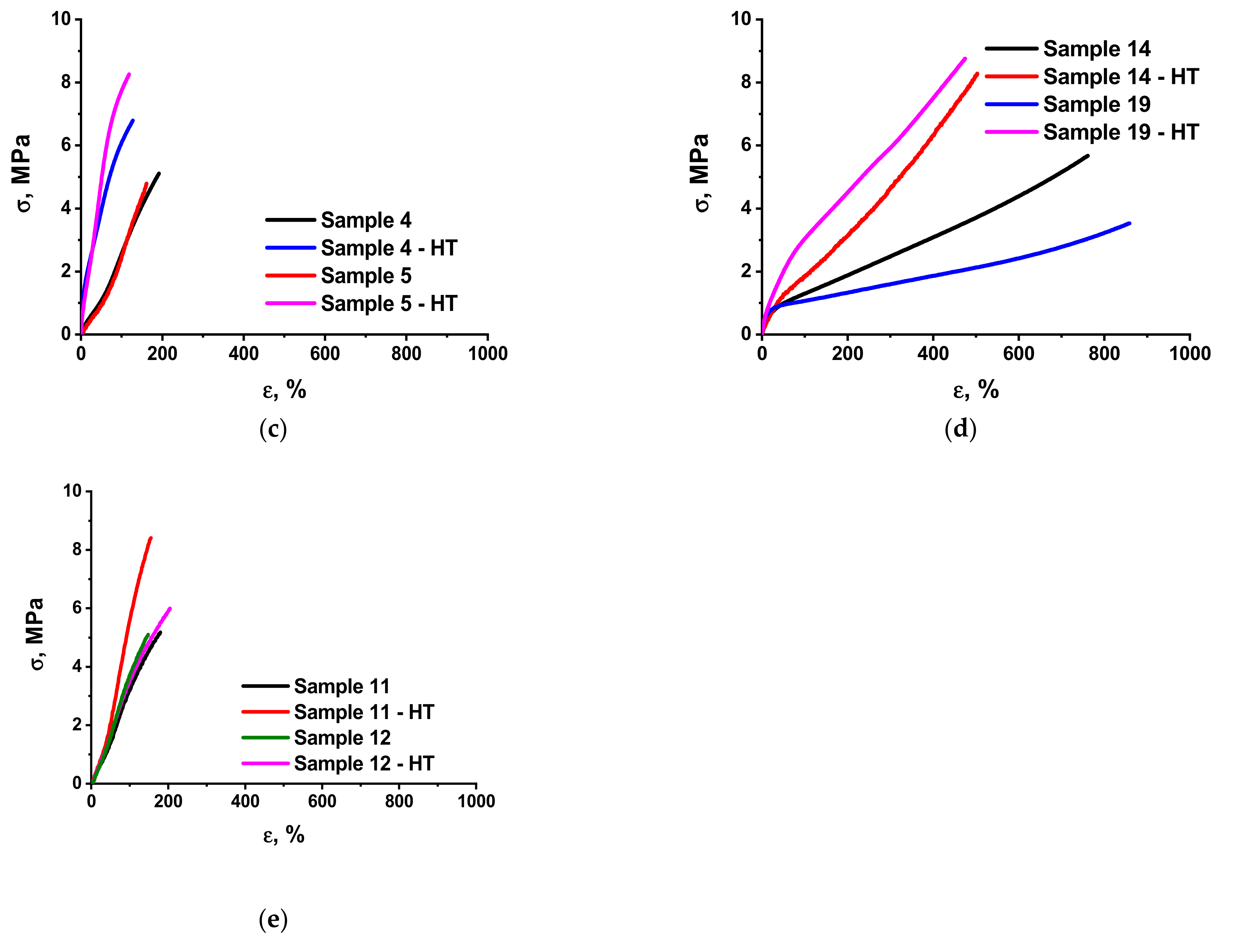
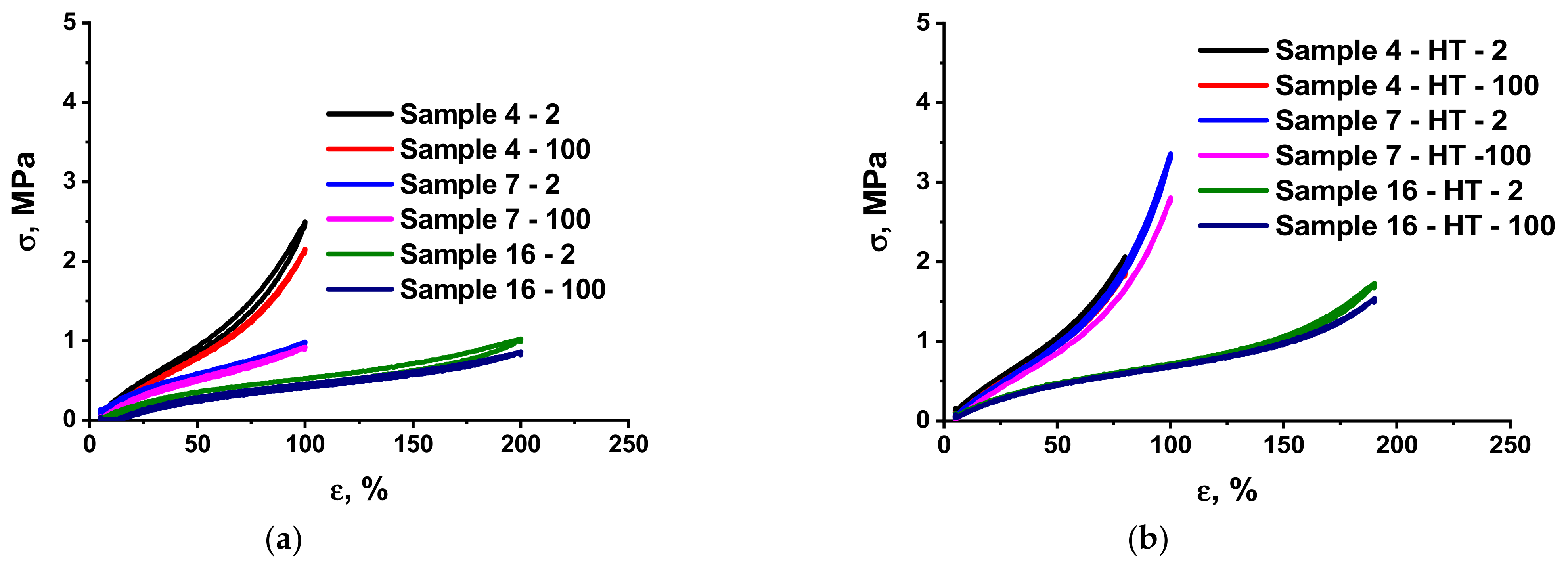
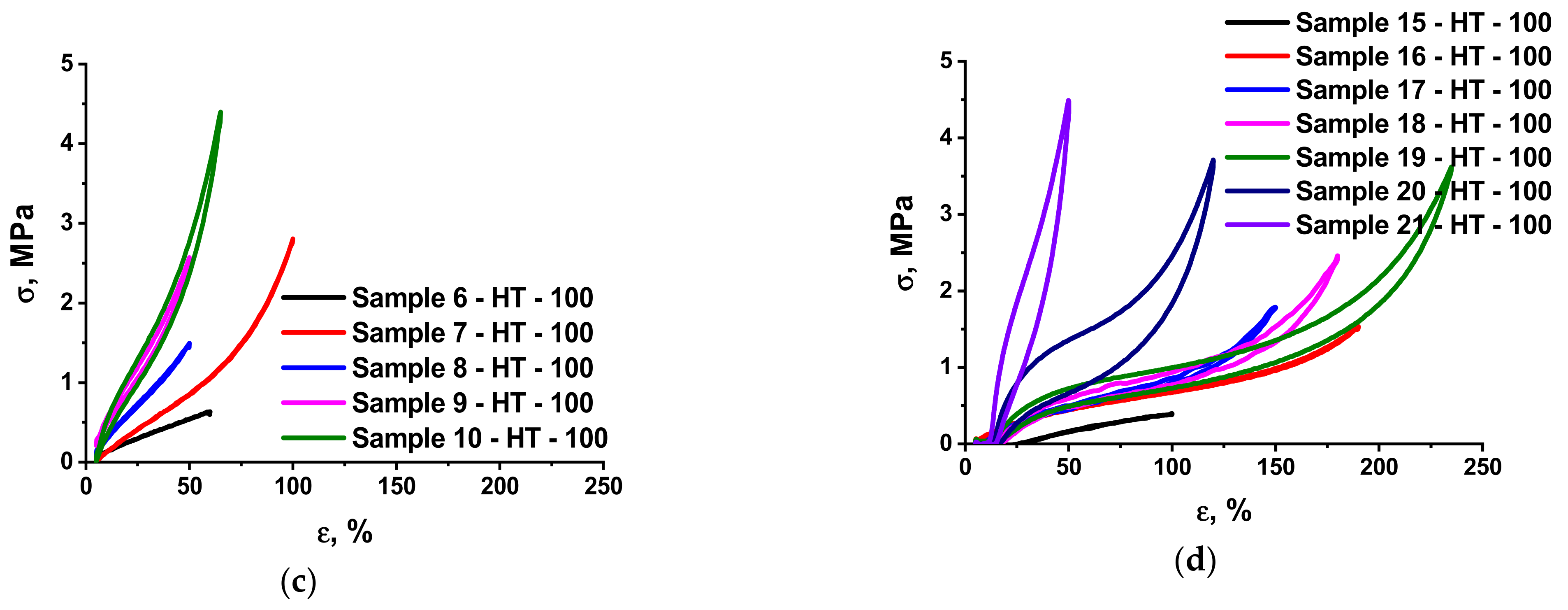
3.2. Mechanical Characteristics of the Elastomer Composites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Liu, X.; Kirlikovali, K.O.; Chen, Z.; Ma, K.; Idrees, K.B.; Cao, R.; Zhang, X.; Islamoglu, T.; Liu, Y.; Farha, O.K. Small Molecules, Big Effects: Tuning Adsorption and Catalytic Properties of Metal–Organic Frameworks. Chem. Mat. 2021, 33, 1444–1454. [Google Scholar] [CrossRef]
- Selezneva, E.V.; Bakirov, A.V.; Sedush, N.G.; Bystrova, A.V.; Chvalun, S.N.; Demco, D.E.; Möller, M. How Shape Memory Effects Can Contribute to Improved Self-Healing Properties in Polymer Materials. Macromolecules 2021, 54, 2506–2517. [Google Scholar] [CrossRef]
- Kumar, V.; Alam, M.N.; Manikkavel, A.; Song, M.; Lee, D.J.; Park, S.S. Silicone Rubber Composites Reinforced by Carbon Nanofillers and Their Hybrids for Various Applications: A Review. Polymers 2021, 13, 2322. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shepelin, N.A.; Sherrell, P.C.; Ellis, A.V. Poly (dimethylsiloxane) for Triboelectricity: From Mechanisms to Practical Strategies. Chem. Mater. 2021, 33, 4304–4327. [Google Scholar] [CrossRef]
- Whelan, A.; Lee, K.S. (Eds.) Developments in Rubber Technology—2: Synthetic Rubbers; Springer: Dordrecht, The Netherlands, 1981. [Google Scholar]
- Shit, S.C.; Shah, P. A review on silicone rubber. Natl. Acad. Sci. Lett. 2013, 36, 355–365. [Google Scholar] [CrossRef]
- Mark, J.E. (Ed.) Polymer Datahandbook, 2nd ed.; Oxford University Press, Inc.: New York, NY, USA, 2009; p. 539. [Google Scholar]
- Mark, J.E.; Pan, S.J. Reinforcement of polydimethylsiloxane networks by in-situ precipitation of silica: A new method for preparation of filled elastomers. Die Makromol. Chem. Rapid Commun. 1982, 3, 681–685. [Google Scholar] [CrossRef]
- Sun, C.C.; Mark, J.E. Comparisons among the reinforcing effects provided by various silica-based fillers in a siloxane elastomer. Polymer 1989, 30, 104–106. [Google Scholar] [CrossRef]
- Schaefer, D.W.; Mark, J.E.; Mccarthy, D.; Jian, L.; Sun, C.C.; Farago, B. Structure of microphase-separated silica/siloxane molecular composites. MRS Online Proc. Libr. Arch. 1989, 171, 57–63. [Google Scholar] [CrossRef]
- Sohoni, G.B.; Mark, J.E. Thermal stability of in situ filled siloxane elastomers. J. Appl. Polym. Sci. 1992, 45, 1763–1775. [Google Scholar] [CrossRef]
- Schaefer, D.W.; Mark, J.E.; Jian, L.; Sun, C.C.; McCarthy, D.W.; Jiang, C.Y.; Spooner, S. Structure-property relationships in silica-siloxane molecular composites. In Ultrastructure Processing of Advanced Materials; Wiley: New York, NY, USA, 1992; pp. 361–375. [Google Scholar]
- Mark, J.E. Hybrid Organic-Inorganic Composites; Lee, C.Y.C., Biancini, P.A., Eds.; American Chemical Society: Washington, DC, USA, 1995; p. 585. [Google Scholar]
- Zhou, W.; Mark, J.E.; Unroe, M.R.; Arnold, F.E. Toughening of a high-temperature polymer by the sol–gel, in situ generation of a rubbery silica–siloxane phase. J. Appl. Polym. Sci. 2001, 79, 2326–2330. [Google Scholar] [CrossRef]
- Schaefer, D.W.; Vu, B.T.; Mark, J.E. The effect of interphase coupling on the structure and mechanical properties of silica-siloxane composites. Rubber Chem. Technol. 2002, 75, 795–810. [Google Scholar] [CrossRef]
- Patwardhan, S.V.; Taori, V.P.; Hassan, M.; Agashe, N.R.; Franklin, J.E.; Beaucage, G.; Clarson, S.J. An investigation of the properties of poly (dimethylsiloxane)-bioinspired silica hybrids. Eur. Polym. J. 2006, 42, 167–178. [Google Scholar] [CrossRef]
- Ibrahim, I.A.M.; Zikry, A.A.F.; Sharaf, M.A.; Mark, J.E.; Jacob, K.; Jasiuk, I.M.; Tannenbaumn, R. Elastic behavior of silica/poly (dimethylsiloxane) nanocomposites: Nano-size effects. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Bedfordshire, UK, 2–4 July 2012; Volume 40, p. 012008. [Google Scholar]
- Tebeneva, N.A.; Meshkov, I.B.; Tarasenkov, A.N.; Polshchikova, N.V.; Kalinina, A.A.; Buzin, M.I.; Serenko, O.A.; Zubavichus, Y.V.; Katsoulis, D.E.; Muzafarov, A.M. Polyfunctional branched metallosiloxane oligomers and composites based on them. J. Organomet. Chem. 2018, 868, 112–121. [Google Scholar] [CrossRef]
- Tarasenkov, A.N.; Tebeneva, N.A.; Parshina, M.S.; Meshkov, I.B.; Vasilenko, N.G.; Cherkaev, G.V.; Goncharuk, G.P.; Katsoulis, D.E.; Muzafarov, A.M. New functional metallosiloxanes with partially siloxy substituted metall atom and their use in silicone compositions. J. Organomet. Chem. 2020, 906, 121034. [Google Scholar] [CrossRef]
- Tatarinova, E.; Vasilenko, N.; Muzafarov, A. Synthesis and Properties of MQ Copolymers: Current State of Knowledge. Molecules 2017, 22, 1768. [Google Scholar] [CrossRef] [Green Version]
- Arkles, B. Commercial applications of sol-gel-derived hybrid materials. MRS Bull. 2001, 26, 402–408. [Google Scholar] [CrossRef]
- Amouroux, N.; Petit, J.; Leger, L. Role of Interfacial Resistance to Shear Stress on Adhesive Peel Strength. Langmuir 2001, 17, 6510–6517. [Google Scholar] [CrossRef]
- Di, M.; He, S.; Li, R.; Yang, D. Radiation effect of 150 keV protons on methyl silicone rubber reinforced with MQ silicone resin. Nucl. Instrum. Method Phys. Res. B 2006, 248, 31–36. [Google Scholar] [CrossRef]
- Xiang, H.; Ge, J.; Cheng, S.; Han, H.; Cui, S. Synthesis and characterization of titania/MQ silicone resin hybrid nanocomposite via sol–gel process. J. Sol-Gel Sci. Technol. 2011, 59, 635–639. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Z.; Yang, Y. Toughening of Poly(l-lactide) with Methyl MQ Silicone Resin. Eur. Polym. J. 2014, 50, 243–248. [Google Scholar] [CrossRef]
- Jia, P.; Liu, H.; Liu, Q.; Cai, X. Thermal degradation mechanism and flame retardancy of MQ silicone/ epoxy resin composites. Polym. Degrad. Stab. 2016, 134, 144–150. [Google Scholar] [CrossRef]
- Voronina, N.V.; Meshkov, I.B.; Myakushev, V.D.; Laptinskaya, T.V.; Papkov, V.S.; Buzin, M.I.; Il’ina, M.N.; Ozerin, A.N.; Muzafarov, A.M. Hybrid organo-inorganic globular nanospecies: Transition from macromolecule to particle. J. Polym. Sci. A Polym. Chem. 2010, 48, 4310–4322. [Google Scholar] [CrossRef]
- Mironova, M.V.; Tatarinova, E.A.; Meshkov, I.B.; Muzafarov, A.M.; Kulichikhin, V.G. Rheological and relaxation properties of MQ copolymers. Polym. Sci. Ser. A 2012, 54, 177–186. [Google Scholar] [CrossRef]
- Vasil’ev, S.G.; Volkov, V.I.; Tatarinova, E.A.; Muzafarov, A.M. A solid-state NMR investigation of MQ silicone copolymers. Appl. Magn. Reson. 2013, 44, 1015–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasil’ev, S.G.; Volkov, V.I.; Tatarinova, E.A.; Muzafarov, A.M. Study of Self-Diffusion of Silicone MQ Resins in Chloroform Solutions by Pulsed Field-Gradient NMR Spectroscopy. Appl. Magn. Reson. 2014, 45, 315–328. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Polyakova, M.Y.; Andrianov, A.V.; Meshkov, I.V.; Muzafarov, A.M. Viscosity and viscoelasticity of liquid nanoparticles with polymeric matrix. Phys. Fluids 2019, 31, 083104. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Polyakova, M.Y.; Subbotin, A.V.; Meshkov, I.B.; Bystrova, A.V.; Kulichikhin, V.G.; Muzafarov, A.M. Molecular liquids formed by nanoparticles. J. Mol. Liq. 2019, 286, 110852. [Google Scholar] [CrossRef]
- Kazakova, V.V.; Zhiltsov, A.S.; Gorbatsevitch, O.B.; Meshkov, I.B.; Pletneva, M.V.; Demchenko, N.V.; Cherkaev, G.V.; Muzafarov, A.M. Synthesis and characterization of hybrid core–shell systems based on molecular silicasols. J. Inorg. Organomet. Polym. Mat. 2012, 22, 564–576. [Google Scholar] [CrossRef]
- Zhiltsov, A.S.; Meshkov, I.B.; Kurkin, T.S.; Gorbatsevich, O.B.; Kazakova, V.V.; Askadskii, A.A.; Serenko, O.A.; Ozerin, A.N.; Muzafarov, A.M. Structure of polylactide-modified silicasol nanocomposites based on thermodynamically compatible components. Nanotechnol. Russ. 2013, 8, 644–654. [Google Scholar] [CrossRef]
- Zhiltsov, A.; Gritsenko, O.; Kazakova, V.; Gorbatsevitch, O.; Bessonova, N.; Askadskii, A.; Serenko, O.A.; Muzafarov, A. Polylactide and hybrid silicasol nanoparticle-based composites. J. Appl. Polym. Sci. 2015, 132, 41894. [Google Scholar] [CrossRef]
- Zhiltsov, A.S.; Boldyrev, K.L.; Gorbatsevitch, O.B.; Kazakova, V.V.; Demchenko, N.V.; Cherkaev, G.V.; Muzafarov, A.M. Synthesis and characterization of Organo-Inorganic nanoobjects based on hyperbranchedpolyethoxysiloxanes. Silicon 2015, 7, 165–176. [Google Scholar] [CrossRef]
- Mironova, M.V.; Meshkov, I.B.; Shabeko, A.A.; Shutov, V.V.; Kulichikhin, V.G.; Tatarinova, E.A. Rheological and Rheokinetic Properties of Compositions Based on a Butyl Rubber, an MQ Copolymer, and Polymethylsilsesquioxane. INEOS OPEN 2020, 3, 29–34. [Google Scholar] [CrossRef]
- Meshkov, I.B.; Kalinina, A.A.; Kazakova, V.V.; Demchenko, A.I. Densely Cross-Linked Polysiloxane Nanogels. INEOS OPEN 2020, 3, 118–132. [Google Scholar] [CrossRef]
- Gorodov, V.V.; Milenin, S.A.; Demchenko, N.V.; Muzafarov, A.M. Carboxyl-containing polydimethylsiloxanes: Synthesis and properties. INEOS OPEN 2020, 3, 43–54. [Google Scholar] [CrossRef]
- Milenin, S.A.; Drozdov, V.; Bezlepkina, K.A.; Majorov, V.Y.; Muzafarov, A.M. Acid-Catalyzed Rearrangement of Aziopropyl-Siloxane Monomers for the Synthesis of Azidopropyl-Polydimethylsiloxane and Their Carboxylic Acid Derivatives. Macromolecules 2021, 54, 2921–2935. [Google Scholar] [CrossRef]
- Liu, L.; Liang, S.; Huang, Y.; Hu, C.; Yang, J. A stretchable polysiloxane elastomer with self-healing capacity at room temperature and solvatochromic properties. Chem. Commun. 2017, 53, 12088–12091. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wang, L.; Wang, L.; Li, L.; Feng, S. Imine-functionalized polysiloxanes for supramolecular elastomers with tunable mechanical properties. Polym. Chem. 2020, 11, 7721–7728. [Google Scholar] [CrossRef]
- Feng, L.; Li, S.; Feng, S. Preparation and characterization of silicone rubber with high modulus via tension spring-type crosslinking. RSC Adv. 2017, 7, 13130–13137. [Google Scholar] [CrossRef] [Green Version]
- Gorodov, V.V.; Demchenko, N.V.; Buzin, M.I.; Vasil’ev, V.G.; Shragin, D.I.; Papkov, V.S.; Muzafarov, A.M. Synthesis and thermal and rheological properties of carboxyl-containing polydimethylsiloxanes. Russ. Chem. Bull. 2017, 66, 1290–1299. [Google Scholar] [CrossRef]
- Kalinina, A.A.; Kholodkov, D.N.; Meshkov, I.B.; Pigaleva, M.A.; Elmanovich, I.V.; Molodtsova, Y.A.; Gallyamov, M.O.; Muzafarov, A.M. Hydrolytic polycondensation of methylalkoxysilanes under pressure. Russ. Chem. Bull. 2016, 65, 1104–1109. [Google Scholar] [CrossRef]
- Godovsky, Y.K. Thermophysical Properties of Polymers; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Schmid, H.; Michel, B. Siloxane polymers for high-resolution, high-accuracy soft lithography. Macromolecules 2000, 33, 3042–3049. [Google Scholar] [CrossRef]
- Vasil’ev, V.G.; Pryakhina, T.A.; Shragin, D.I.; Kononevich, Y.N.; Papkov, V.S.; Muzafarov, A.M. Formation of a physical crosslinked structure in polydimethylsiloxanes modified with long-chain hydrocarbon substituents with polar fragments. Polym. Sci. Ser. B 2017, 59, 320–327. [Google Scholar] [CrossRef]
- Vasil’ev, V.G.; Gorodov, V.V.; Buzin, M.I.; Shragin, D.I.; Papkov, V.S. Physical Crosslinking in Statistical and Telechelic Carboxyl-Containing Polydimethylsiloxanes. Polym. Sci. Ser. A 2021, 63, 15–23. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Composite’s Abbreviation a | Components Content in Composites, wt.% | Gel Fraction b, wt.% | ||||
|---|---|---|---|---|---|---|---|
| P1 | P2 | M2 (M2*) | M3 | P3 | |||
| 1 | P1/85-P3/15 | 85.00 | - | - | - | 15.00 | 96.6 |
| 2 | P1/100-M2/3.5-M2*/50 | 65.15 | - | 2.12 (32.57) | - | 79.2/68.8 c | |
| 3 | P1/100-M2/3.5 | 96.62 | - | 3.38 | - | - | 92.3/85.8 c |
| 4 | P1/100-M2/20 | 83.33 | - | 16.67 | - | - | 99.1/95.4 c |
| 5 | P1/100-M2/50 | 66.67 | - | 33.33 | - | - | 98.7 |
| 6 | P1/85-M2/10-P3/15 | 77.27 | - | 9.09 | - | 13.64 | 99.5 |
| 7 | P1/85-M2/20-P3/15 | 70.83 | - | 16.67 | - | 12.50 | 97.9/94.1 c |
| 8 | P1/85-M2/30-P3/15 | 65.38 | - | 23.08 | - | 11.54 | 99.5 |
| 9 | P1/85-M2/40-P3/15 | 60.72 | - | 28.57 | - | 10.71 | 99.4 |
| 10 | P1/85-M2/50-P3/15 | 56.67 | - | 33.33 | - | 10.00 | 100 |
| 11 | P1/100-M3/20 | 83.33 | - | - | 16.67 | - | 99.4 |
| 12 | P1/85-M3/20-P3/15 | 70.83 | - | - | 16.67 | 12.50 | 95.7 |
| 13 | P2/85-P3/15 | - | 85.00 | - | - | 15.00 | 74.4/5.8 c |
| 14 | P2/100-M2/50 | - | 66.67 | 33.33 | - | - | 99.2/95.4 c |
| 15 | P2/85-M2/10-P3/15 | - | 77.27 | 9.09 | - | 13.64 | 100/77.4 c |
| 16 | P2/85-M2/20-P3/15 | - | 70.83 | 16.67 | - | 12.5 | 98.2 |
| 17 | P2/85-M2/30-P3/15 | - | 65.38 | 23.08 | - | 11.54 | 100 |
| 18 | P2/85-M2/40-P3/15 | - | 60.72 | 28.57 | - | 10.71 | 97.4 |
| 19 | P2/85-M2/50-P3/15 | - | 56.67 | 33.33 | - | 10.00 | 98.6 |
| 20 | P2/85-M2/75-P3/15 | - | 48.57 | 42.86 | - | 8.57 | 96.9 |
| 21 | P2/85-M2/100-P3/15 | - | 42.50 | 50.00 | - | 7.50 | 98.0 |
| Sample | Composite’s Abbreviation | σmax, MPa | εb, % | E, MPa | |||
|---|---|---|---|---|---|---|---|
| before | after | before | after | before | after | ||
| 1 | P1/85-P3/15 | 0.2 ± 0.01 | 0.6 ± 0.04 | 174 ± 14 | 197 ± 21 | 0.3 ± 0.01 | 0.4 ± 0.01 |
| 2 | P1/100-M2/3.5-M2*/50 | 0.9 ± 0.1 | 1.6 ± 0.1 | 257 ± 16 | 331 ± 10 | 0.5 ± 0.05 | 0.5 ± 0.05 |
| 3 | P1/100-M2/3.5 | 0.8 ± 0.1 | 0.9 ± 0.1 | 229 ± 11 | 229 ± 11 | 0.5 ± 0.04 | 0.6 ± 0.05 |
| 4 | P1/100-M2/20 | 5.1 ± 0.1 | 4.8 ± 0.1 | 192 ± 21 | 162 ± 11 | 2.8 ± 0.1 | 2.0 ± 0.1 |
| 5 | P1/100-M2/50 | 6.8 ± 0.3 | 8.2 ± 0.4 | 127 ± 11 | 118 ± 9 | 25.7 ± 0.2 | 16.3 ± 0.3 |
| 6 | P1/85-M2/10-P3/15 | 0.9 ± 0.1 | 2.0 ± 0.1 | 112 ± 8 | 121 ± 8 | 1.2 ± 0.05 | 1.1 ± 0.04 |
| 7 | P1/85-M2/20-P3/15 | 2.5 ± 0.1 | 4.5 ± 0.1 | 194 ± 16 | 113 ± 7 | 2.5 ± 0.1 | 2.4 ± 0.1 |
| 8 | P1/85-M2/30-P3/15 | 4.6 ± 0.2 | 4.5 ± 0.1 | 193 ± 12 | 96 ± 12 | 6.5 ± 0.2 | 3.1 ± 0.1 |
| 9 | P1/85-M2/40-P3/15 | 4.9 ± 0.2 | 7.0 ± 0.3 | 194 ± 18 | 101 ± 9 | 12.8 ± 0.2 | 9.1 ± 0.2 |
| 10 | P1/85-M2/50-P3/15 | 6.2 ± 0.2 | 10.0 ± 0.4 | 339 ± 24 | 132 ± 11 | 16.0 ± 0.4 | 10.8 ± 0.2 |
| 11 | P1/100-M3/20 | 5.2 ± 0.2 | 8.4 ± 0.3 | 179 ± 10 | 155 ± 10 | 3.2 ± 0.1 | 3.7 ± 0.1 |
| 12 | P1/85-M3/20-P3/15 | 6.0 ± 0.3 | 5.1 ± 0.2 | 205 ± 13 | 148 ± 9 | 3.5 ± 0.1 | 3.3 ± 0.1 |
| Sample | Composite’s Abbreviation | σmax, MPa | εb, % | E, MPa | |||
|---|---|---|---|---|---|---|---|
| before | after | before | after | before | after | ||
| 13 | P2/85-P3/15 | 0.2 ± 0.01 | 0.1 ± 0.01 | 900 ± 51 | 198 ± 12 | 0.1 ± 0.01 | 0.1 ± 0.01 |
| 14 | P2/100-M2/50 | 5.7 ± 0.2 | 8.3 ± 0.3 | 761 ± 44 | 503 ± 24 | 3.8 ± 0.2 | 3.4 ± 0.2 |
| 15 | P2/85-M2/10-P3/15 | 2.4 ± 0.2 | 1.8 ± 0.1 | 582 ± 32 | 302 ± 12 | 0.7 ± 0.1 | 0.7 ± 0.1 |
| 16 | P2/85-M2/20-P3/15 | 1.9 ± 0.1 | 4.2 ± 0.2 | 393 ± 19 | 378 ± 20 | 1.7 ± 0.1 | 1.3 ± 0.1 |
| 17 | P2/85-M2/30-P3/15 | 2.9 ± 0.2 | 5.8 ± 0.3 | 498 ± 22 | 437 ± 14 | 2.3 ± 0.1 | 2.2 ± 0.1 |
| 18 | P2/85-M2/40-P3/15 | 3.1 ± 0.2 | 8.0 ± 0.3 | 560 ± 24 | 539 ± 22 | 5.2 ± 0.2 | 5.1 ± 0.2 |
| 19 | P2/85-M2/50-P3/15 | 3.5 ± 0.2 | 8.6 ± 0.2 | 858 ± 39 | 453 ± 20 | 7.0 ± 0.2 | 6.5 ± 0.2 |
| 20 | P2/85-M2/75-P3/15 | 5.7 ± 0.2 | 7.2 ± 0.3 | 575 ± 15 | 240 ± 11 | 24.4 ± 0.9 | 24.0 ± 0.7 |
| 21 | P2/85-M2/100-P3/15 | 5.9 ± 0.3 | 6.6 ± 0.2 | 525 ± 19 | 121 ± 9 | 51.0 ± 2.1 | 47.3 ± 1.6 |
| Sample | Composite’s Abbreviation | MQ Content a, wt.% | Hysteresis, % | |||
|---|---|---|---|---|---|---|
| before | after | |||||
| 2nd Cycle | 100th Cycle | 2nd Cycle | 100th Cycle | |||
| 4 | P1/100-M2/20 | 16.67 | 7.12 | 2.10 | 3.93 | 1.21 |
| 6 | P1/85-M2/20-P3/15 | 16.67 | 7.26 | 5.62 | 3.49 | 0.97 |
| 11 | P1/100-M3/20 | 16.67 | 9.61 | 3.49 | 6.73 | 2.83 |
| 12 | P1/85-M3/20-P3/15 | 16.67 | 16.72 | 5.22 | 6.95 | 2.42 |
| 8 | P1/85-M2/30-P3/15 | 23.08 | 17.07 | 13.19 | 4.32 | 3.34 |
| 5 | P1/100-M2/50 | 33.33 | 17.80 | 14.27 | 13.49 | 10.13 |
| 10 | P1/85-M2/50-P3/15 | 33.33 | 39.72 | 29.79 | 17.67 | 15.71 |
| 14 | P2/100-M2/50 | 33.33 | 29.78 | 17.43 | 22.09 | 12.64 |
| 19 | P2/85-M2/50-P3/15 | 33.33 | 35.50 | 22.99 | 27.68 | 19.46 |
| 16 | P2/85-M2/20-P3/15 | 16.67 | 13.01 | 8.48 | 4.29 | 2.46 |
| 2 | P1/100-M2/3.5-M2*/50 | 2.12/32.57 b | 5.12 | 4.45 | 3.91 | 3.15 |
| 3 | P1/100-M2/3.5 | 3.38 | 2.05 | 0.91 | 1.59 | 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meshkov, I.B.; Kalinina, A.A.; Gorodov, V.V.; Bakirov, A.V.; Krasheninnikov, S.V.; Chvalun, S.N.; Muzafarov, A.M. New Principles of Polymer Composite Preparation. MQ Copolymers as an Active Molecular Filler for Polydimethylsiloxane Rubbers. Polymers 2021, 13, 2848. https://doi.org/10.3390/polym13172848
Meshkov IB, Kalinina AA, Gorodov VV, Bakirov AV, Krasheninnikov SV, Chvalun SN, Muzafarov AM. New Principles of Polymer Composite Preparation. MQ Copolymers as an Active Molecular Filler for Polydimethylsiloxane Rubbers. Polymers. 2021; 13(17):2848. https://doi.org/10.3390/polym13172848
Chicago/Turabian StyleMeshkov, Ivan B., Aleksandra A. Kalinina, Vadim V. Gorodov, Artem V. Bakirov, Sergey V. Krasheninnikov, Sergei N. Chvalun, and Aziz M. Muzafarov. 2021. "New Principles of Polymer Composite Preparation. MQ Copolymers as an Active Molecular Filler for Polydimethylsiloxane Rubbers" Polymers 13, no. 17: 2848. https://doi.org/10.3390/polym13172848