
Metabolomics and Transcriptomics Provide Insights into Lipid Biosynthesis in the Embryos of Walnut (Juglans regia L.)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Analysis of Walnut Fruit Morphological Characteristics and Oil Content
2.2. Analysis of the Principal Component Analysis (PCA)
2.3. Analysis of Differentially Expressed Genes (DEGs) across Different Developmental Periods
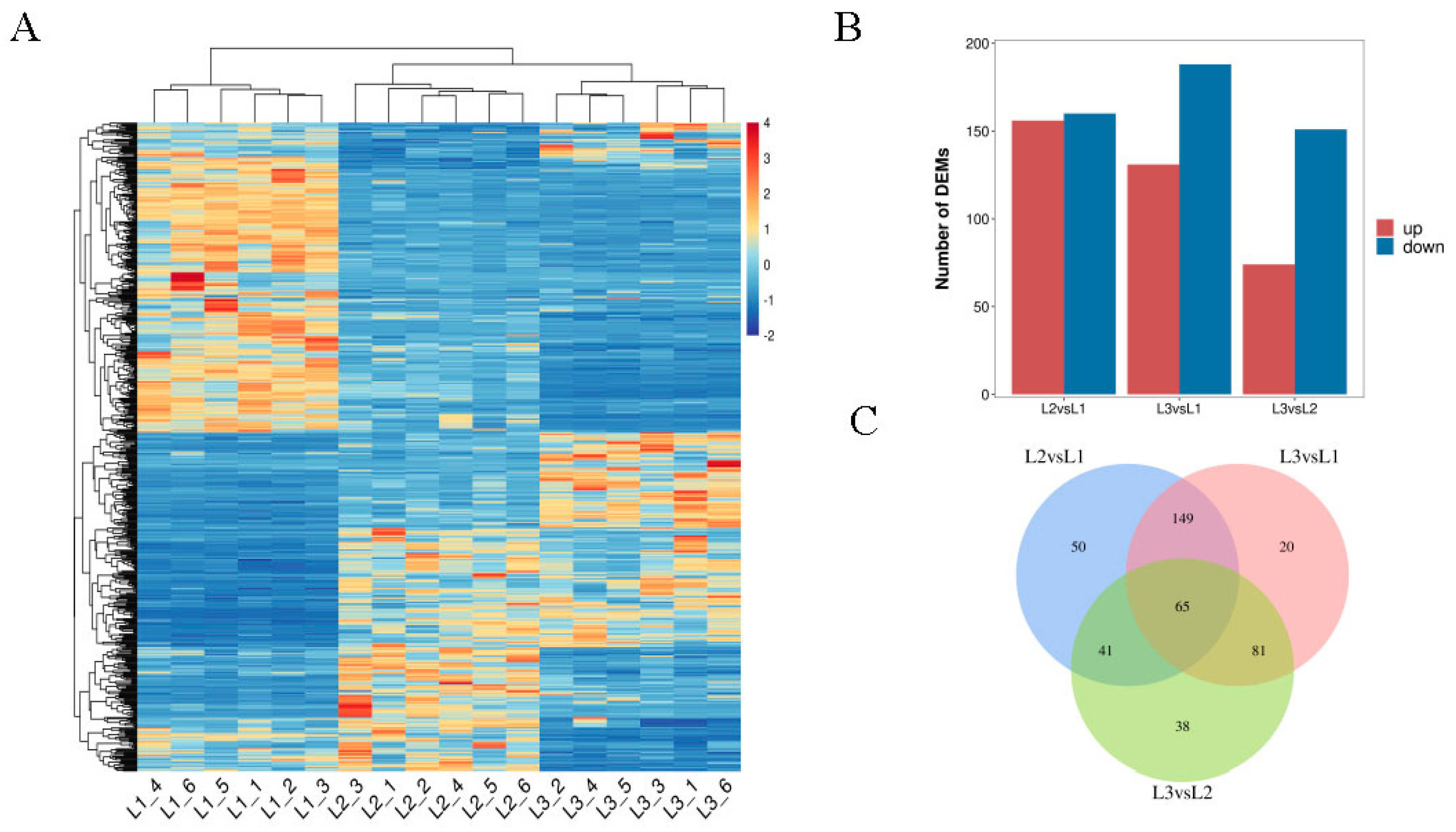
2.4. Analysis of Differentially Expressed Metabolites (DEMs) across Different Developmental Periods
2.5. Analysis of Transcription Factors
2.6. Integrated Analysis of the Transcriptomic and Metabolomic Data Pertaining to Lipid Biosynthesis across Walnut Fruit Developmental Periods
2.7. Identification of TFs Related to Oil Biosynthesis
2.8. Integrated Analysis of the Transcriptome and Metabolome of the Glycolysis and TCA Cycle Biosynthesis Pathways across Different Developmental Periods
2.8.1. Analysis of the Glycolysis Pathway
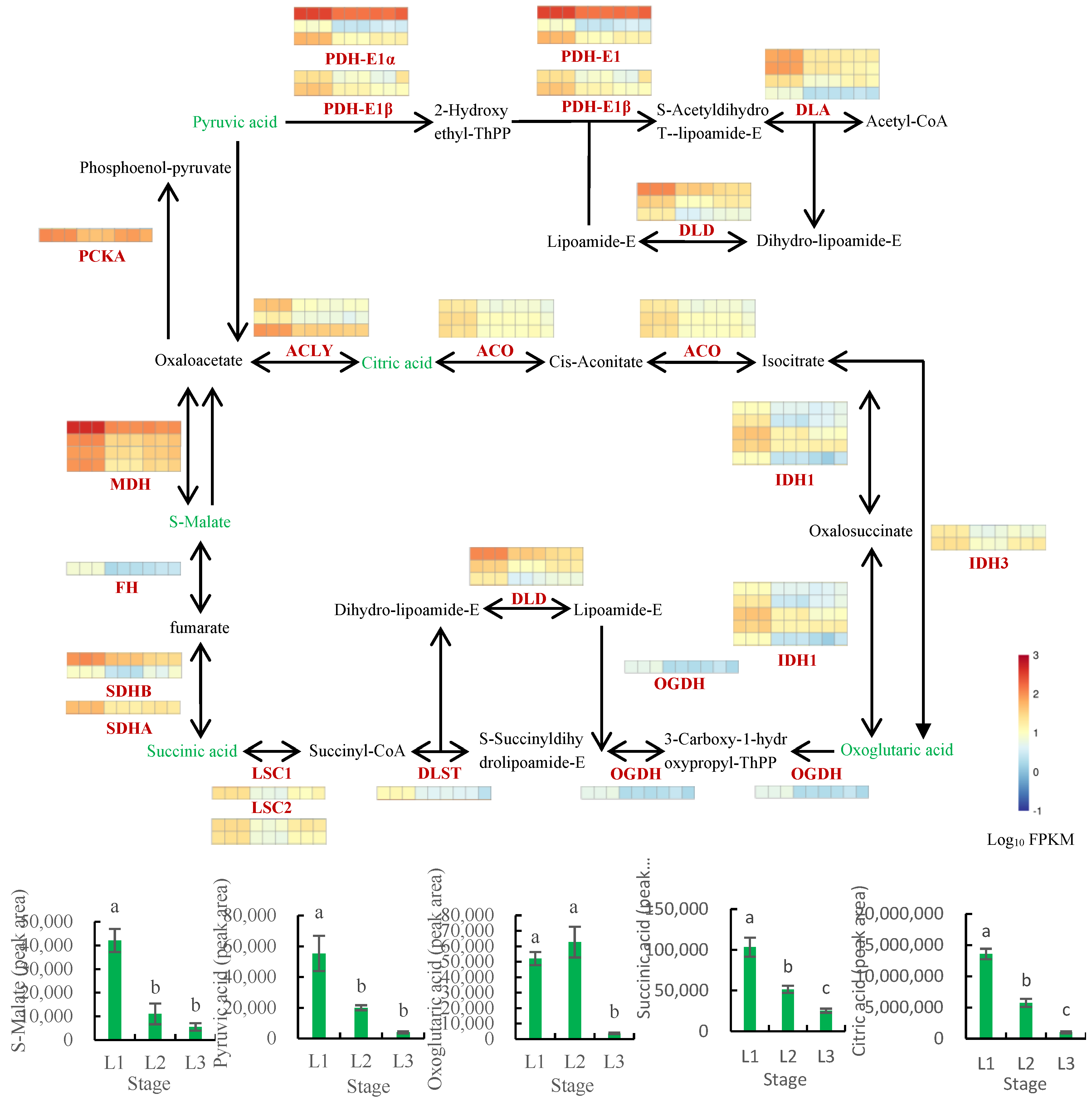
2.8.2. Analysis of the TCA Cycle Pathway
2.9. RT-qPCR Validation of DEGs in Transcriptomic Data
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Physiological Indices during Fruit Development
4.3. RNA Extraction and Library Construction
4.4. Bioinformatic Analysis of RNA-Seq Data
4.5. Quantitative Analysis
4.6. Metabolite Extraction
4.7. LC-MS Analysis
4.8. Metabolomics Data Processing
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, Q.; Bu, D.; Zhang, J.; Wu, Y.; Pei, D. The transcriptome landscape of walnut interspecies hybrid (Juglans hindsii × Juglans regia) and regulation of cambial activity in relation to grafting. Front. Genet. 2019, 10, 577. [Google Scholar] [CrossRef]
- Guasch-Ferré, M.; Li, J.; Hu, F.B.; Salas-Salvadó, J.; Tobias, D.K. Effects of walnut consumption on blood lipids and other cardiovascular disease risk factors: An updated meta-analysis and systematic review of controlled trials. Am. J. Clin. Nutr. 2018, 108, 174–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zibaeenezhad, M.J.; Farhadi, P.; Attar, A.; Mosleh, A.; Amirmoezi, F.; Azimi, A. Effects of walnut oil on lipid profiles in hyperlipidemic type 2 diabetic patients: A randomized, double-blind, placebo-controlled trial. Nutr. Diabetes. 2017, 7, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hama, J.R.; Fitzsimmons-Thoss, V. Determination of unsaturated fatty acids composition in walnut (Juglans regia L.) oil using NMR spectroscopy. Food Anal. Methods 2022, 15, 1226–1236. [Google Scholar] [CrossRef]
- Li, M.; Liu, Y.; Sun, C. Research advance about nutrients and medicinal value of walnut. J. Chin. Cereals Oils Assoc. 2009, 24, 166–170. [Google Scholar]
- Sun, S.; Wang, Z.; Song, K. Research advancement on nutritional value and functional activity of walnut. Food Nutr. China 2013, 19, 72–74. [Google Scholar]
- Bates, P.D.; Stymne, S.; Ohlrogge, J.B. Biochemical pathways in seed oil synthesis. Curr. Opin. Plant Biol. 2013, 16, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Zhang, D.; Ma, Z. Transcriptome analysis of genes involved in fatty acid and lipid biosynthesis in developing walnut (Juglans regia L.) seed kernels from Qinghai Plateau. Plants 2022, 11, 3207. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Su, Y.; Chen, Y.; Liu, Y.; Sun, M.; Qi, G. Transcriptome analysis reveals dynamic fat accumulation in the walnut kernel. Int. J. Genom. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Cui, W.; Liang, Y.; Tian, W.; Ji, M.; Ma, X. Regulating effect of beta-ketoacyl synthase domain of fatty acid synthase on fatty acyl chain length in de novo fatty acid synthesis. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Zheng, X.; Zhang, Z.; Wang, Z.; Xia, H.; Changming, L.U.; Shoulai, G.U. Long chain Acyl-coenzyme A synthetase 4 (BnLACS4) gene from brassica napus enhances the yeast lipid contents. J. Integr. Agric. 2014, 13, 54–62. [Google Scholar] [CrossRef]
- Kim, S.; Yamaoka, Y.; Ono, H.; Kim, H.; Shim, D.; Maeshima, M.; Martinoia, E.; Cahoon, E.B.; Nishida, I.; Lee, Y. AtABCA9 transporter supplies fatty acids for lipid synthesis to the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2013, 110, 773–778. [Google Scholar] [CrossRef]
- Baud, S. Seeds as oil factories. Plant Reprod. 2018, 31, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Li-Beisson, Y.; Shorrosh, B.; Beisson, F.; Andersson, M.X.; Arondel, V.; Bates, P.D.; Baud, S.; Bird, D.; Debono, A.; Durrett, T.P.; et al. Acyl-lipid metabolism. Arab. Book. 2013, 11, e0161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elahi, N.; Duncan, R.W.; Stasolla, C. Modification of oil and glucosinolate content in canola seeds with altered expression of Brassica napus LEAFY COTYLEDON1. Plant Physiol. Biochem. 2016, 100, 52–63. [Google Scholar] [CrossRef]
- Elahi, N.; Duncan, R.W.; Stasolla, C. Decreased seed oil production in FUSCA3 Brassica napus mutant plants. Plant Physiol. Biochem. 2015, 96, 222–230. [Google Scholar] [CrossRef]
- Kagaya, Y.; Toyoshima, R.; Okuda, R.; Usui, H.; Yamamoto, A.; Hattori, T. LEAFY COTYLEDON1 controls seed storage protein genes through its regulation of FUSCA3 and ABSCISIC ACID INSENSITIVE3. Plant Cell Physiol. 2005, 46, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manan, S.; Ahmad, M.Z.; Zhang, G.; Chen, B.; Haq, B.U.; Yang, J.; Zhao, J. Soybean LEC2 regulates subsets of genes involved in controlling the biosynthesis and catabolism of seed storage substances and seed development. Front. Plant Sci. 2017, 8, 1604. [Google Scholar] [CrossRef] [Green Version]
- Baud, S.; Guyon, V.; Kronenberger, J.; Wuilleme, S.; Miquel, M.; Caboche, M.; Lepiniec, L.; Rochat, C. Multi-functional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 2003, 33, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Q.; Yuan, L.; Ma, W. WRINKLED1, a “Master Regulator” in transcriptional control of plant oil biosynthesis. Plants 2019, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Maeo, K.; Tokuda, T.; Ayame, A.; Mitsui, N.; Kawai, T.; Tsukagoshi, H.; Ishiguro, S.; Nakamura, K. An AP2-type transcription factor, WRINKLED1, of Arabidopsis thaliana binds to the AW-box sequence conserved among proximal upstream regions of genes involved in fatty acid synthesis. Plant J. 2009, 60, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, G.; Liu, X.; Yu, Z.; Peng, S. Integrated analysis of seed microRNA and mRNA transcriptome reveals important functional genes and microRNA-targets in the process of walnut (Juglans regia) seed oil accumulation. Int. J. Mol. Sci. 2020, 21, 9093. [Google Scholar] [CrossRef]
- Huang, R.; Zhou, Y.; Zhang, J.; Ji, F.; Jin, F.; Fan, W.; Pei, D. Transcriptome analysis of walnut (Juglans regia L.) embryos reveals key developmental stages and genes involved in lipid biosynthesis and polyunsaturated fatty acid metabolism. J. Agric. Food Chem. 2021, 69, 377–396. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Yang, G.; Liu, C.; Yu, Z.; Zhai, M. The complete chloroplast genome of the Juglans regia (Juglandales: Julandaceae). Mitochondrial DNA 2017, 28, 407–408. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Jia, Q. Transcriptome analysis reveals candidate genes for petroselinic acid biosynthesis in fruits of Coriandrum sativum L. J. Agric. Food Chem. 2020, 68, 5507. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Yuan, G. Comparative transcriptome analysis provides insights into the distinct germination in sheep grass (Leymus chinensis) during seed development. Plant Physiol. Biochem. 2019, 139, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Barros, E.; Lezar, S.; Anttonen, M.J.; Dijk, J.P.V.; Röhlig, R.M.; Kok, E.J.; Engel, K.H. Comparison of two GM maize varieties with a near-isogenic non-GM variety using transcriptomics, proteomics and metabolomics. Plant Biotechnol. J. 2010, 8, 436–451. [Google Scholar] [CrossRef]
- Jia, Z.; Chen, F.; Hong, L. Metabolic profiling of transgenic rice with cryIAc and sck genes: An evaluation of unintended effects at metabolic level by using GC-FID and GC-MS. J. Chromatogr. B 2009, 877, 725–732. [Google Scholar]
- Meng, J.; Wang, B.; He, G.; Wang, Y.; Tang, X.; Wang, S. Metabolomics integrated with transcriptomics reveals redirection of the phenylpropanoids metabolic flux in Ginkgo biloba. J. Agric. Food Chem. 2019, 67, 3284–3291. [Google Scholar] [CrossRef]
- Li, D.; Chen, G.; Ma, B.; Zhong, C.; He, N. Metabolic profiling and transcriptome analysis of mulberry leaves provide insights into flavonoid biosynthesis. J. Agric. Food Chem. 2020, 68, 1494–1504. [Google Scholar] [CrossRef]
- Yan, S.; Wang, X.; Yang, C.; Wang, J.; Wang, Y.; Wu, B.; Qiao, L.; Zhao, J.; Mohammad, P.; Zheng, X. Insights into walnut lipid metabolism from metabolome and transcriptome analysis. Front. Genet. 2021, 12, 1334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, M.; Zhang, B.; Shrestha, P.; Petrie, J.R.; Green, A.G.; Singh, S.P. Genetic enhancement of palmitic acid accumulation in cotton seed oil through RNAi down-regulation of ghKAS2 encoding β-ketoacyl-ACP synthase II (KASII). Plant Biotechnol. J. 2017, 15, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjay, P.; Vidya, S.; Ashwini, V.; Prakash, B.; Narendra, Y.; Abhay, M. Differential transcriptional activity of SAD, FAD2 and FAD3 desaturase genes in developing seeds of linseed contributes to varietal variation in α-linolenic acid content. Phytochemistry 2014, 98, 41–53. [Google Scholar]
- Aznarmoreno, J.A.; Venegascaleron, M.; Martinezforce, E.; Garces, R.; Salas, J.J. Acyl carrier proteins from sunflower (Helianthus nannuus L.) seeds and their influence on FatA and FatB acyl-ACP thioesterase activities. Planta 2016, 244, 479–490. [Google Scholar] [CrossRef]
- Baud, S.; Lepiniec, L. Regulation of de novo fatty acid synthesis in maturing oilseeds of Arabidopsis. Plant Physiol. Biochem. 2009, 47, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Okuley, J.; Lightner, J.; Feldmann, K.; Yadav, N.; Lark, E.; Browse, J. Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 1994, 6, 147–158. [Google Scholar]
- Marmon, S.; Sturtevant, D.; Herrfurth, C.; Chapman, K.; Stymne, S.; Feussner, I. Two acyltransferases contribute differently to linolenic acid levels in seed oil. Plant Physiol. 2017, 173, 2081–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Rodrigo, M.; Siloto, P.; Aruna, D.; Wickramarathna, E. Identification of a pair of phospholipid:diacylglycerol acyltransferases from developing flax (Linum usitatissimum L.) seed catalyzing the selective production of trilinolenin. J. Biol. Chem. 2013, 288, 24173–24188. [Google Scholar] [CrossRef] [Green Version]
- Troncosoponce, M.A.; Kilaru, A.; Cao, X. Comparative deep transcriptional profiling of four developing oilseeds. Plant J. 2011, 68, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhao, S.; Wang, S.; Wang, H.; Zhang, Z. Identification and analysis of the FAD gene family in walnuts (Juglans regia L.) based on transcriptome data. BMC Genom. 2020, 21, 299. [Google Scholar] [CrossRef] [Green Version]
- Minic, Z.; Jamet, E.; SanClemente, H.; Pelletier, S.; Renou, J.P.; Rihouey, C.; Okinyo, D.P.; Lerouge, P.; Jouanin, L. Transcriptomic analysis of Arabidopsis developing stems: A close-up on cell wall genes. BMC Plant Biol. 2009, 9, 6. [Google Scholar] [CrossRef]
- Zhang, M.; Fan, J.; Taylor, D.C.; Ohlrogge, J.B. DGAT1 and PDAT1 acyltransferases have overlapping functions in Arabidopsis triacylglycerol biosynthesis and are essential for normal pollen and seed development. Plant Cell 2010, 21, 3885–3901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Lou, H.; Chen, W.; Zhou, Y.; Zhang, Z.; Xiao, M.; Wang, Z.; Yang, Y.; Yang, L.; Zhang, F.; et al. Comparative transcriptome analysis of the genes involved in lipid biosynthesis pathway and regulation of oil body formation in Torreya grandis kernels. Ind. Crop. Prod. 2020, 145, 112051. [Google Scholar] [CrossRef]
- Li, Y.; Sun, C.; Luo, H. Transcriptome characterization for Salvia miltiorrhiza using 454 GS FLX. Acta Pharm. Sin. 2010, 45, 524–529. [Google Scholar]
- He, C.; Poysa, V.; Yu, K. Development and characterization of Simple Sequence Repeat (SSR) markers and their use in determining relationships among Lycopersicon esculentum cultivars. Theor. Appl. Genet. 2003, 106, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Hovav, R.; Faigenboimdoron, A.; Kadmon, N. Atranscriptome profile for developing seed of polyploid cotton. Plant Genom. 2015, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ståhl, U.; Carlsson, A.S.; Lenman, M.; Dahlqvist, A.; Huang, B.; Banas, W.; Banas, A.; Stymne, S. Cloning and functional characterization of a phospholipid:diacylglycerol acyltransferase from Arabidopsis. Plant Physiol. 2004, 135, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Cronan, J.E.; Waldrop, G.L. Multi-subunit acetyl-CoA carboxylases. Prog. Lipid Res. 2002, 41, 407–435. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Tan, H.; Zheng, Q. LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol. 2008, 148, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sun, Q.; Zhao, S. Plant Physiological and Biochemical Principles and Experimental Techniques; Higher Education Press (HEP): Beijing, China, 2000; pp. 184–185. [Google Scholar]
- Wei, J.; Wu, C.; Jiang, Y. Sample preparation optimization for determination of soluble sugar in red jujube fruits by anthrone method. Food Sci. 2014, 35, 136–140. [Google Scholar]
- GB/T 5511-2008; Cereals and Pulses. Determination of the Nitrogen Content and Calculation of the Protein Content—Kjeldahl Method. ISO: Geneva, Switzerland, 2006.
- Thompson, O.; Meyenn, F.; Hewitt, Z.; Alexander, J.; Wood, A.; Weightman, R.; Gregory, S.; Krueger, F.; Andrews, S.; Barbaric, I.; et al. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nat. Commun. 2020, 23, 1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Tian, F.; Yang, D.; Meng, Y.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, 1040–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Luo, X.; Zhan, X.; Hao, J.; Zhang, L.; Song, Y.; Shen, C.; Dong, M. Comparative metabolomics reveals the metabolic variations between two endangered Taxus species (T. fuana and T. yunnanensis) in the Himalayas. BMC Plant Biol. 2018, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fang, J.; Qi, X.; Lin, M.; Zhong, Y.; Sun, L.; Cui, W. Combined Analysis of the Fruit Metabolome and Transcriptome Reveals Candidate Genes Involved in Flavonoid Biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, M.; Zhang, X.; Dong, Q.; Li, H.; Guo, S.; Luan, H.; Jia, P.; Yang, M.; Qi, G. Metabolomics and Transcriptomics Provide Insights into Lipid Biosynthesis in the Embryos of Walnut (Juglans regia L.). Plants 2023, 12, 538. https://doi.org/10.3390/plants12030538
Liang M, Zhang X, Dong Q, Li H, Guo S, Luan H, Jia P, Yang M, Qi G. Metabolomics and Transcriptomics Provide Insights into Lipid Biosynthesis in the Embryos of Walnut (Juglans regia L.). Plants. 2023; 12(3):538. https://doi.org/10.3390/plants12030538
Chicago/Turabian StyleLiang, Manman, Xuemei Zhang, Qinglong Dong, Han Li, Suping Guo, Haoan Luan, Peng Jia, Minsheng Yang, and Guohui Qi. 2023. "Metabolomics and Transcriptomics Provide Insights into Lipid Biosynthesis in the Embryos of Walnut (Juglans regia L.)" Plants 12, no. 3: 538. https://doi.org/10.3390/plants12030538