Relationship between Degree of Polymeric Ionisation and Hydrolytic Degradation of Eudragit® E Polymers under Extreme Acid Conditions

 , and
, and
Abstract
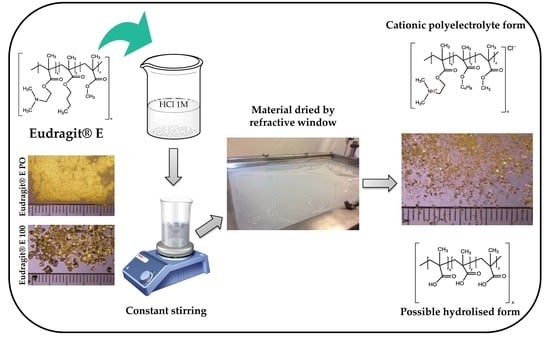
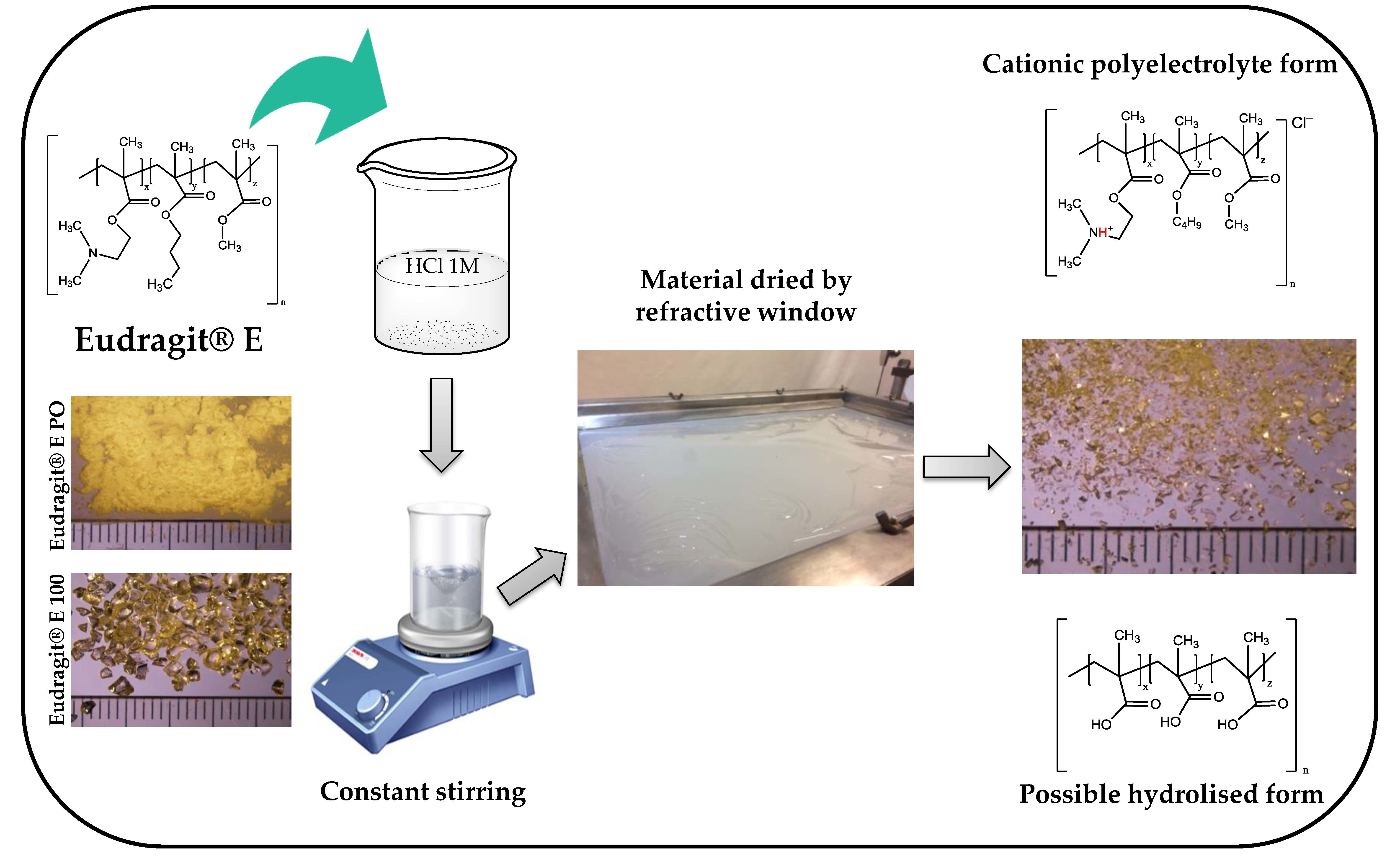
:
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Obtention of Polymeric Materials
Determination of Ionisation Degree and Zeta Potential
2.3. Structural Characterisations of Polymeric Materials
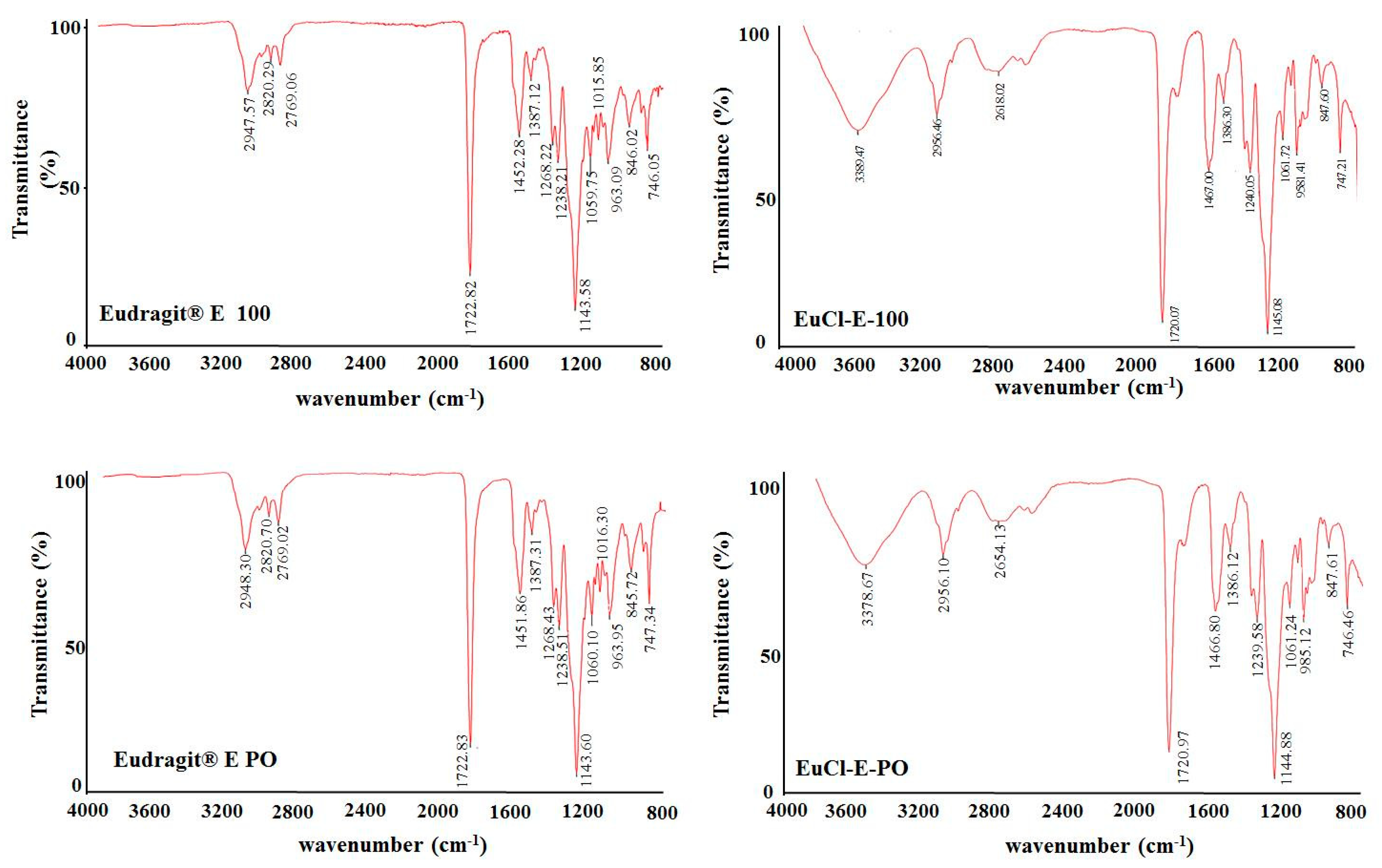
2.3.1. FT-IR Characterisation
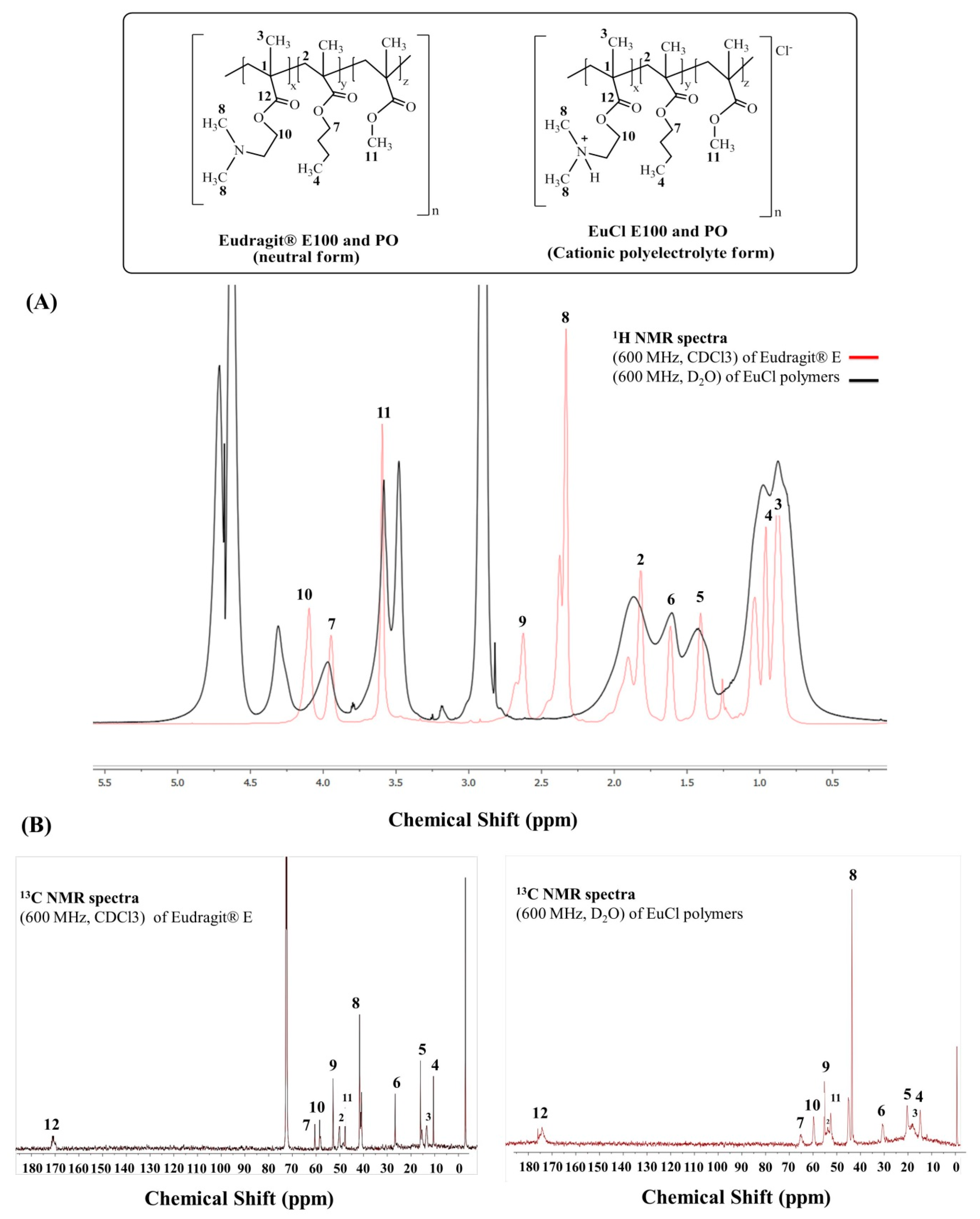
2.3.2. NMR Characterisation
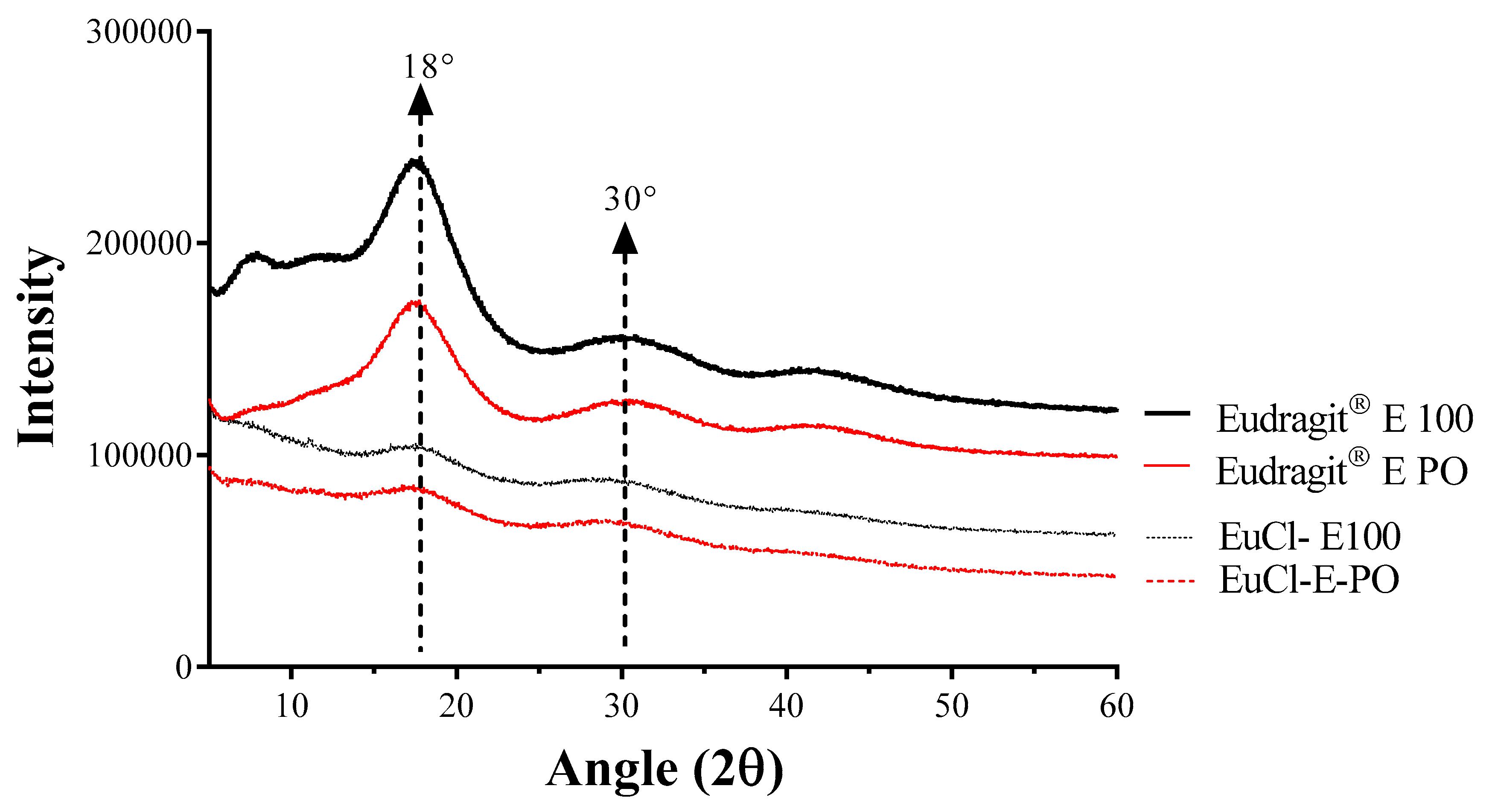
2.3.3. X-ray Diffraction (XRD)
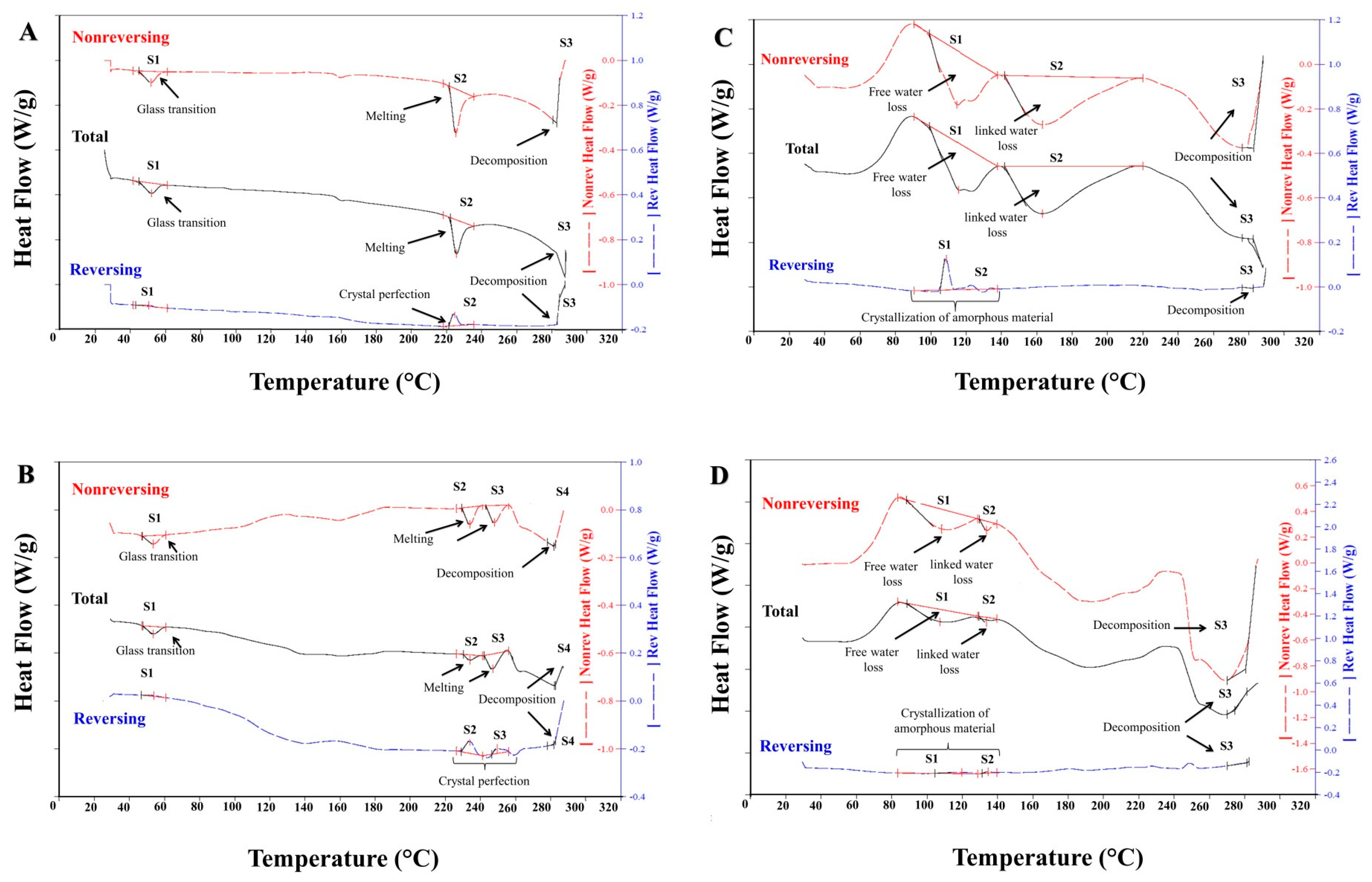
2.3.4. Thermal Analysis
2.4. Physical Characterisation of Polymeric Materials
2.4.1. External Morphology Description
2.4.2. Particle Characterisation
2.4.3. Disintegration and Dissolution Test
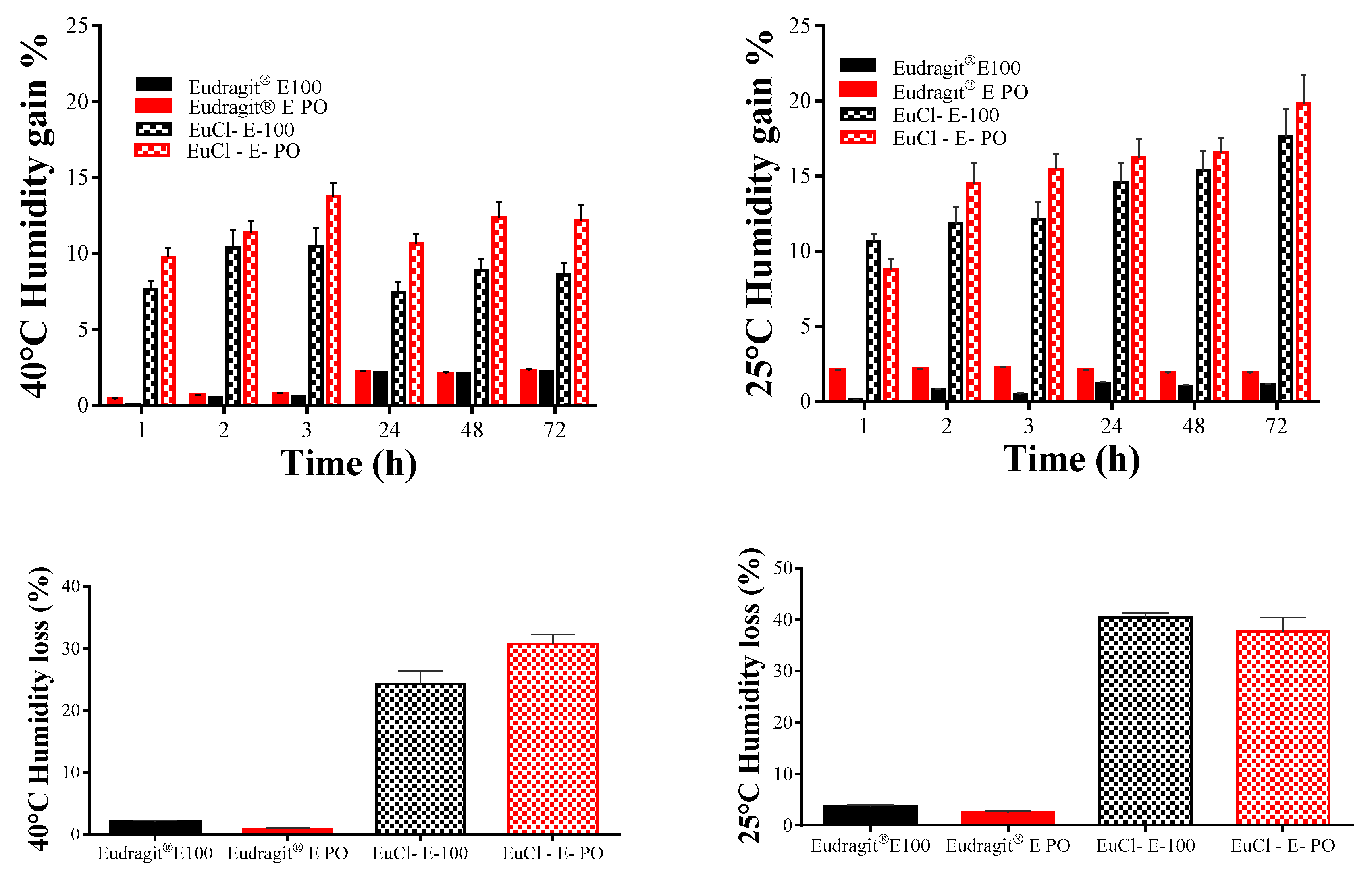
2.4.4. Humidity Loss and Gain Studies
2.5. Thermodynamics Surface Characterisation
2.5.1. Contact Angle Measurements
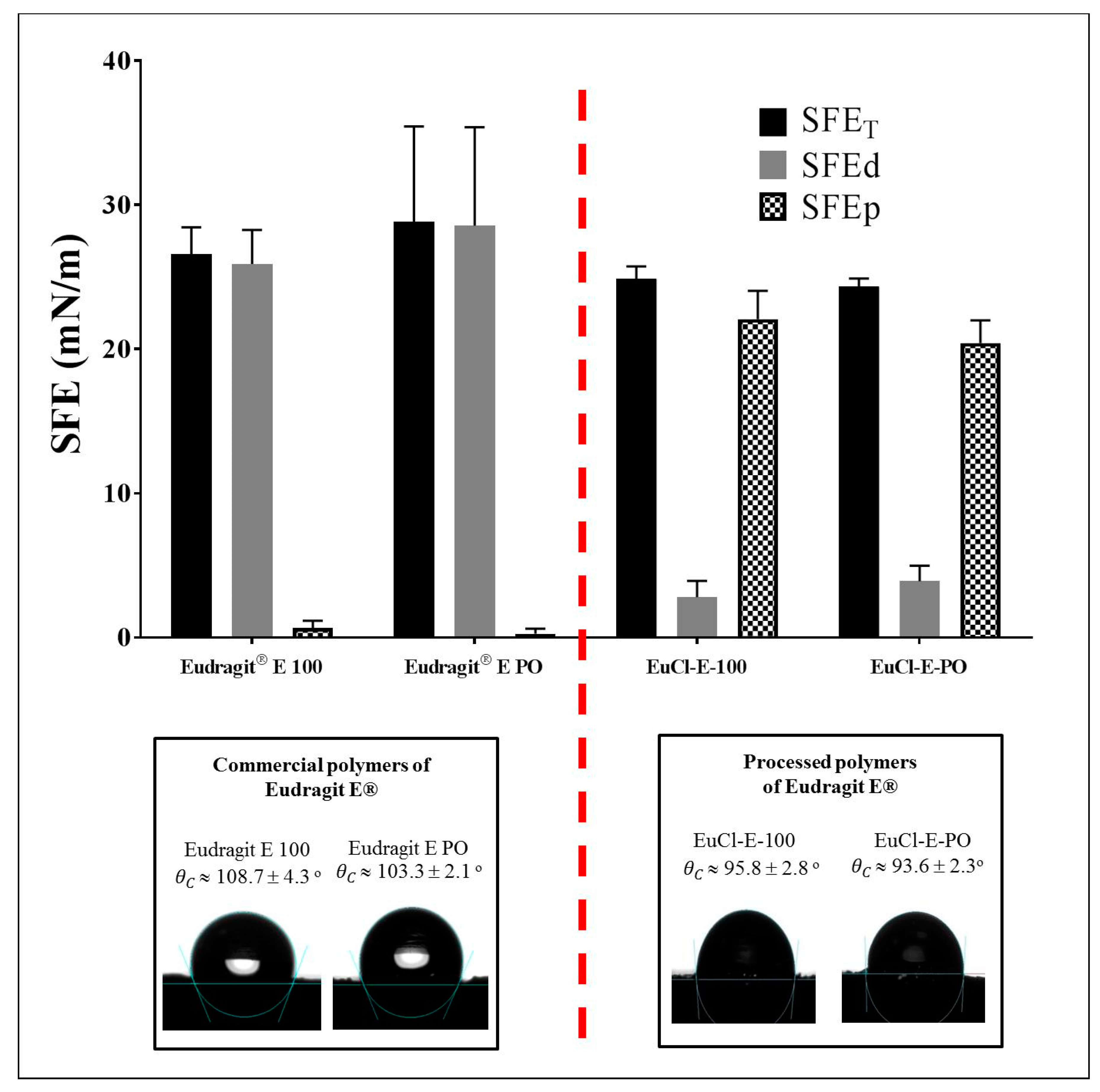
2.5.2. Surface Free Energy (SFE) Determination
3. Results and Discussion
3.1. Obtention of Polymeric Materials
Determination of Ionisation Degree and Zeta Potential
3.2. Structural Characterisation of Polymer Materials
3.2.1. FT-IR Characterisation
3.2.2. NMR Characterisation
3.2.3. XRD and DSC Analyses
3.3. Characterisation of Powder Polymeric Materials
3.3.1. Powder Morphology and Shape
3.3.2. Flowability Assays
3.3.3. Humidity Loss and Gain Study
3.3.4. Disintegration and Dissolution Test
3.4. Surface Polymer Characterisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kadian, S.S.; Harikumar, S.L. Eudragit and its Pharmaceutical Significance. Roorke Coll. Pharm. 2009, 1, 1–16. [Google Scholar]
- Nikam, V.K.; Kotade, K.B.; Gaware, V.M.; Dolas, R.T.; Dhamak, K.B.; Somwanshi, S.B.; Khadse, A.N.; Kashid, V.A. Eudragit a versatile Polymer: a Review. Pharmacologyonline 2011. [Google Scholar]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit®: a technology evaluation. Expert Opin. Drug Deliv. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sonje, A.; Chandra, A. Comprehensive Review On Eudragit Polymers. Int. Res. J. Pharm. 2013. [Google Scholar] [CrossRef]
- Ghose, D.; Patra, C.N.; Swain, S.; Panigrahi, K.C.; Priya, R.; Kumar Jena, G. Pharmaceutical significance of Eudragit: A review. Futur. J. Pharm. Sci. 2017. [Google Scholar]
- Karn, P.R.; Vanić, Z.; Pepić, I.; Skalko-Basnet, N. Mucoadhesive liposomal delivery systems: the choice of coating material. Drug Dev. Ind. Pharm. 2011, 37, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Gad, S.C. United States Pharmacopoeia (USP). In Encyclopedia of Toxicology (Third Edition); Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 881–882. ISBN 978-0-12-386455-0. [Google Scholar]
- European Pharmacopoeia Commission, Dissolution test for Solid Dosage Forms in European Pharmacopoeia, 6th ed.; Council of Europe: Strasbourg, France, 2007; pp. 263–275.
- Berg, M.C.; Zhai, L.; Cohen, R.E.; Rubner, M.F. Controlled drug release from porous polyelectrolyte multilayers. Biomacromolecules 2006, 7, 357–364. [Google Scholar] [CrossRef]
- Oh, K.T.; Lee, H.-G.; Piao, Z.-Z.; Lee, B.-J.; Lee, J.; Lee, K.-H.; Kim, D.-J. Comparison of Release-Controlling Efficiency of Polymeric Coating Materials Using Matrix-type Casted Films and Diffusion-Controlled Coated Tablet. AAPS PharmSciTech 2010. [Google Scholar]
- Hu, L.D.; Liu, Y.; Tang, X.; Zhang, Q. Preparation and in vitro/in vivo evaluation of sustained-release metformin hydrochloride pellets. Eur. J. Pharm. Biopharm. 2006. [Google Scholar] [CrossRef]
- Chiappetta, D.A.; Carcaboso, A.M.; Bregni, C.; Rubio, M.; Bramuglia, G.; Sosnik, A. Indinavir-loaded pH-sensitive microparticles for taste masking: toward extemporaneous pediatric anti-HIV/AIDS liquid formulations with improved patient compliance. AAPS PharmSciTech 2009. [Google Scholar] [CrossRef]
- Naresh Kumar, K.; Sandeep, D. Formulating taste-masked orally disintegrating tablets of a bitter drug ibuprofen. Int. Res. J. Pharm. 2013, 86, 275–284. [Google Scholar]
- Hasanovic, A.; Hollick, C.; Fischinger, K.; Valenta, C. Improvement in physicochemical parameters of DPPC liposomes and increase in skin permeation of aciclovir and minoxidil by the addition of cationic polymers. Eur. J. Pharm. Biopharm. 2010, 75, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Choi, S.G.; Davaa, E.; Park, J.S. Encapsulation enhancement and stabilization of insulin in cationic liposomes. Int. J. Pharm. 2011, 415, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xu, N.; Ji, H.; Liu, H.; Wang, Z.; Wu, L. Eudragit nanoparticles containing genistein: formulation, development, and bioavailability assessment. Int. J. Nanomedicine 2011. [Google Scholar]
- Glaessl, B.; Siepmann, F.; Tucker, I.; Rades, T.; Siepmann, J. Deeper insight into the drug release mechanisms in Eudragit RL-based delivery systems. Int. J. Pharm. 2010, 389, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Barea, M.J.; Jenkins, M.J.; Gaber, M.H.; Bridson, R.H. Evaluation of liposomes coated with a pH responsive polymer. Int. J. Pharm. 2010, 402, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Six, K.; Verreck, G.; Peeters, J.; Brewster, M.; Van Den Mooter, G. Increased Physical Stability and Improved Dissolution Properties of Itraconazole, a Class II Drug, by Solid Dispersions that Combine Fast- and Slow-Dissolving Polymers. J. Pharm. Sci. 2004. [Google Scholar] [CrossRef]
- Adibkia, K.; Javadzadeh, Y.; Dastmalchi, S.; Mohammadi, G.; Niri, F.K.; Alaei-Beirami, M. Naproxen-eudragit® RS100 nanoparticles: Preparation and physicochemical characterization. Colloids Surf. B Biointerfaces 2011. [Google Scholar] [CrossRef]
- Salamanca, C.H.; Castillo, D.F.; Villada, J.D.; Rivera, G.R. Physicochemical characterization of in situ drug-polymer nanocomplex formed between zwitterionic drug and ionomeric material in aqueous solution. Mater. Sci. Eng. C 2017. [Google Scholar] [CrossRef]
- Arévalo, L.M.; Yarce, C.J.; Oñate-Garzón, J.; Salamanca, C.H. Decrease of antimicrobial resistance through polyelectrolyte-coated nanoliposomes loaded with β-lactam drug. Pharmaceuticals 2019, 12, 1. [Google Scholar] [CrossRef]
- Cantor, S.; Vargas, L.; Rojas, O.E.A.; Yarce, C.J.; Salamanca, C.H.; Oñate-Garzón, J. Evaluation of the antimicrobial activity of cationic peptides loaded in surface-modified nanoliposomes against foodborne bacteria. Int. J. Mol. Sci. 2019, 20, 680. [Google Scholar] [CrossRef]
- Ghanem, T.H. Modeling of refractance window film dryer for liquids. Misr J. Agric. Eng. 2010. [Google Scholar]
- Grey, R.O.; Beddow, J.K. On the Hausner Ratio and its relationship to some properties of metal powders. Powder Technol. 1969, 2, 323–326. [Google Scholar] [CrossRef]
- Saw, H.Y.; Davies, C.E.; Paterson, A.H.J.; Jones, J.R. Correlation between powder flow properties measured by shear testing and Hausner ratio. Procedia Eng. 2015, 102, 218–225. [Google Scholar]
- Leuenberger, H. The compressibility and compactibility of powder systems. Int. J. Pharm. 1982, 12, 41–55. [Google Scholar] [CrossRef]
- Salamanca, C.H.; Yarce, C.J.; Zapata, C.A.; Giraldo, J.A. Relationship between the polymeric ionization degree and powder and surface properties in materials derived from poly(maleic anhydride-alt-octadecene). Molecules 2018, 23, 320. [Google Scholar] [CrossRef] [PubMed]
- Yarce, C.J.; Pineda, D.; Correa, C.E.; Salamanca, C.H. Relationship between surface properties and in Vitro drug release from a compressed matrix containing an amphiphilic polymer material. Pharmaceuticals 2016, 9, 34. [Google Scholar] [CrossRef]
- Janssen, D.; De Palma, R.; Verlaak, S.; Heremans, P.; Dehaen, W. Static solvent contact angle measurements, surface free energy and wettability determination of various self-assembled monolayers on silicon dioxide. Thin Solid Films 2006. [Google Scholar] [CrossRef]
- Owens, D.K.; Wendt, R.C. Estimation of the surface free energy of polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. [Google Scholar] [CrossRef]
- Yarce, C.J.C.; Echeverri, J.J.D.; Palacio, M.M.A.; Rivera, C.C.A.; Salamanca, C.H.C. Relationship between Surface Properties and In Vitro Drug Release from Compressed Matrix Containing Polymeric Materials with Different Hydrophobicity Degrees. Pharmaceuticals 2017, 10, 15. [Google Scholar] [CrossRef]
- Salamanca, C.H.; Yarce, C.J.; Roman, Y.; Davalos, A.F.; Rivera, G.R. Application of nanoparticle technology to reduce the anti-microbial resistance through β-lactam antibiotic-polymer inclusion nano-complex. Pharmaceuticals 2018, 11, 19. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, X.; Suwardie, H.; Wang, P.; Gogos, C.G. Miscibility Studies of Indomethacin and Eudragit® E PO by Thermal, Rheological, and Spectroscopic Analysis. J. Pharm. Sci. 2012, 101, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Ngai, K.L.; Floudas, G.; Plazek, D.J.; Rizos, A.K. Amorphous Polymers. Encycl. Polym. Sci. Technol. 2002, 5, 63–111. [Google Scholar]
- Schönhals, A.; Kremer, F. Amorphous Polymers. In Polymer Science: A Comprehensive Reference; 2012; pp. 201–226. ISBN 9780080878621. [Google Scholar]
- Zhao, J.; Fan, Q. Amorphous phase in atactic polystyrene. Polym. Bull. 2001, 47, 91–97. [Google Scholar] [CrossRef]
- Schick, C. Differential scanning calorimetry (DSC) of semicrystalline polymers. Anal. Bioanal. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.S.; Sauerbrunn, S.R.; Reading, M. Modulated differential scanning calorimetry. J. Therm. Anal. 1993. [Google Scholar] [CrossRef]
- Alasino, R.V.; Leonhard, V.; Bianco, I.D.; Beltramo, D.M. Eudragit E100 surface activity and lipid interactions. Colloids Surf. B Biointerfaces 2012, 91, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Zhang, L.; Ma, Y.; Zhu, J.; Xiao, W. A novel electrostatic dry coating process for enteric coating of tablets with Eudragit® L100-55. Eur. J. Pharm. Biopharm. 2013. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymeric System | DMAE Groups ± SD (%) | Carboxylic Acid Percentage (%) | Ionisation Degree (%) | Zeta Potential ± SD (mV) |
|---|---|---|---|---|
| Eudragit® E 100 | 20.9 ± 0.1 | 0 | 0 | undetermined |
| Eudragit® E PO | 19.5 ± 0.2 | 0 | 0 | undetermined |
| EuCl-E-100 | 7.9 ± 0.1 | 0 | 38 | +47.7 ± 1.0 |
| EuCl-E-PO | 7.5 ± 0.3 | 0 | 38 | +47.9 ± 0.3 |
| Position | Eudragit® E100 and PO δ (ppm) CDCl3 | Form EuCl-E-100 and PO δ (ppm) D2O | ||
|---|---|---|---|---|
| 1H | 13C | 1H | 13C | |
| 1 | - | - | - | 74.2 |
| 2 | 1.81 br s | 54.2 | 1.86 br s | 56.1 |
| 3 | 0.87 br s | 16.7 | 0.80 br s | 19.4 |
| 4 | 0.96 br s | 13.8 | 0.87 br s | 16.0 |
| 5 | 1.40 br s | 19.3 | 1.35 br s | 21.6 |
| 6 | 1.61 br s | 30.2 | 1.59 br s | 32.2 |
| 7 | 3.94 br s | 64.8 | 3.97 br s | 67.9 |
| 8 | 2.34 (br s. N-CH3)2 | 45.1 | 2.89 br s | 45.7 |
| 9 | 2.63 br s | 57.0 | 4.30 br s | 62.3 |
| 10 | 4.10 br s | 62.7 | 3.47 br s | 57.7 |
| 11 | 3.59 s | 51.8 | 3.58 br s | 54.9 |
| 12 | - | 177.9 | - | 180.4 |
| Polymer Material | Heat Flow (W/g) | Signal | Signal Type | Onset (°C) | Peak (°C) | Enthalpy (J/g) |
|---|---|---|---|---|---|---|
| Eudragit® E 100 | Total | S1 | glass transition | 44.93 | 51.89 | 4.28 |
| S2 | melting point | 222.11 | 225.64 | 10.32 | ||
| S3 | decomposition | 287.84 | - | - | ||
| Reversible | S1 | glass transition | 42.90 | 50.53 | 0.35 | |
| S2 | crystal perfection | 221.24 | 224.60 | 2.89 | ||
| S3 | decomposition | 284.33 | - | - | ||
| Non-reversible | S1 | glass transition | 44.78 | 51.76 | 4.63 | |
| S2 | melting point | 221.62 | 225.26 | 13.20 | ||
| S3 | decomposition | 282.73 | - | - | ||
| Eudragit® E PO | Total | S1 | glass transition | 47.59 | 53.60 | 2.63 |
| S2 | melting amorphous | 230.24 | 234.10 | 1.36 | ||
| S3 | melting crystal | 242.14 | 246.98 | 4.73 | ||
| Reversible | S4 | decomposition | 282.66 | - | - | |
| S1 | glass transition | 46.72 | 54.02 | 0.34 | ||
| S2 | crystallization | 229.01 | 233.97 | 3.54 | ||
| Non-reversible | S3 | crystal perfection | 246.51 | 249.55 | 0.94 | |
| S4 | decomposition | 271.73 | - | - | ||
| S1 | glass transition | 47.51 | 53.64 | 2.95 | ||
| EuCl-E-100 | Total | S1 | free water loss | 99.24 | 115.67 | 55.92 |
| S2 | linked water loss | 141.79 | 163.15 | 98.05 | ||
| S3 | decomposition | 282.42 | - | - | ||
| Reversible | S1 | crystal perfection | 105.31 | 108.74 | 7.70 | |
| S2 | crystallization | 124.27 | 129.13 | 1.85 | ||
| S3 | decomposition | 282.40 | - | - | ||
| Non-reversible | S1 | free water loss | 99.19 | 114.83 | 63.59 | |
| S2 | linked water loss | 141.79 | 163.35 | 102.30 | ||
| S3 | decomposition | 282.71 | - | - | ||
| EuCl-E-PO | Total | S1 | free water loss | 88.67 | 107.38 | 42.70 |
| S2 | linked water loss | 129.40 | 133.76 | 3.80 | ||
| S3 | decomposition | 274.42 | - | - | ||
| S1 | crystal perfection | 104.42 | 119.76 | 2.01 | ||
| Reversible | S2 | crystallization | 131.19 | 134.52 | 0.62 | |
| S3 | decomposition | 281.21 | - | - | ||
| S1 | free water loss | 88.35 | 108.39 | 44.71 | ||
| S2 | linked water loss | 129.70 | 133.76 | 4.42 | ||
| Non-reversible | S3 | decomposition | 280.54 | - | - | |
| S1 | free water loss | 88.67 | 107.38 | 42.70 | ||
| S2 | linked water loss | 129.40 | 133.76 | 3.80 | ||
| S3 | decomposition | 274.42 | - | - |
| Polymer Material | Repose Angle (°) ± SD | Carr Index (%) ± SD | Hausner Index (%) ± SD |
|---|---|---|---|
| Eudragit® E 100 | 28 ± 0.5 | 24.0 ± 0.8 | 1.3 ± 0.05 |
| Eudragit® E PO | 7.4 ± 1.3 | 5.9 ± 0.2 | 1.1 ± 0.03 |
| EuCl-E-100 | 15.2 ± 0.6 | 10.9 ± 0.2 | 1.1 ± 0.04 |
| EuCl-E-PO | 13.3 ± 1.1 | 10.8 ± 0.3 | 1.1 ± 0.03 |
| Polymer Material | Applied Pressure (psi) | Tablet Hardness (kp) | Disintegration Time (h:min) | Formation of a Homogeneous Phase |
|---|---|---|---|---|
| Eudragit® E 100 | 200 | >20 | >8h | No |
| 300 | ||||
| 400 | ||||
| Eudragit® E PO | 200 | 2.17 ± 0.13 | 2:26 | No |
| 300 | 2.57 ± 0.25 | 4:75 | ||
| 400 | 3.69 ± 0.43 | 6:50 | ||
| EuCl-E-100 | 200 | 10.50 ± 0.20 | 00:08: | Yes |
| 300 | 10.58 ± 1.11 | 00:10 | ||
| 400 | 11.06 ± 0.17 | 00:11 | ||
| EuCl-E-PO | 200 | 5.40 ± 0.47 | 00:07 | Yes |
| 300 | 5.52 ± 0.37 | 00:10 | ||
| 400 | 5.59 ± 0.47 | 00:11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linares, V.; Yarce, C.J.; Echeverri, J.D.; Galeano, E.; Salamanca, C.H. Relationship between Degree of Polymeric Ionisation and Hydrolytic Degradation of Eudragit® E Polymers under Extreme Acid Conditions. Polymers 2019, 11, 1010. https://doi.org/10.3390/polym11061010
Linares V, Yarce CJ, Echeverri JD, Galeano E, Salamanca CH. Relationship between Degree of Polymeric Ionisation and Hydrolytic Degradation of Eudragit® E Polymers under Extreme Acid Conditions. Polymers. 2019; 11(6):1010. https://doi.org/10.3390/polym11061010
Chicago/Turabian StyleLinares, Valentina, Cristhian J. Yarce, Juan D. Echeverri, Elkin Galeano, and Constain H. Salamanca. 2019. "Relationship between Degree of Polymeric Ionisation and Hydrolytic Degradation of Eudragit® E Polymers under Extreme Acid Conditions" Polymers 11, no. 6: 1010. https://doi.org/10.3390/polym11061010