

Thermosensitive Polymeric Nanoparticles for Drug Co-Encapsulation and Breast Cancer Treatment
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Peptide Synthesis
2.3. Nanoparticle Generation
2.4. SILY Peptide Attachment
2.5. NP-SILY Characterization
2.6. NP-SILY In Vitro Collagen-Binding Assay
2.7. Drug Loading and Release from NP-SILY
2.8. In Vitro Nanoparticle Efficacy and Uptake in Cells and Spheroid
2.8.1. Cytotoxicity of Nanoparticles in Cell Monolayers
2.8.2. Cytotoxicity of Nanoparticles in Spheroids
2.8.3. Assessment of Nanoparticle Uptake
2.9. In Vitro Inhibition of MK2
2.10. Retention in Mammary Tissue
2.11. In Vivo Proof of Concept of Nanoparticle Efficacy
2.12. Data Analysis
3. Results
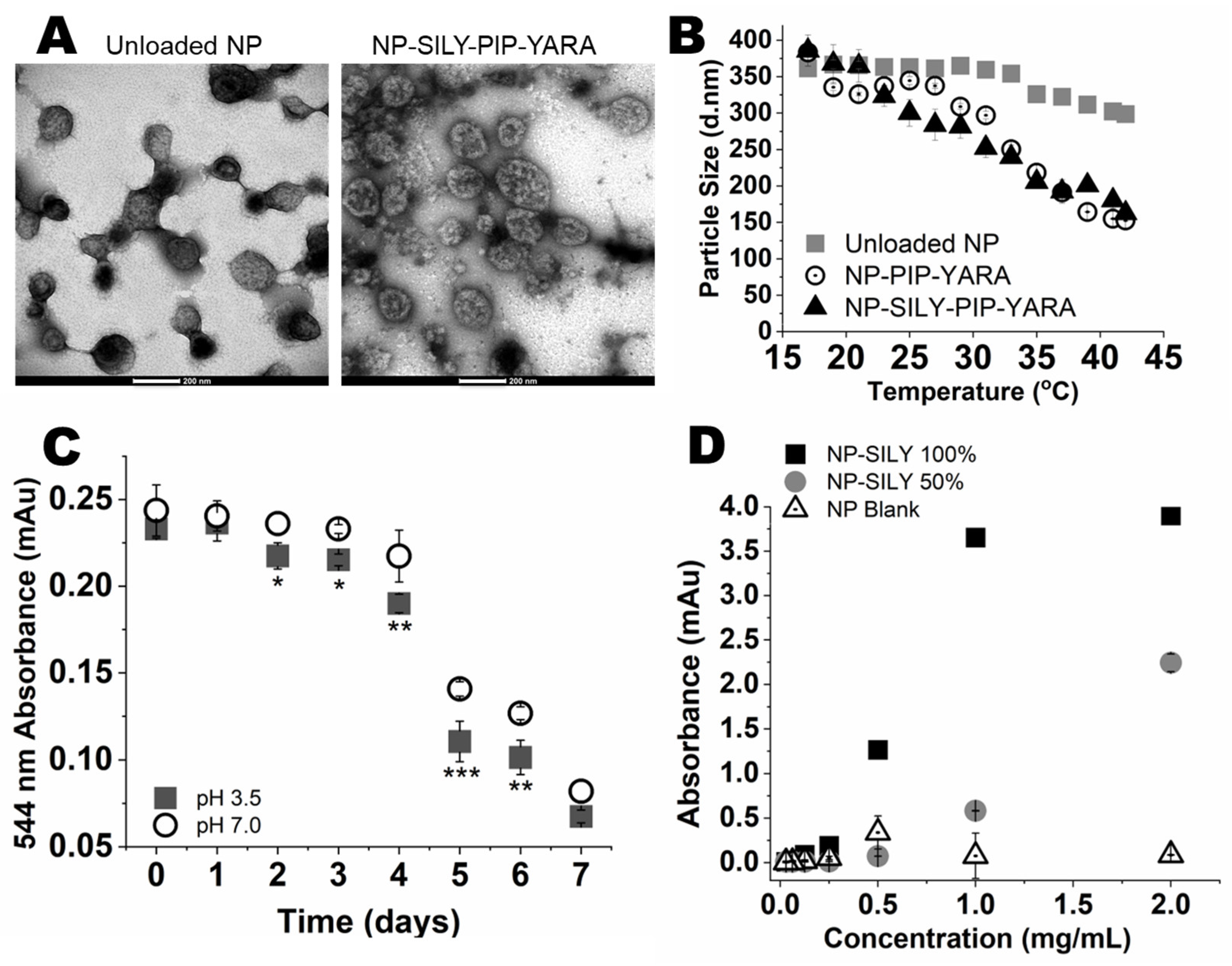
3.1. Nanoparticle Characterization
3.2. In Vitro Collagen Binding
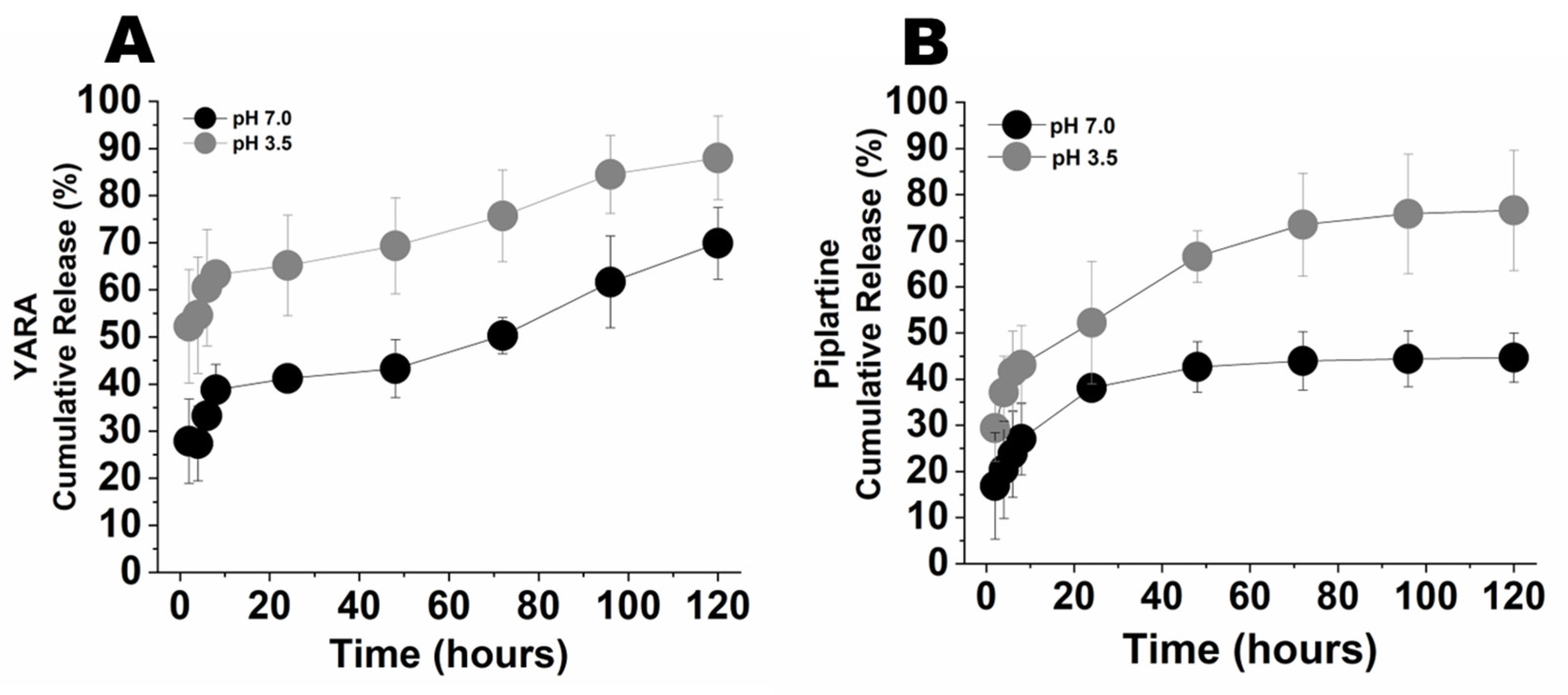
3.3. Drug Loading and Release
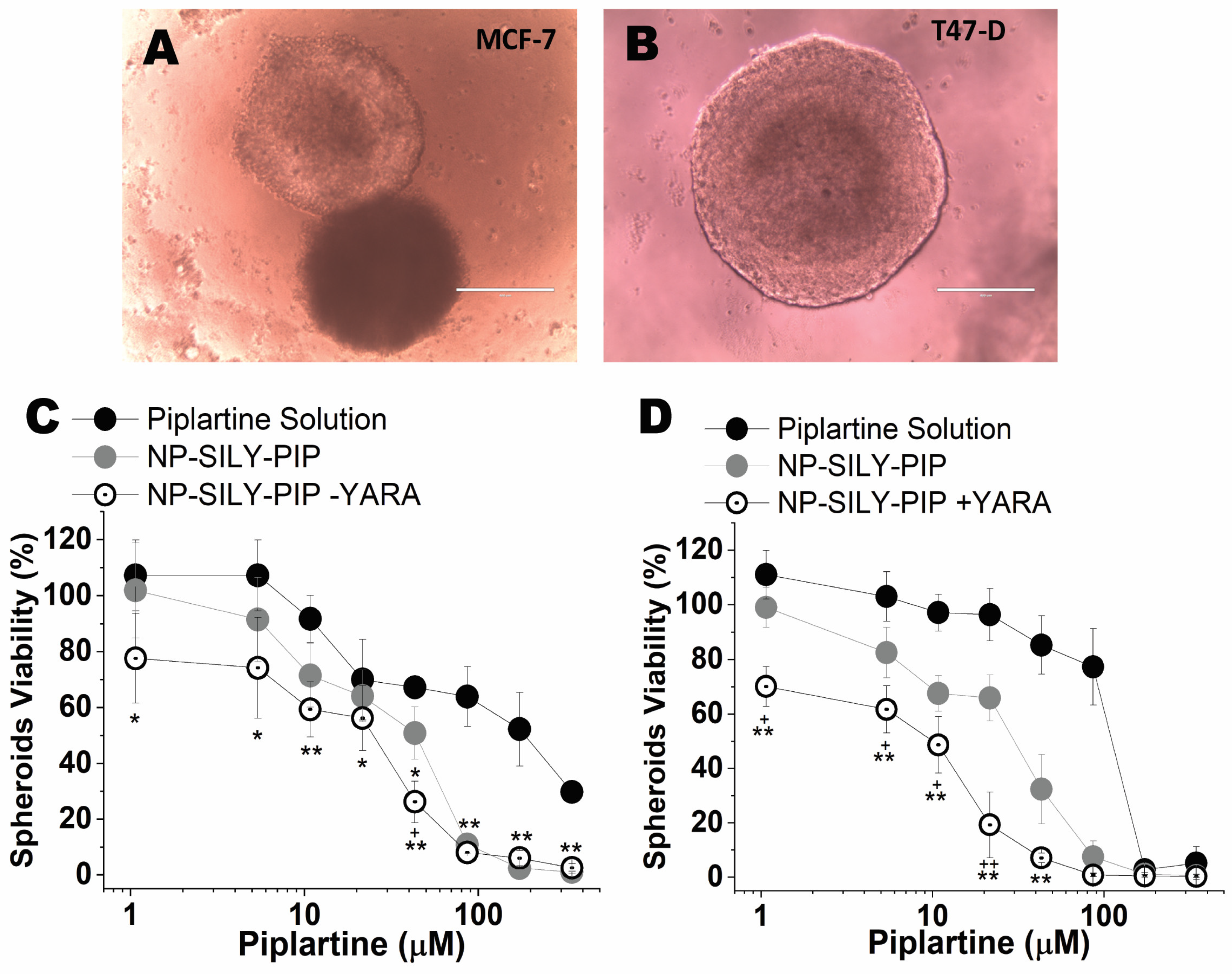
3.4. In Vitro Nanoparticle Effects on the Viability of Cell Monolayers and Spheroids
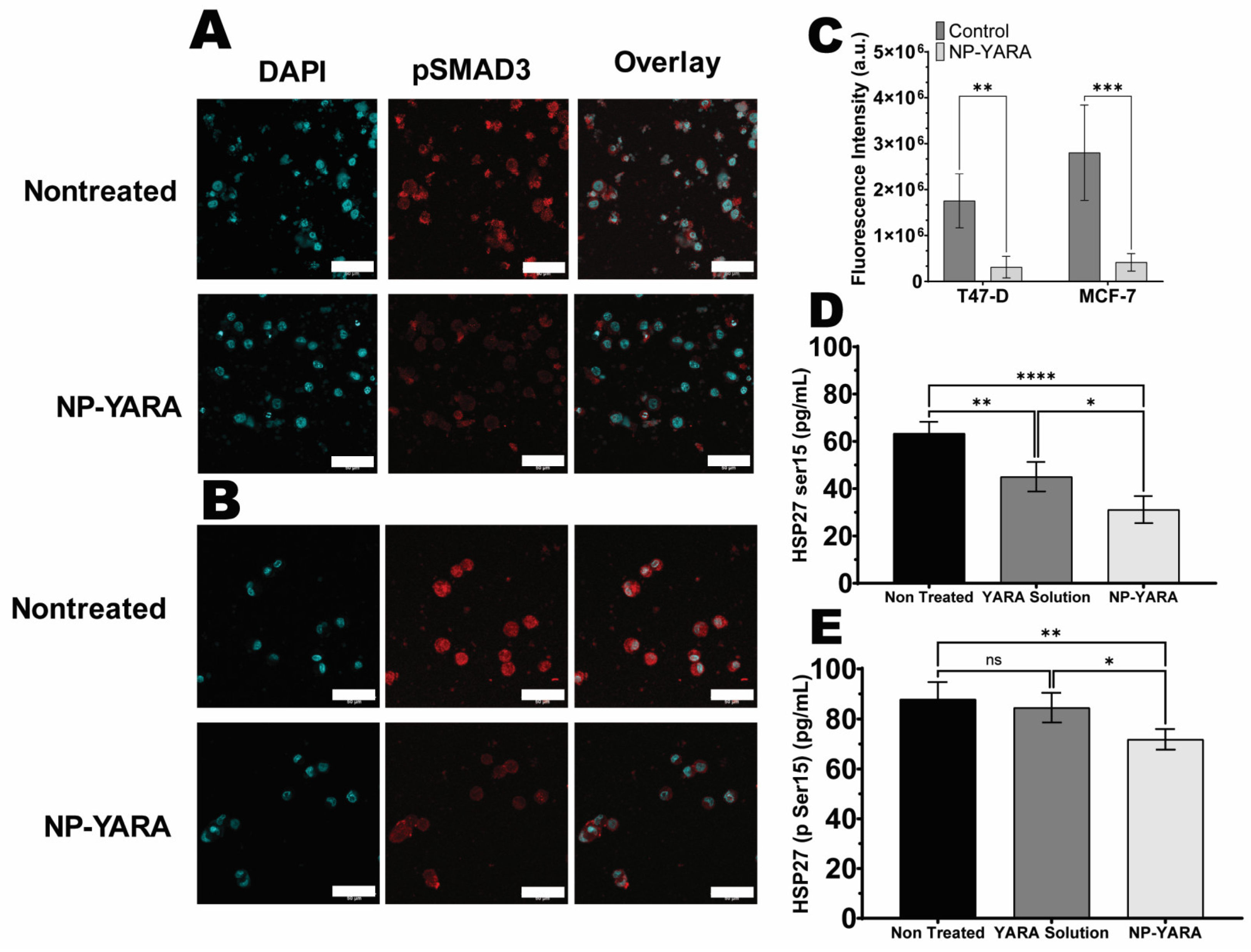
3.4.1. MK2 Inhibition by YARA-Loaded Nanoparticles
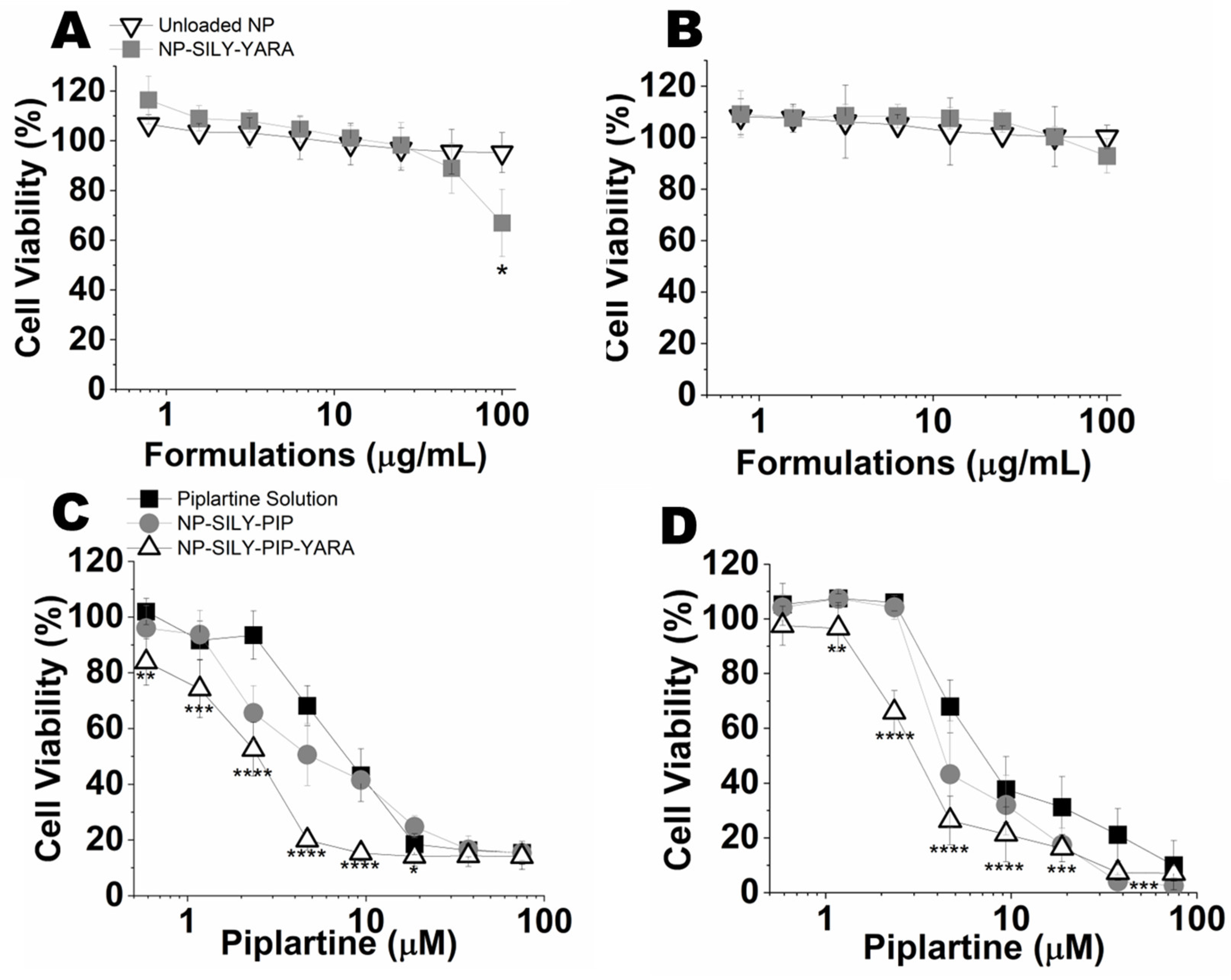
3.4.2. Evaluation of Cell Viability (2D Model)
3.4.3. Evaluation of the Viability of Spheroids (3D Model)
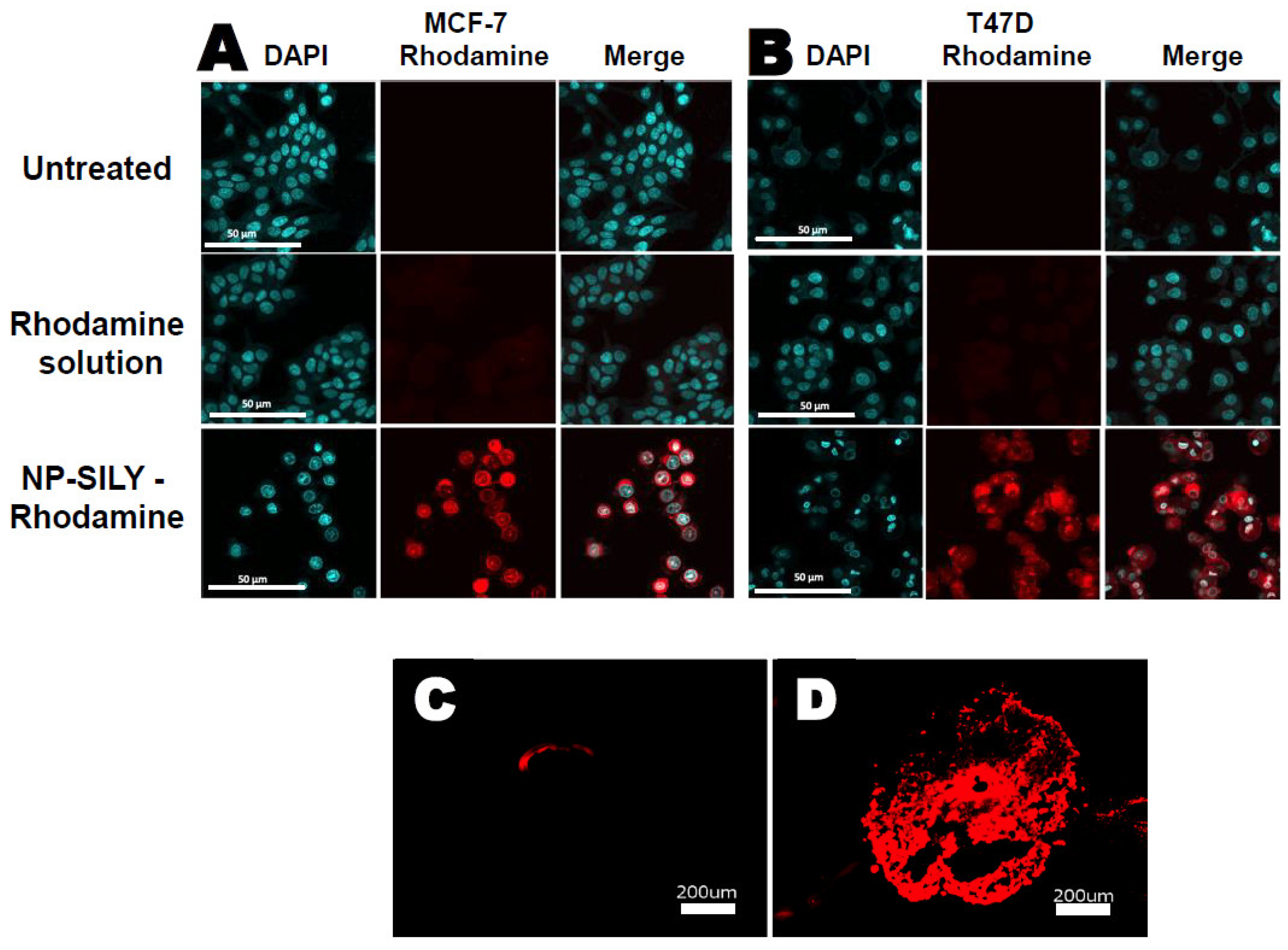
3.5. Nanoparticle Uptake Studies
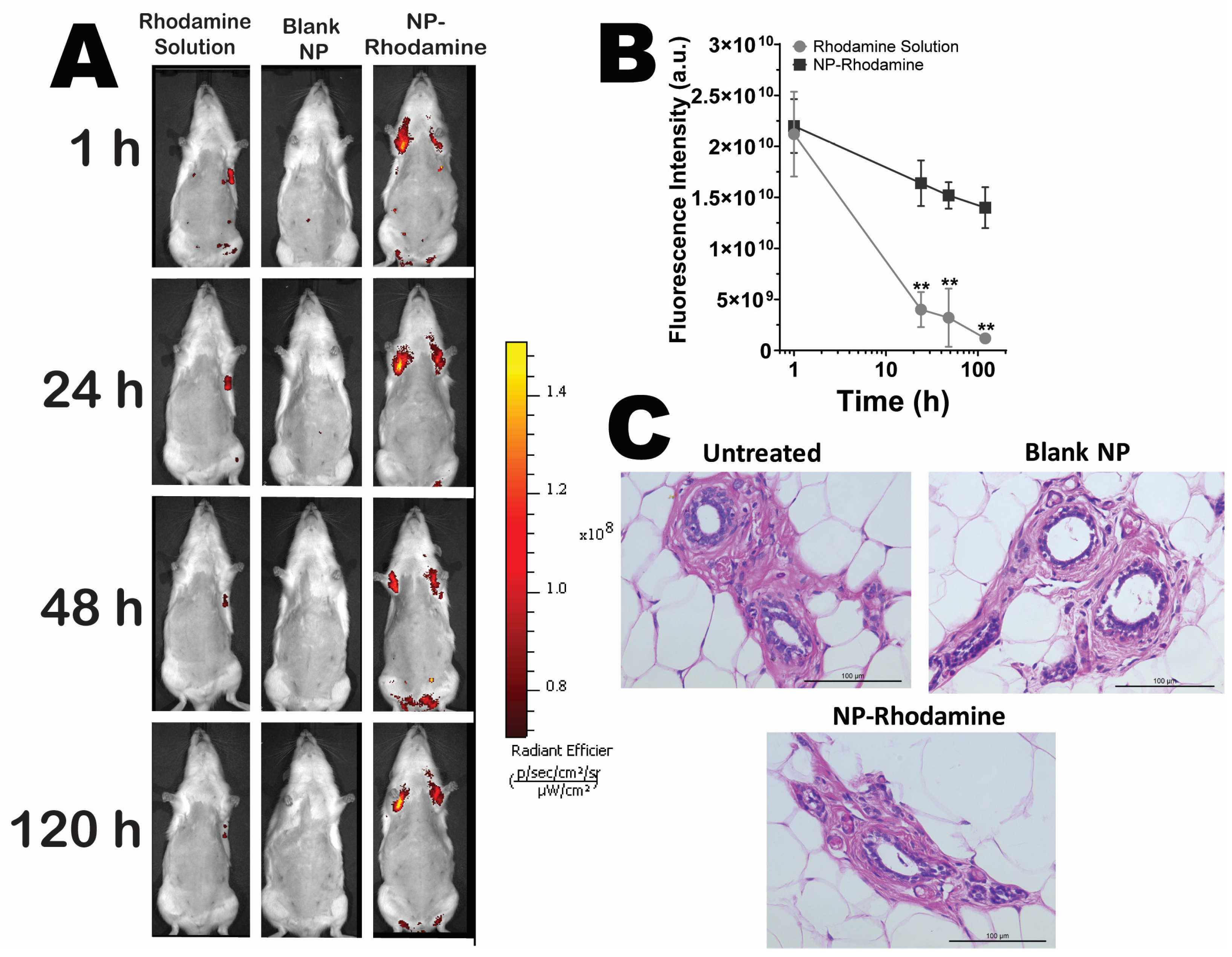
3.6. Retention of Nanoparticles in Mammary Tissue upon Intraductal Administration
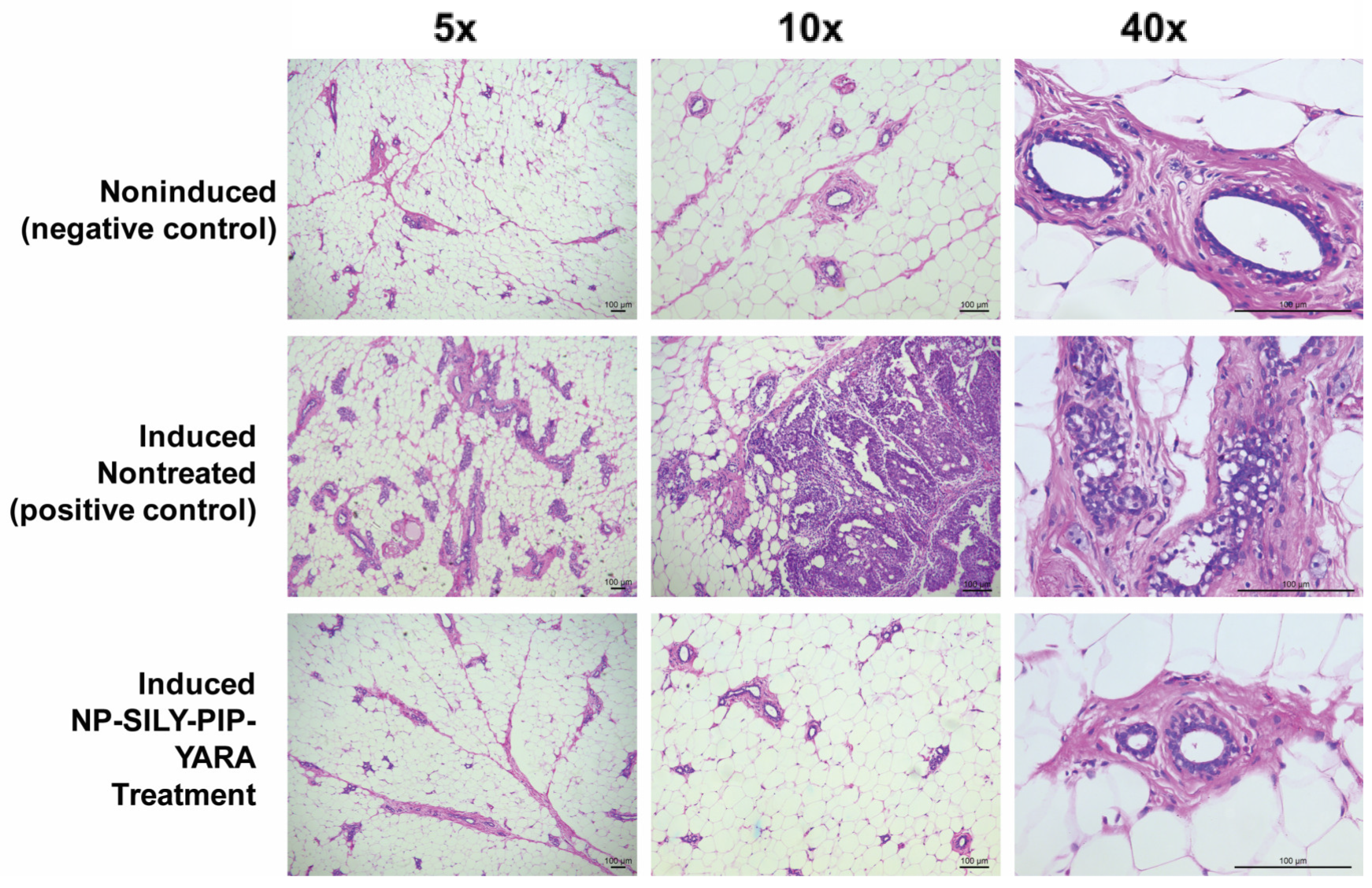
3.7. Effectiveness of Nanoparticles in an In Vivo Carcinogenesis Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wellings, S.R.; Jensen, H.M.; Marcum, R.G. An atlas of subgross pathology of the human breast with special reference to possible precancerous lesions. JNCI J. Natl. Cancer Inst. 1975, 55, 231–273. [Google Scholar] [CrossRef]
- Zhang, B.; Love, S.M.; Chen, G.; Wang, J.; Gao, J.; Xu, X.; Wang, Z.; Wang, X. The safety parameters of the study on intraductal cytotoxic agent delivery to the breast before mastectomy. Chin. J. Cancer Res. 2014, 26, 579–587. [Google Scholar] [CrossRef]
- Ward, E.M.; DeSantis, C.E.; Lin, C.C.; Kramer, J.L.; Jemal, A.; Kohler, B.; Brawley, O.W.; Gansler, T. Cancer statistics: Breast cancer in situ. CA Cancer J. Clin. 2015, 65, 481–495. [Google Scholar] [CrossRef]
- Benson, J.R.; Jatoi, I.; Toi, M. Treatment of low-risk ductal carcinoma in situ: Is nothing better than something? Lancet Oncol. 2016, 17, e442–e451. [Google Scholar] [CrossRef]
- Yuan, Y.; Liu, H.; Sahin, A.; Le Dai, J. Reactivation of SYK expression by inhibition of DNA methylation suppresses breast cancer cell invasiveness. Int. J. Cancer 2004, 113, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Nakhlis, F.; Morrow, M. Ductal carcinoma in situ. Surg. Clin. N. Am. 2003, 83, 821–839. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, L.; Francis, A. Overtreatment of Low-Grade Ductal Carcinoma In Situ. JAMA Oncol. 2016, 2, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Elshof, L.E.; Visser, L.L.; Rutgers, E.J.T.; Winter-Warnars, H.A.; Lips, E.H.; Wesseling, J. Finding the balance between over- and under-treatment of ductal carcinoma in situ (DCIS). Breast 2016, 31, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.E.; Gordon, E.J.; Rao, J.Y.; Jin, Y.; Hylton, N.; Love, S.M. Intraductal therapy of ductal carcinoma in situ: A presurgery study. Clin. Breast Cancer 2013, 13, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, H.; Yamamoto, D.; Uemura, Y.; Sakaida, N.; Tanano, A.; Tanaka, K.; Kamiyama, Y. Effect of perductal paclitaxel exposure on the development of MNU-induced mammary carcinoma in female S–D rats. Breast Cancer Res. Treat. 2005, 91, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Kominsky, S.L.; Vali, M.; Zhang, Z.; Garrett-Mayer, E.; Korz, D.; Huso, D.; Baker, S.D.; Barber, J.; Jaffee, E.; et al. Ductal access for prevention and therapy of mammary tumors. Cancer Res. 2006, 66, 638–645. [Google Scholar] [CrossRef]
- Chun, Y.S.; Bisht, S.; Chenna, V.; Pramanik, D.; Yoshida, T.; Hong, S.-M.; de Wilde, R.F.; Zhang, Z.; Huso, D.L.; Zhao, M.; et al. Intraductal administration of a polymeric nanoparticle formulation of curcumin (NanoCurc) significantly attenuates incidence of mammary tumors in a rodent chemical carcinogenesis model: Implications for breast cancer chemoprevention in at-risk populations. Carcinogenesis 2012, 33, 2242–2249. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.; Brock, A.; Ingber, D.E. Intraductal injection for localized drug delivery to the mouse mammary gland. J. Vis. Exp. 2013, 80, 50692. [Google Scholar]
- Stearns, V.; Mori, T.; Jacobs, L.K.; Khouri, N.F.; Gabrielson, E.; Yoshida, T.; Kominsky, S.L.; Huso, D.L.; Jeter, S.; Powers, P.; et al. Preclinical and clinical evaluation of intraductally administered agents in early breast cancer. Sci. Transl. Med. 2011, 3, 106ra108. [Google Scholar] [CrossRef] [PubMed]
- Love, S.M.; Zhang, W.; Gordon, E.J.; Rao, J.; Yang, H.; Li, J.; Zhang, B.; Wang, X.; Chen, G.; Zhang, B. A feasibility study of the intraductal administration of chemotherapy. Cancer Prev. Res. 2013, 6, 51–58. [Google Scholar] [CrossRef]
- Lee, O.; Khan, S.A. Novel routes for administering chemoprevention: Local transdermal therapy to the breasts. Semin. Oncol. 2016, 43, 107–115. [Google Scholar] [CrossRef]
- Carvalho, V.F.; Salata, G.C.; de Matos, J.K.; Costa-Fernandez, S.; Chorilli, M.; Steiner, A.A.; de Araujo, G.L.; Silveira, E.R.; Costa-Lotufo, L.V.; Lopes, L.B. Optimization of composition and obtainment parameters of biocompatible nanoemulsions intended for intraductal administration of piplartine (piperlongumine) and mammary tissue targeting. Int. J. Pharm. 2019, 567, 118460. [Google Scholar] [CrossRef]
- Migotto, A.; Carvalho, V.F.M.; Salata, G.C.; da Silva, F.W.M.; Yan, C.Y.I.; Ishida, K.; Costa-Lotufo, L.V.; Steiner, A.A.; Lopes, L.B. Multifunctional nanoemulsions for intraductal delivery as a new platform for local treatment of breast cancer. Drug Deliv. 2018, 25, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Gao, D.; Gu, Z.; Li, S.; Rivera, K.A.; Stein, S.; Love, S.; Sinko, P.J. Influence of molecular size on the retention of polymeric nanocarrier diagnostic agents in breast ducts. Pharm. Res. 2012, 29, 2377–2388. [Google Scholar] [CrossRef]
- Al-Zubaydi, F.; Gao, D.; Kakkar, D.; Li, S.; Adler, D.; Holloway, J.; Szekely, Z.; Gu, Z.; Chan, N.; Kumar, S.; et al. Breast intraductal nanoformulations for treating ductal carcinoma in situ I: Exploring metal-ion complexation to slow ciclopirox release, enhance mammary persistence and efficacy. J. Control. Release 2020, 323, 71–82. [Google Scholar] [CrossRef]
- Dartora, V.F.; Salata, G.C.; Passos, J.S.; Branco, P.C.; Silveira, E.; Steiner, A.A.; Costa-Lotufo, L.V.; Lopes, L.B. Hyaluronic acid nanoemulsions improve piplartine cytotoxicity in 2D and 3D breast cancer models and reduce tumor development after intraductal administration. Int. J. Biol. Macromol. 2022, 219, 84–95. [Google Scholar] [CrossRef]
- Passos, J.S.; Dartora, V.F.; Salata, G.C.; Malagó, I.D.; Lopes, L.B. Contributions of nanotechnology to the intraductal drug delivery for local treatment and prevention of breast cancer. Int. J. Pharm. 2023, 635, 122681. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Shymborska, Y.; Budkowski, A.; Raczkowska, J.; Donchak, V.; Melnyk, Y.; Vasiichuk, V.; Stetsyshyn, Y. Switching it Up: The Promise of Stimuli-Responsive Polymer Systems in Biomedical Science. Chem. Rec. 2023, e202300217. [Google Scholar] [CrossRef] [PubMed]
- Alsuraifi, A.; Curtis, A.; Lamprou, D.A.; Hoskins, C. Stimuli Responsive Polymeric Systems for Cancer Therapy. Pharmaceutics 2018, 10, 136. [Google Scholar] [CrossRef]
- Nagase, K.; Yamato, M.; Kanazawa, H.; Okano, T. Poly(N-isopropylacrylamide)-based thermoresponsive surfaces provide new types of biomedical applications. Biomaterials 2018, 153, 27–48. [Google Scholar] [CrossRef]
- Poh, S.; Lin, J.B.; Panitch, A. Release of anti-inflammatory peptides from thermosensitive nanoparticles with degradable cross-links suppresses pro-inflammatory cytokine production. Biomacromolecules 2015, 16, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.K.; Martin, J.R.; Werfel, T.A.; Shen, T.; Page, J.M.; Duvall, C.L. Cell protective, ABC triblock polymer-based thermoresponsive hydrogels with ROS-triggered degradation and drug release. J. Am. Chem. Soc. 2014, 136, 14896–14902. [Google Scholar] [CrossRef]
- McMasters, J.; Panitch, A. Collagen-binding nanoparticles for extracellular anti-inflammatory peptide delivery decrease platelet activation, promote endothelial migration, and suppress inflammation. Acta Biomater. 2017, 49, 78–88. [Google Scholar] [CrossRef]
- Deloney, M.; Smart, K.; Christiansen, B.A.; Panitch, A. Thermoresponsive, hollow, degradable core-shell nanoparticles for intra-articular delivery of anti-inflammatory peptide. J. Control. Release 2020, 323, 47–58. [Google Scholar] [CrossRef]
- McMasters, J.; Poh, S.; Lin, J.B.; Panitch, A. Delivery of anti-inflammatory peptides from hollow PEGylated poly(NIPAM) nanoparticles reduces inflammation in an ex vivo osteoarthritis model. J. Control. Release 2017, 258, 161–170. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Militão, G.C.G.; de Castro, F.O.; Pessoa, C.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Elmiro, F.J.M.; Costa-Lotufo, L.V. Piplartine induces inhibition of leukemia cell proliferation triggering both apoptosis and necrosis pathways. Toxicol. In Vitro 2007, 21, 1–8. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Moura, D.J.; Rosa, R.M.; de Vasconcellos, M.C.; e Silva, A.C.R.; de Moraes, M.O.; Silveira, E.R.; Lima, M.A.S.; Henriques, J.A.P.; Costa-Lotufo, L.V.; et al. Evaluation of the genotoxicity of piplartine, an alkamide of Piper tuberculatum, in yeast and mammalian V79 cells. Mutat. Res. Toxicol. Environ. Mutagen. 2008, 652, 164–174. [Google Scholar] [CrossRef]
- Razzak, M. Basic research: MK2 and p53—A lethal pairing. Nat. Rev. Clin. Oncol. 2014, 11, 3. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Lee, M.K.; Mazzoletti, M.; Broggini, M.; Sabapathy, K. Cancer-derived p53 mutants suppress p53-target gene expression--potential mechanism for gain of function of mutant p53. Nucleic Acids Res. 2007, 35, 2093–2104. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Morandell, S.; Reinhardt, H.C.; Cannell, I.G.; Kim, J.S.; Ruf, D.M.; Mitra, T.; Couvillon, A.D.; Jacks, T.; Yaffe, M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.Y.; Vidnovic, N.; Ellisen, L.W.; Leong, C.-O. Mutant p53 mediates survival of breast cancer cells. Br. J. Cancer 2009, 101, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Tang, S.; Qi, Y.-K.; Wang, Z.-P.; Liu, L. Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat. Protoc. 2013, 8, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.; Seal, B.L.; Brophy, C.M.; Panitch, A. Design of a bioactive cell-penetrating peptide: When a transduction domain does more than transduce. J. Pept. Sci. 2009, 15, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fraylich, M.; Saunders, B.R. Thermoresponsive copolymers: From fundamental studies to applications. Colloid Polym. Sci. 2009, 287, 627–643. [Google Scholar] [CrossRef]
- McMasters, J.; Panitch, A. Prevention of Collagen-Induced Platelet Binding and Activation by Thermosensitive Nanoparticles. AAPS J. 2015, 17, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Dubbert, J.; Honold, T.; Pedersen, J.S.; Radulescu, A.; Drechsler, M.; Karg, M.; Richtering, W. How hollow are thermoresponsive hollow nanogels? Macromolecules 2014, 47, 8700–8708. [Google Scholar] [CrossRef]
- Carvalho, V.F.M.; Giacone, D.V.; Costa-Lotufo, L.V.; Silveira, E.R.; Lopes, L.B. Development of a method for quantitative determination of the cytotoxic agent piplartine (piperlongumine) in multiple skin layers. Biomed. Chromatogr. 2018, 33, e4386. [Google Scholar] [CrossRef] [PubMed]
- do Amaral, J.B.; Urabayashi, M.S.; Machado-Santelli, G.M. Cell death and lumen formation in spheroids of MCF-7 cells. Cell Biol. Int. 2010, 34, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.B.D.; Rezende-Teixeira, P.; Freitas, V.M.; Machado-Santelli, G.M. MCF-7 cells as a three-dimensional model for the study of human breast cancer. Tissue Eng. Part C Methods 2011, 17, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.P.; Dean, D.M.; Man, A.J.; Youssef, J.; Ho, D.N.; Rago, A.P.; Lech, M.P.; Morgan, J.R.; Lee, J.W.; Sung, J.S.; et al. Scaffold-free three-dimensional cell culture utilizing micromolded nonadhesive hydrogels. BioTechniques 2007, 43, 494–500. [Google Scholar] [CrossRef]
- Pereira, P.M.R.; Berisha, N.; Bhupathiraju, N.V.S.D.K.; Fernandes, R.; Tomé, J.P.C.; Drain, C.M. Cancer cell spheroids are a better screen for the photodynamic efficiency of glycosylated photosensitizers. PLoS ONE 2017, 12, e0177737. [Google Scholar] [CrossRef]
- Wenzel, C.; Riefke, B.; Gründemann, S.; Krebs, A.; Christian, S.; Prinz, F.; Osterland, M.; Golfier, S.; Räse, S.; Ansari, N.; et al. 3D high-content screening for the identification of compounds that target cells in dormant tumor spheroid regions. Exp. Cell Res. 2014, 323, 131–143. [Google Scholar] [CrossRef]
- Soni, S.; Anand, P.; Padwad, Y.S. MAPKAPK2: The master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J. Exp. Clin. Cancer Res. 2019, 38, 121. [Google Scholar] [CrossRef]
- Vittal, R.; Fisher, A.; Gu, H.; Mickler, E.A.; Panitch, A.; Lander, C.; Cummings, O.W.; Sandusky, G.E.; Wilkes, D.S. Peptide-mediated inhibition of mitogen-activated protein kinase-activated protein kinase-2 ameliorates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 47–57. [Google Scholar] [CrossRef]
- Thompson, H.J.; Adlakha, H. Dose-responsive induction of mammary gland carcinomas by the intraperitoneal injection of 1-methyl-1-nitrosourea. Cancer Res. 1991, 51, 3411–3415. [Google Scholar] [PubMed]
- Maffini, M.V.; Soto, A.M.; Calabro, J.M.; Ucci, A.A.; Sonnenschein, C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004, 117 Pt 8, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.H.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [PubMed]
- Salata, G.C.; Malagó, I.D.; Dartora, V.F.M.C.; Pessoa, A.F.M.; Fantini, M.C.D.A.; Costa, S.K.P.; Machado-Neto, J.A.; Lopes, L.B. Microemulsion for Prolonged Release of Fenretinide in the Mammary Tissue and Prevention of Breast Cancer Development. Mol. Pharm. 2021, 18, 3401–3417. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.L.; Medow, M.R.; Panitch, A.; Seal, B. Hemocompatible poly(NIPAm-MBA-AMPS) colloidal nanoparticles as carriers of anti-inflammatory cell penetrating peptides. Biomacromolecules 2012, 13, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.; Lyon, L.A. Tunable swelling kinetics in core—Shell hydrogel nanoparticles. J. Am. Chem. Soc. 2001, 123, 7511–7517. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Ding, Y.; Zhang, G. Microcalorimetric Investigation on the lower critical solution temperature behavior of N-isopropycrylamide-co-acrylic acid copolymer in aqueous solution. J. Phys. Chem. B 2006, 110, 11813–11817. [Google Scholar] [CrossRef]
- Jain, A.K.; Gund, M.G.; Desai, D.C.; Borhade, N.; Senthilkumar, S.P.; Dhiman, M.; Mangu, N.K.; Mali, S.V.; Dubash, N.P.; Halder, S.; et al. Mutual prodrugs containing bio-cleavable and drug releasable disulfide linkers. Bioorganic Chem. 2013, 49, 40–48. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolás, F.J.; Hill, C.S. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell 2002, 10, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Akel, S.; Bertolette, D.; Ruscetti, F.W. Crosstalk between the Smad and the Mitogen-Activated Protein Kinase Pathways is Essential for Erythroid Differentiation of Erythroleukemia Cells Induced by TGF-β, Activin, Hydroxyurea and Butyrate. J. Leuk. 2013, 1, 109. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.B.; Flynn, C.; Komalavilas, P.; Panitch, A.; Brophy, C.M.; Seal, B.L. Inhibition of HSP27 phosphorylation by a cell-permeant MAPKAP Kinase 2 inhibitor. Biochem. Biophys. Res. Commun. 2009, 382, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Brugnano, J.L.; Chan, B.K.; Seal, B.L.; Panitch, A. Cell-penetrating peptides can confer biological function: Regulation of inflammatory cytokines in human monocytes by MK2 inhibitor peptides. J. Control. Release 2011, 155, 128–133. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.; Wang, X.; Linding, R.; Ong, S.-E.; Weaver, D.; Carr, S.A.; et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, srep19103. [Google Scholar] [CrossRef]
- Kimlin, L.C.; Casagrande, G.; Virador, V.M. In vitro three-dimensional (3D) models in cancer research: An update. Mol. Carcinog. 2011, 52, 167–182. [Google Scholar] [CrossRef]
- Montanez-Sauri, S.I.; Sung, K.E.; Berthier, E.; Beebe, D.J. Enabling screening in 3D microenvironments: Probing matrix and stromal effects on the morphology and proliferation of T47D breast carcinoma cells. Integr. Biol. 2013, 5, 631–640. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Madhavan, M.; Jaiswal, D.; Karlberg, S.; Duggan, A.; Almarshad, H.A.; Claffey, K.P.; Hoshino, K. Electron microscopy imaging and mechanical characterization of T47D multicellular tumor spheroids–Older spheroids reduce interstitial space and become stiffer. PLoS ONE 2023, 18, e0286291. [Google Scholar] [CrossRef]
- Masso-Welch, P.A.; Darcy, K.M.; Stangle-Castor, N.C.; Ip, M.M. A developmental atlas of rat mammary gland histology. J. Mammary Gland. Biol. Neoplasia 2000, 5, 165–185. [Google Scholar] [CrossRef]
- Perše, M.; Cerar, A.; Injac, R.; Štrukelj, B. N-methylnitrosourea induced breast cancer in rat, the histopathology of the resulting tumours and its drawbacks as a model. Pathol. Oncol. Res. 2008, 15, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef]
- Bielawski, K.; Bielawska, A.; Muszyńska, A.; Popławska, B.; Czarnomysy, R. Cytotoxic activity of G3 PAMAM-NH2 dendrimer-chlorambucil conjugate in human breast cancer cells. Environ. Toxicol. Pharmacol. 2011, 32, 364–372. [Google Scholar] [CrossRef]
- Passos, J.S.; Lopes, L.B.; Panitch, A. Collagen-Binding Nanoparticles for Paclitaxel Encapsulation and Breast Cancer Treatment. ACS Biomater. Sci. Eng. 2023, 9, 6805–6820. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef]
- Sprague, B.L.; Vacek, P.M.; Mulrow, S.E.; Evans, M.F.; Trentham-Dietz, A.; Herschorn, S.D.; James, T.A.; Surachaicharn, N.; Keikhosravi, A.; Eliceiri, K.W.; et al. Collagen Organization in Relation to Ductal Carcinoma In Situ Pathology and Outcomes. Cancer Epidemiol. Biomark. Prev. 2020, 30, 80–88. [Google Scholar] [CrossRef]
- Karimi, M.; Zangabad, P.S.; Ghasemi, A.; Amiri, M.; Bahrami, M.; Malekzad, H.; Asl, H.G.; Mahdieh, Z.; Bozorgomid, M.; Ghasemi, A.; et al. Temperature-Responsive Smart Nanocarriers for Delivery of Therapeutic Agents: Applications and Recent Advances. ACS Appl. Mater. Interfaces 2016, 8, 21107–21133. [Google Scholar] [CrossRef] [PubMed]
- García-Peñas, A.; Biswas, C.S.; Liang, W.; Wang, Y.; Yang, P.; Stadler, F.J. Effect of Hydrophobic Interactions on Lower Critical Solution Temperature for Poly(N-isopropylacrylamide-co-dopamine Methacrylamide) Copolymers. Polymers 2019, 11, 991. [Google Scholar] [CrossRef] [PubMed]
- Ristroph, K.D.; Prud’Homme, R.K. Hydrophobic ion pairing: Encapsulating small molecules, peptides, and proteins into nanocarriers. Nanoscale Adv. 2019, 1, 4207–4237. [Google Scholar] [CrossRef] [PubMed]
- García-Salinas, M.J.; Romero-Cano, M.S.; de las Nieves, F.J. Colloidal stability of a temperature-sensitive poly(N-isopropylacrylamide/2-acrylamido-2-methylpropanesulphonic acid) microgel. J. Colloid Interface Sci. 2002, 248, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Juettner, V.V.; Hong, S. Biomolecular corona on nanoparticles: A survey of recent literature and its implications in targeted drug delivery. Front. Chem. 2014, 2, 108. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Feng, F.; Meng, F.; Deng, C.; Feijen, J.; Zhong, Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 2011, 152, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Meng, F.; Ma, S.; Xu, H.; Liu, H.; Jing, X.; Zhong, Z. Reduction and temperature dual-responsive crosslinked polymersomes for targeted intracellular protein delivery. J. Mater. Chem. 2011, 21, 19013–19020. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, E.-J.; Lim, J.H.; Cho, H.K.; Hong, W.J.; Jeon, H.H.; Chung, B.G. Dual Stimuli-Triggered Nanogels in Response to Temperature and pH Changes for Controlled Drug Release. Nanoscale Res. Lett. 2019, 14, 77. [Google Scholar] [CrossRef]
- Bhattacharjee, S. Understanding the burst release phenomenon: Toward designing effective nanoparticulate drug-delivery systems. Ther. Deliv. 2021, 12, 21–36. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Bai, X.; Smith, Z.L.; Wang, Y.; Butterworth, S.; Tirella, A. Sustained Drug Release from Smart Nanoparticles in Cancer Therapy: A Comprehensive Review. Micromachines 2022, 13, 1623. [Google Scholar] [CrossRef]
- Thirupathi, K.; Phan, T.T.V.; Santhamoorthy, M.; Ramkumar, V.; Kim, S.-C. pH and Thermoresponsive PNIPAm-co-Polyacrylamide Hydrogel for Dual Stimuli-Responsive Controlled Drug Delivery. Polymers 2022, 15, 167. [Google Scholar] [CrossRef]
- Vikulina, A.S.; Feoktistova, N.A.; Balabushevich, N.G.; von Klitzing, R.; Volodkin, D. Cooling-Triggered Release from Mesoporous Poly(N-isopropylacrylamide) Microgels at Physiological Conditions. ACS Appl. Mater. Interfaces 2020, 12, 57401–57409. [Google Scholar] [CrossRef]
- Lima, L.H.; Morales, Y.; Cabral, T. Ocular Biocompatibility of Poly-N-Isopropylacrylamide (pNIPAM). J. Ophthalmol. 2016, 2016, 5356371. [Google Scholar] [CrossRef]
- Dartora, V.F.C. Desenvolvimento de Nanocarreadores Multifuncionais Para Co-Localização Cutânea de Agentes Quimioterápicos. Master’s Thesis, Universidade de São Paulo, São Paulo, Brazil, 2016. [Google Scholar]
- Valencia, P.M.; Pridgen, E.M.; Perea, B.; Gadde, S.; Sweeney, C.; Kantoff, P.W.; Bander, N.H.; Lippard, S.J.; Langer, R.; Karnik, R.; et al. Synergistic cytotoxicity of irinotecan and cisplatin in dual-drug targeted polymeric nanoparticles. Nanomedicine 2013, 8, 687–698. [Google Scholar] [CrossRef]
- Li, Y.; Köpper, F.; Dobbelstein, M. Inhibition of MAPKAPK2/MK2 facilitates DNA replication upon cancer cell treatment with gemcitabine but not cisplatin. Cancer Lett. 2018, 428, 45–54. [Google Scholar] [CrossRef]
- Guo, M.; Sun, D.; Fan, Z.; Yuan, Y.; Shao, M.; Hou, J.; Zhu, Y.; Wei, R.; Zhu, Y.; Qian, J.; et al. Targeting MK2 Is a Novel Approach to Interfere in Multiple Myeloma. Front. Oncol. 2019, 9, 722. [Google Scholar] [CrossRef] [PubMed]
- Sanzari, I.; Buratti, E.; Huang, R.; Tusan, C.G.; Dinelli, F.; Evans, N.D.; Prodromakis, T.; Bertoldo, M. Poly(N-isopropylacrylamide) based thin microgel films for use in cell culture applications. Sci. Rep. 2020, 10, 6126. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Ma, C.; Li, C.; Wang, T.; Tang, Y.; Wang, H.; Mou, X.; Chen, Z.; He, N. Influence of nanoparticle shape, size, and surface functionalization on cellular uptake. J. Nanosci. Nanotechnol. 2013, 13, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Sanità, G.; Carrese, B.; Lamberti, A. Nanoparticle Surface Functionalization: How to Improve Biocompatibility and Cellular Internalization. Front. Mol. Biosci. 2020, 7, 587012. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanocarrier | Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|
| Unloaded NPs | 370.7 ± 11.5 | 0.15 ± 0.007 | −31.3 ± 8.4 |
| NP-PIP-YARA | 380.6 ± 13.4 | 0.21 ± 0.06 | −25.9 ± 2.05 |
| NP-SILY-PIP-YARA | 387.1 ± 12.8 | 0.22 ± 0.02 | −8.9 ± 0.38 |
| Nanocarrier | YARA Loading (mg/mg) | Piplartine Loading (mg/mg) |
|---|---|---|
| Non-modified NPs | 0.753 ± 0.05 | 0.921 ± 0.30 |
| NP-SILY | 1.262 ± 0.10 | 0.733 ± 0.04 |
| Treatment | Cell Line and IC50 (μM) | 95% CI (µM) | ||
|---|---|---|---|---|
| MCF-7 | T47-D | MCF-7 | T47-D | |
| Piplartine solution | 10.6 | 9.5 | 5.2–20.6 | 4.9–15.8 |
| NP-SILY-PIP | 6.6 | 6.1 | 3.2–12.0 | 4.1–10.4 |
| NP-SILY-PIP-YARA | 4.0 | 2.3 | 2.2–8.0 | 0.7–3.4 |
| Treatment | Cell Line and IC50 (μM) | 95% CI (µM) | ||
|---|---|---|---|---|
| MCF-7 | T47-D | MCF-7 | T47-D | |
| Piplartine solution | 133.8 | 104.8 | 113.4–158.1 | 84.7–130.0 |
| NP-SILY-PIP | 27.2 | 22.7 | 25.5–34.3 | 19.9–25.6 |
| NP-SILY-PIP-YARA | 16.6 | 6.7 | 14.1–19.4 | 5.7–7.7 |
| Group | Induced Nontreated | NP-SILY-PIP-YARA | Control Uninduced |
|---|---|---|---|
| Incidence (%) | 75 | 14.2 | 0.0 |
| Multiplicity | 1.83 ± 0.9 | 0.14 ± 0.3 | 0.0 |
| Size (mm3) | 28.3 ± 8.2 | 4.6 ± 1.8 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dartora, V.F.C.; Passos, J.S.; Costa-Lotufo, L.V.; Lopes, L.B.; Panitch, A. Thermosensitive Polymeric Nanoparticles for Drug Co-Encapsulation and Breast Cancer Treatment. Pharmaceutics 2024, 16, 231. https://doi.org/10.3390/pharmaceutics16020231
Dartora VFC, Passos JS, Costa-Lotufo LV, Lopes LB, Panitch A. Thermosensitive Polymeric Nanoparticles for Drug Co-Encapsulation and Breast Cancer Treatment. Pharmaceutics. 2024; 16(2):231. https://doi.org/10.3390/pharmaceutics16020231
Chicago/Turabian StyleDartora, Vanessa Franco Carvalho, Julia S. Passos, Leticia V. Costa-Lotufo, Luciana B. Lopes, and Alyssa Panitch. 2024. "Thermosensitive Polymeric Nanoparticles for Drug Co-Encapsulation and Breast Cancer Treatment" Pharmaceutics 16, no. 2: 231. https://doi.org/10.3390/pharmaceutics16020231