
A Single-Run HPLC–MS Multiplex Assay for Therapeutic Drug Monitoring of Relevant First- and Second-Line Antibiotics in the Treatment of Drug-Resistant Tuberculosis

 , , , , , and add
Show full author list
, , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Definition of an Assay Target Profile
2.2. Chemicals
2.3. Sample Preparation
2.4. HPLC–MS/MS
2.4.1. MS/MS
2.4.2. HPLC
2.5. Extraction Procedure
2.6. Validation
2.6.1. Calibration
2.6.2. Carry-Over, Selectivity, and Specificity
2.6.3. Recovery and Matrix Effect
2.6.4. Accuracy and Precision
2.7. Clinical Application
2.7.1. Patient Histories
2.7.2. Sample Collection and Management
3. Results
3.1. Definition of an Assay Target Profile
3.2. HPLC-MS/MS
3.2.1. MS/MS
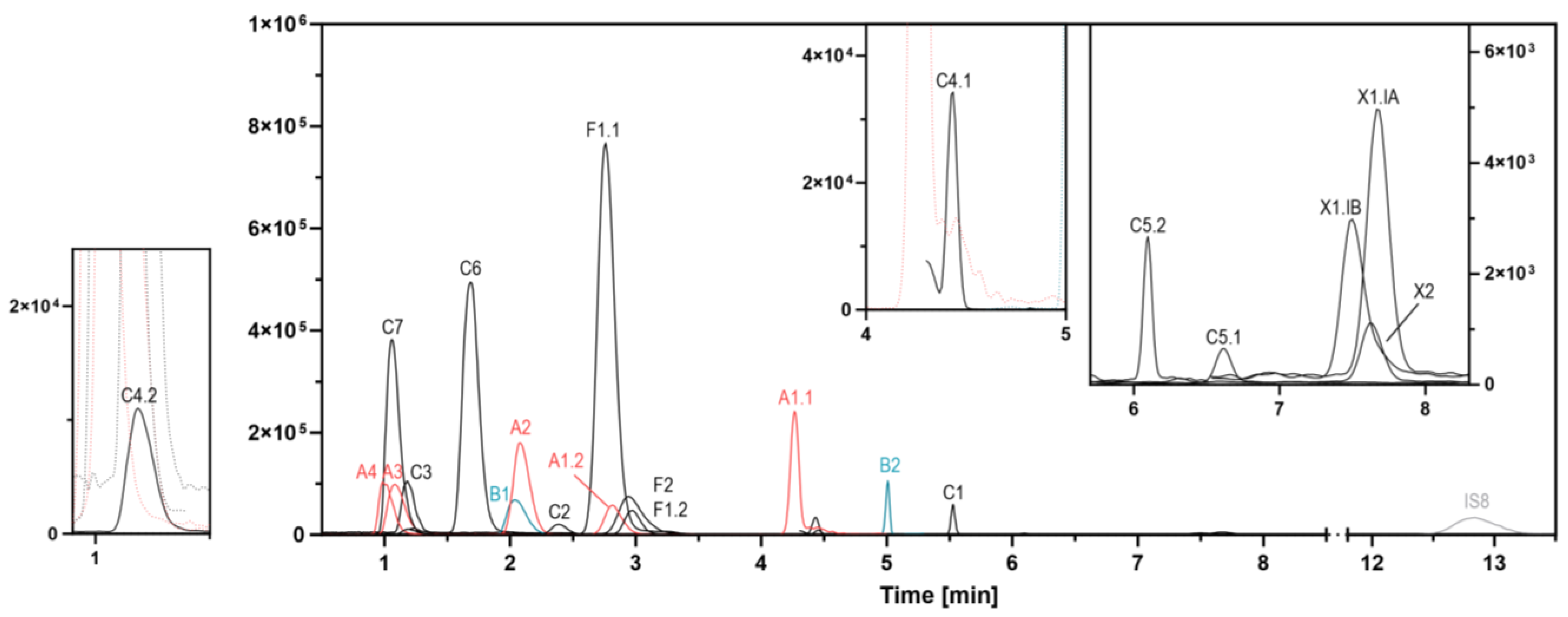
3.2.2. HPLC
3.3. Validation
3.3.1. Calibration
3.3.2. Carry-Over, Selectivity, and Specificity
3.3.3. Recovery and Matrix Effect
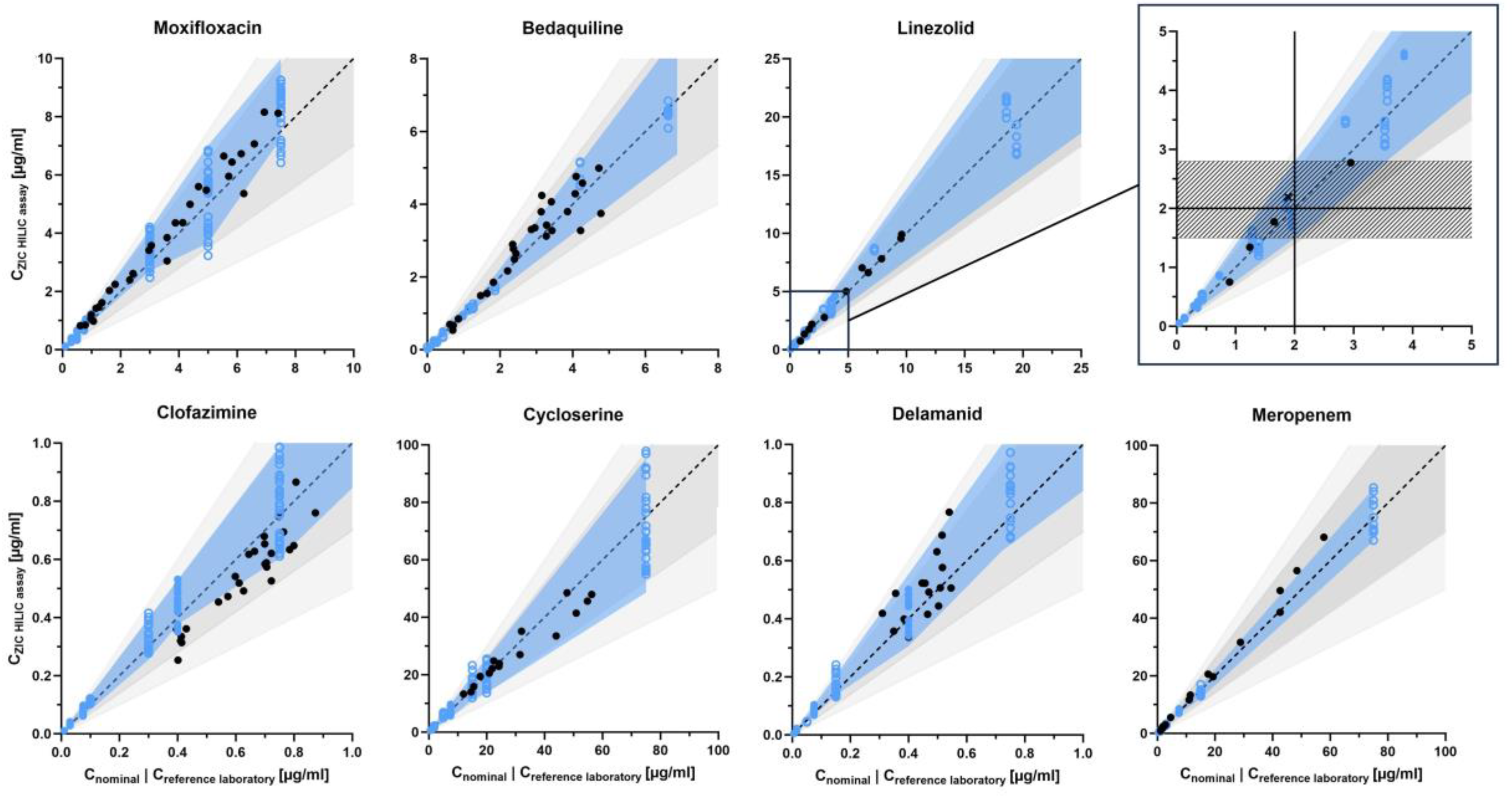
3.3.4. Accuracy and Precision
- Linezolid, delamanid, meropenem, and prothionamide were strictly within the EMA/FDA-recommended range of 85–115% accuracy and 0–15% within-day and between day precision [14,15]. The expected deviation from the nominal concentration in the form of the 80% beta-expectation tolerance interval was up to ±30% in meropenem and up to approximately ±40% in linezolid, delamanid, and prothionamide (linezolid: −19.3% to +40.4%, ethambutol: −9.7% to +41.0%, both at QClow).
- Moxifloxacin, clofazimine, cycloserine, and ethambutol showed accuracy of 80–120% and precision of approximately 0–20% (moxifloxacin between-day precision: 20.6%, ethambutol accuracy: 123.4%, both at QCmed). 80% beta-expectation tolerance intervals were within approximately ±40% (moxifloxacin: −26.9% to +40.3% at QCmed, ethambutol: −9.7% to +41.0% at QClow).
- Rifampicin, rifabutin, levofloxacin, bedaquiline, pyrazinamide, and PAS showed accuracy of approximately 80–120% and precision of approximately 0–20% with higher deviations at QClow: rifampicin with an accuracy of 122.2%, rifabutin with a between-day precision of 20.8%, and bedaquiline with a between-day precision of 24.4%. Correspondingly, rifampicin, rifabutin, levofloxacin, bedaquiline, pyrazinamide, and PAS showed higher expected deviations at low concentrations, with an 80% beta-expectation tolerance interval of up to ±60% at QClow and ±30% at QCmed and QChigh for bedaquiline and pyrazinamide, as well as ±40% at QCmed and QChigh for rifampicin, rifabutin, levofloxacin, and PAS (PAS: −23.3% to +42.9% and −40.3% to +21.9% at QCmed and QChigh, respectively).
- Amikacin was evaluated based on only one set of QClow, QCmed, and QChigh, hence stratified between-day precision could not be calculated and overall accuracy (93.5%), within-day (6.3%), and between-day precision (6.3%) were determined instead. The overall 80% beta-expectation tolerance interval was −15.2% to +2.2%.
- Isoniazid, pretomanid, streptomycin, capreomycin IB and IA, as well as kanamycin showed inadequate accuracy and/or within- and between-day precision. 80% beta-expectation tolerance intervals of isoniazid, pretomanid, streptomycin, capreomycin IB and IA, and kanamycin partly exceeded 100%. Quantification of isoniazid showed systematic deviations, with accuracies of 141.2% to 160.3%.
3.4. Clinical Application
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. Meeting Report of the WHO Expert Consultation on the Definition of Extensively Drug-Resistant Tuberculosis, 27–29 October 2020; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis: Module 4: Treatment: Drug-Resistant Tuberculosis Treatment, 2022nd ed.; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Srivastava, S.; Pasipanodya, J.G.; Meek, C.; Leff, R.; Gumbo, T. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J. Infect. Dis. 2011, 204, 1951–1959. [Google Scholar] [CrossRef]
- Song, T.; Lee, M.; Jeon, H.S.; Park, Y.; Dodd, L.E.; Dartois, V.; Follman, D.; Wang, J.; Cai, Y.; Goldfeder, L.C.; et al. Linezolid Trough Concentrations Correlate with Mitochondrial Toxicity-Related Adverse Events in the Treatment of Chronic Extensively Drug-Resistant Tuberculosis. EBioMedicine 2015, 2, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Court, R.; Centner, C.M.; Chirehwa, M.; Wiesner, L.; Denti, P.; de Vries, N.; Harding, J.; Gumbo, T.; Maartens, G.; McIlleron, H. Neuropsychiatric toxicity and cycloserine concentrations during treatment for multidrug-resistant tuberculosis. Int. J. Infect. Dis. 2021, 105, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.; Bangert, S. Clinical Chemistry, 6th ed.; Mosby Elsevier: Edinburgh, Scotland; London, UK, 2008. [Google Scholar]
- Raju, K.S.R.; Gundeti, M.; Malik, M.Y.; Kadian, N.; Rashid, M.; Taneja, I.; Singh, S.P.; Wahajuddin, M. Bioanalysis of antitubercular drugs using liquid chromatography. J. Pharm. Biomed. Anal. 2017, 134, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Margineanu, I.; Akkerman, O.; Cattaneo, D.; Goletti, D.; Marriott, D.J.E.; Migliori, G.B.; Mirzayev, F.; Peloquin, C.A.; Stienstra, Y.; Alffenaar, J.W. Practices of therapeutic drug monitoring in tuberculosis: An international survey. Eur. Respir. J. 2022, 59, 2102787. [Google Scholar] [CrossRef]
- Alffenaar, J.W.C.; Stocker, S.L.; Forsman, L.D.; Garcia-Prats, A.; Heysell, S.K.; Aarnoutse, R.E.; Akkerman, O.W.; Aleksa, A.; van Altena, R.; de Onata, W.A.; et al. Clinical standards for the dosing and management of TB drugs. Int. J. Tuberc. Lung Dis. 2022, 26, 483–499. [Google Scholar] [CrossRef]
- World Health Organization. WHO Operational Handbook on Tuberculosis: Module 4: Treatment: Drug-Susceptible Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Kim, H.J.; Seo, K.A.; Kim, H.M.; Jeong, E.S.; Ghim, J.L.; Lee, S.H.; Lee, Y.M.; Kim, D.H.; Shin, J.G. Simple and accurate quantitative analysis of 20 anti-tuberculosis drugs in human plasma using liquid chromatography-electrospray ionization-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 102, 9–16. [Google Scholar] [CrossRef]
- Mercier, T.; Desfontaine, V.; Cruchon, S.; Da Silva Pereira Clara, J.A.; Briki, M.; Mazza-Stalder, J.; Kajkus, A.; Burger, R.; Suttels, V.; Buclin, T.; et al. A battery of tandem mass spectrometry assays with stable isotope-dilution for the quantification of 15 anti-tuberculosis drugs and two metabolites in patients with susceptible-, multidrug-resistant- and extensively drug-resistant tuberculosis. J. Chromatogr. B 2022, 1211, 123456. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. M10 Bioanalytical Method Validation and Study Sample Analysis–Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m10-bioanalytical-method-validation-and-study-sample-analysis (accessed on 31 July 2023).
- European Medicines Agency; Committee for Medicinal Products for Human Use; International Council for Harmonisation of Technical Requirements Registration Pharmaceuticals Human Use. ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. Available online: https://www.ema.europa.eu/en/ich-m10-bioanalytical-method-validation-scientific-guideline (accessed on 31 July 2023).
- Aarnoutse, R.E.; Sturkenboom, M.G.; Robijns, K.; Harteveld, A.R.; Greijdanus, B.; Uges, D.R.; Touw, D.J.; Alffenaar, J.W. An interlaboratory quality control programme for the measurement of tuberculosis drugs. Eur. Respir. J. 2015, 46, 268–271. [Google Scholar] [CrossRef]
- McGregor, M.M.; Olliaro, P.; Wolmarans, L.; Mabuza, B.; Bredell, M.; Felten, M.K.; Fourie, P.B. Efficacy and safety of rifabutin in the treatment of patients with newly diagnosed pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 1996, 154, 1462–1467. [Google Scholar] [CrossRef]
- Zuur, M.A.; Bolhuis, M.S.; Anthony, R.; den Hertog, A.; van der Laan, T.; Wilffert, B.; de Lange, W.; van Soolingen, D.; Alffenaar, J.C. Current status and opportunities for therapeutic drug monitoring in the treatment of tuberculosis. Expert Opin. Drug Metab. Toxicol. 2016, 12, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Bao, Z.; Davies Forsman, L.; Paues, J.; Werngren, J.; Niward, K.; Schön, T.; Bruchfeld, J.; Alffenaar, J.-W.; Hu, Y. Population pharmacokinetics and model-based dosing evaluation of bedaquiline in multidrug-resistant tuberculosis patients. Front. Pharmacol. 2023, 14, 1022090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, Y.; Davies Forsman, L.; Paues, J.; Werngren, J.; Niward, K.; Schon, T.; Bruchfeld, J.; Alffenaar, J.W.; Hu, Y. Population pharmacokinetics and dose evaluations of linezolid in the treatment of multidrug-resistant tuberculosis. Front. Pharmacol. 2022, 13, 1032674. [Google Scholar] [CrossRef] [PubMed]
- Martson, A.G.; Burch, G.; Ghimire, S.; Alffenaar, J.C.; Peloquin, C.A. Therapeutic drug monitoring in patients with tuberculosis and concurrent medical problems. Expert Opin. Drug Metab. Toxicol. 2021, 17, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Aarnoutse, R.E.; Alffenaar, J.W.C.; Bothamley, G.; Brinkmann, F.; Costa, J.; Chesov, D.; van Crevel, R.; Dedicoat, M.; Dominguez, J.; et al. Management of patients with multidrug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2019, 23, 645–662. [Google Scholar] [CrossRef]
- Wise, R.; Logan, M.; Cooper, M.; Ashby, J.; Andrews, J. Meropenem pharmacokinetics and penetration into an inflammatory exudate. Antimicrob. Agents Chemother. 1990, 34, 1515–1517. [Google Scholar] [CrossRef]
- Adam, D.; De Visser, I.; Koeppe, P. Pharmacokinetics of amoxicillin and clavulanic acid administered alone and in combination. Antimicrob. Agents Chemother. 1982, 22, 353–357. [Google Scholar] [CrossRef]
- Lange, C.; Abubakar, I.; Alffenaar, J.W.; Bothamley, G.; Caminero, J.A.; Carvalho, A.C.; Chang, K.C.; Codecasa, L.; Correia, A.; Crudu, V.; et al. Management of patients with multidrug-resistant/extensively drug-resistant tuberculosis in Europe: A TBNET consensus statement. Eur. Respir. J. 2014, 44, 23–63. [Google Scholar] [CrossRef]
- World Health Organization. Rapid Communication: Key Changes to Treatment of Multidrug- and Rifampicin-Resistant Tuberculosis (MDR/RR-TB); World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Han, M.; Jun, S.H.; Lee, J.H.; Park, K.U.; Song, J.; Song, S.H. Method for simultaneous analysis of nine second-line anti-tuberculosis drugs using UPLC-MS/MS. J. Antimicrob. Chemother. 2013, 68, 2066–2073. [Google Scholar] [CrossRef]
- Waters Corporation. Waters Micromass Quattro Premier Mass Spectrometer Operator’s Guide. Available online: https://www.waters.com/webassets/cms/support/docs/71500073002rb.pdf (accessed on 15 September 2023).
- Snyder, L.R.; Kirkland, J.J.; Dolan, J.W. Basic Concepts and the Control of Separation. In Introduction to Modern Liquid Chromatography; John Wiley & Sons: Hoboken, NJ, USA, 2009; pp. 19–86. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Bohring, H. MassBank—High Quality Mass Spectral Database. Available online: https://massbank.eu/MassBank/ (accessed on 1 October 2023).
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D. DrugBank. Available online: https://www.drugbank.com/ (accessed on 1 October 2023).
- European Medicines Agency; Committee for Medicinal Products for Human Use. Guideline on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/en/bioanalytical-method-validation-scientific-guideline (accessed on 15 September 2023).
- Rozet, E.; Hubert, C.; Ceccato, A.; Dewe, W.; Ziemons, E.; Moonen, F.; Michail, K.; Wintersteiger, R.; Streel, B.; Boulanger, B.; et al. Using tolerance intervals in pre-study validation of analytical methods to predict in-study results. The fit-for-future-purpose concept. J. Chromatogr. A 2007, 1158, 126–137. [Google Scholar] [CrossRef]
- Van Ewijk-Beneken Kolmer, E.W.J.; Teulen, M.J.A.; van den Hombergh, E.C.A.; van Erp, N.E.; Te Brake, L.H.M.; Aarnoutse, R.E. Determination of protein-unbound, active rifampicin in serum by ultrafiltration and Ultra Performance Liquid Chromatography with UV detection. A method suitable for standard and high doses of rifampicin. J. Chromatogr. B 2017, 1063, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.; Musshoff, F.; Aebi, B.; Auwärter, V.; Krämer, T.; Peters, F.; Skopp, G.; Aderjan, R.; Herbold, M.; Schmitt, G. Richtlinie der GTFCh zur Qualitätssicherung bei forensisch-toxikologischen Untersuchungen. Toxichem Krimtech 2009, 76, 142–176. [Google Scholar]
- Hubert, P.; Nguyen-Huu, J.J.; Boulanger, B.; Chapuzet, E.; Cohen, N.; Compagnon, P.A.; Dewe, W.; Feinberg, M.; Laurentie, M.; Mercier, N.; et al. Harmonization of strategies for the validation of quantitative analytical procedures. A SFSTP proposal—Part III. J. Pharm. Biomed. Anal. 2007, 45, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Koehler, N.; Andres, S.; Merker, M.; Dreyer, V.; John, A.; Kuhns, M.; Krieger, D.; Choong, E.; Verougstraete, N.; zur Wiesch, P.A.; et al. Pretomanid-Resistant Tuberculosis. J. Infect. 2023, 86, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Heyckendorf, J.; Marwitz, S.; Reimann, M.; Avsar, K.; DiNardo, A.R.; Günther, G.; Hoelscher, M.; Ibraim, E.; Kalsdorf, B.; Kaufmann, S.H.E.; et al. Prediction of anti-tuberculosis treatment duration based on a 22-gene transcriptomic model. Eur. Respir. J. 2021, 58, 2003492. [Google Scholar] [CrossRef] [PubMed]
- Wicha, S.G. TDMx—Model-Informed Precision Dosing Made Simple. Available online: https://tdmx.eu/ (accessed on 31 July 2023).
- Wicha, S.G.; Kees, M.G.; Solms, A.; Minichmayr, I.K.; Kratzer, A.; Kloft, C. TDMx: A novel web-based open-access support tool for optimising antimicrobial dosing regimens in clinical routine. Int. J. Antimicrob. Agents 2015, 45, 442–444. [Google Scholar] [CrossRef]
- Li, C.; Kuti, J.L.; Nightingale, C.H.; Nicolau, D.P. Population pharmacokinetic analysis and dosing regimen optimization of meropenem in adult patients. J. Clin. Pharmacol. 2006, 46, 1171–1178. [Google Scholar] [CrossRef]
- Guo, Z.-Q.; Wang, J.-H.; Fang, D.-M.; Wu, Z.-J. Fragmentation of Moxifloxacin and Its Analogs by Electrospray Ionization Time-of-Flight Mass Spectrometry. Anal. Lett. 2014, 47, 2882–2890. [Google Scholar] [CrossRef]
- Chopra, S.; Pendela, M.; Hoogmartens, J.; Van Schepdael, A.; Adams, E. Impurity profiling of capreomycin using dual liquid chromatography coupled to mass spectrometry. Talanta 2012, 100, 113–122. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Analytical Procedures and Methods Validation for Drugs and Biologics—Guidance for INDUSTRY. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/analytical-procedures-and-methods-validation-drugs-and-biologics (accessed on 15 September 2023).
- Eimer, J.; Frechet-Jachym, M.; Le Du, D.; Caumes, E.; El-Helali, N.; Marigot-Outtandy, D.; Mechai, F.; Peytavin, G.; Pourcher, V.; Rioux, C.; et al. Association Between Increased Linezolid Plasma Concentrations and the Development of Severe Toxicity in Multidrug-Resistant Tuberculosis Treatment. Clin. Infect. Dis. 2023, 76, e947–e956. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Fujii, S.; Shigemi, A.; Takesue, Y. A meta-analysis of the target trough concentration of gentamicin and amikacin for reducing the risk of nephrotoxicity. J. Infect. Chemother. 2021, 27, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Delattre, I.K.; Musuamba, F.T.; Nyberg, J.; Taccone, F.S.; Laterre, P.-F.; Verbeeck, R.K.; Jacobs, F.; Wallemacq, P.E. Population pharmacokinetic modeling and optimal sampling strategy for Bayesian estimation of amikacin exposure in critically ill septic patients. Ther. Drug Monit. 2010, 32, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Paquette, F.; Bernier-Jean, A.; Brunette, V.; Ammann, H.; Lavergne, V.; Pichette, V.; Troyanov, S.; Bouchard, J. Acute Kidney Injury and Renal Recovery with the Use of Aminoglycosides: A Large Retrospective Study. Nephron 2015, 131, 153–160. [Google Scholar] [CrossRef]
- ALSACHIM. [2H6]-Pretomanid. Available online: https://www.alsachim.com/en/stable-labeled-standards/6354-202310--2H6-Pretomanid.html#/5-size-1_mg (accessed on 31 July 2023).
- Cai, J.; Yan, Z. Re-Examining the Impact of Minimal Scans in Liquid Chromatography-Mass Spectrometry Analysis. J. Am. Soc. Mass Spectrom. 2021, 32, 2110–2122. [Google Scholar] [CrossRef]
- Ford, A.C.; Achkar, J.P.; Khan, K.J.; Kane, S.V.; Talley, N.J.; Marshall, J.K.; Moayyedi, P. Efficacy of 5-aminosalicylates in ulcerative colitis: Systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 601–616. [Google Scholar] [CrossRef]
- Helleberg, L. Clinical Pharmacokinetics of indomethacin. Clin. Pharmacokinet. 1981, 6, 245–258. [Google Scholar] [CrossRef]
- Schäcke, H.; Döcke, W.-D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Müller-Lissner, S. Pharmacokinetic and pharmacodynamic considerations for the current chronic constipation treatments. Expert Opin. Drug Metab. Toxicol. 2013, 9, 391–401. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criterion | Sufficient and Ideal Performance |
|---|---|
| Analyte panel | |
| Platform |
|
| Throughput |
|
| Sensitivity | LLOQ: cover pharmacokinetics from lower Cmax over five half-lives: ULOQ:
|
| Validation |
|
| WHO Group | Analyte | Panel | TDM Recommended [10] | TDM Priority | Toxicity Target [10] [µg/mL] | Efficacy Target [10] | Cmax [10] [µg/mL] | Target Calibration LLOQtarget-ULOQtarget [µg/mL] | QClow [µg/mL] | QCmed [µg/mL] | QChigh [µg/mL] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| First-line | rifampicin | 2 | toxicity/efficacy | ++ | - | AUC/MIC > 271 | 8–24 | 0.2–10 | 0.6 | 1.5 | 7.5 |
| rifabutin | 5 | toxicity [17] | ++ | - | - | 0.45–0.9 [18] | 0.01–0.5 | 0.03 | 0.075 | 0.4 | |
| isoniazid | 2 | toxicity/efficacy | ++ | - | AUC/MIC > 567 | 3–6 | 0.1–5 | 0.3 | 0.75 | 3.75 | |
| Group A | levofloxacin | 1, 5 | efficacy | +++ | - | AUC/MIC > 146 | 8–13 | 0.25–2.5 | 0.75 | 1.25 | 2 |
| moxifloxacin | 1, 3 | efficacy | +++ | - | fAUC/MIC > 53 | 3–5 | 0.1–1 | 0.3 | 0.5 | 0.75 | |
| bedaquiline | 1, 4 | toxicity/efficacy [19] | +++ | - | AUC/MIC > 74.6 * [19] | 0.9–4.3 | 0.01–1 | 0.03 | 0.15 | 0.75 | |
| linezolid | 1, 4 | toxicity/efficacy [20] | +++ | Cmin > 2–2.5 | fAUC/MIC > 125 [20] | 12–26 | 0.1–5 | 0.3 | 0.75 | 4 | |
| Unclassified | pretomanid | 5 | - | ++ | - | - | 1.4–4.3 [21] | 0.01–1 | 0.03 | 0.15 | 0.75 |
| Group B | clofazimine | 1, 3 | - | ++ | - | - | 0.5–2 | 0.01–0.5 | 0.03 | 0.075 | 0.4 |
| cycloserine | 1, 3 | toxicity/efficacy | +++ | - | %T > MIC > 30% | 20–35 | 0.5–10 | 1.5 | 2 | 7.5 | |
| Group C | ethambutol | 1, 2, 3 | toxicity | ++ | - | AUC/MIC > 119 | 2–6 | 0.062–2.5 | 0.18 | 0.4 | 1.9 |
| delamanid | 1, 5 | - | ++ | - | - | 0.3–0.9 [22] | 0.005–0.5 | 0.015 | 0.075 | 0.4 | |
| pyrazinamide | 1, 2 | toxicity/efficacy | +++ | - | AUC/MIC > 8.42 | 20–60 | 0.5–10 | 1.5 | 2 | 7.5 | |
| meropenem | 1, 4 | - | ++ | - | %T > MIC > 60% [22] | 50–100 [23] | 0.1–10 | 0.3 | 1.5 | 7.5 | |
| clavulanic acid | 1, 4 | - | ++ | - | - | 2.5–4 [24] | 1–10 | 1.5 | 2 | 7.5 | |
| amikacin | 1, 4 | toxicity/efficacy | ++ | Cmin > 2 | Cmax/MIC > 75 | 35–45 | 1–10 | 3 | 5 | 7.5 | |
| streptomycin | 2, 3 | - | + | - | fCmax/MIC > 20 [25] | 35–45 [25] | 1–10 | 3 | 5 | 7.5 | |
| prothionamide | 1, 3 | - | ++ | - | AUC/MIC > 56.2 | 2–5 | 0.05–1 | 0.15 | 0.2 | 0.75 | |
| PAS | 1, 5 | - | + | - | fCmin > 1 | 20–60 | 0.5–10 | 1.5 | 2 | 7.5 | |
| Excluded | capreomycin | 3 | - | + | - | fCmax/MIC > 20 [25] | 35–45 [25] | 1–10 | 3 | 5 | 7.5 |
| kanamycin | 5 | - | + | - | fCmax/MIC > 20 [25] | 35–45 [25] | 1–10 | 3 | 5 | 7.5 |
| WHO Group | Analyte | Precursor [m/z] | → | Fragment [m/z] | Ion Mode | Cone Voltage [eV] | Collision Energy [eV] | Retention Time [min] Median (90% RT Range) | Channel Start [min] | Channel Stop [min] | Dwell [s] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| First-line | rifampicin | 823.46 | → | 791.01 | [M + H]+ | 30 | 20 | 2.60 (2.43–2.89) | 1.75 | 4.5 | 0.05 |
| D-rifampicin | 830.25 | → | 798.69 | [M + H]+ | 30 | 20 | 2.61 (2.44–2.89) | 1.75 | 4.5 | 0.05 | |
| rifabutin | 847.02 | → | 815.52 | [M + H]+ | 40 | 35 | 2.91 (2.79–3.61) | 1.75 | 3.75 | 0.05 | |
| D-rifabutin | 852.59 | → | 821.16 | [M + H]+ | 40 | 35 | 2.92 (2.79–3.62) | 1.75 | 3.75 | 0.05 | |
| isoniazid | 137.68 | → | 78.50 | [M + H]+ | 25 | 25 | 2.81 (2.70–3.40) | 2 | 4.5 | 0.10 | |
| D-isoniazid | 141.72 | → | 82.60 | [M + H]+ | 25 | 25 | 2.89 (2.78–3.47) | 2 | 4.5 | 0.10 | |
| Group A | levofloxacin | 361.82 | → | 261.17 | [M + H]+ | 40 | 30 | 4.25 (4.08–4.51) | 2 | 5 | 0.05 |
| D-levofloxacin | 366.04 | → | 261.04 | [M + H]+ | 40 | 30 | 4.25 (3.63–4.42) | 2 | 5 | 0.05 | |
| moxifloxacin | 401.67 | → | 364.21 | [M + H]+ | 40 | 35 | 3.18 (2.77–4.20) | 2 | 4.8 | 0.05 | |
| D-moxifloxacin | 407.30 | → | 369.08 | [M + H]+ | 40 | 35 | 3.20 (2.77–4.20) | 2 | 4.8 | 0.05 | |
| bedaquiline | 555.14 | → | 58.16 | [M + H]+ | 30 | 35 | 2.52 (2.08–3.15) | 1.5 | 4 | 0.05 | |
| D-bedaquiline | 561.13 | → | 64.13 | [M + H]+ | 30 | 35 | 2.52 (2.08–3.16) | 1.5 | 4 | 0.05 | |
| linezolid | 337.17 | → | 195.20 | [M + H]+ | 40 | 25 | 1.06 (0.96–1.10) | 0.5 | 1.5 | 0.05 | |
| D-linezolid | 345.10 | → | 203.20 | [M + H]+ | 40 | 25 | 1.07 (1.03–1.10) | 0.5 | 1.5 | 0.05 | |
| Un-clssfd | pretomanid | 359.00 | → | 174.82 | [M + H]+ | 35 | 25 | 1.00 (0.95–1.05) | 0 | 2 | 0.05 |
| Group B | clofazimine | 471.61 | → | 395.25 | [M + H]+ | 60 | 45 | 2.41 (2.02–2.88) | 1.5 | 4 | 0.05 |
| D-clofazimine | 480.00 | → | 396.15 | [M + H]+ | 60 | 45 | 2.29 (2.02–2.87) | 1.5 | 4 | 0.05 | |
| cycloserine | 102.88 | → | 75.03 | [M + H]+ | 25 | 10 | 4.99 (4.92–5.10) | 4.6 | 5.3 | 0.05 | |
| D-cycloserine | 106.22 | → | 78.85 | [M + H]+ | 25 | 10 | 4.99 (4.92–5.10) | 4.6 | 5.3 | 0.05 | |
| Group C | ethambutol | 204.78 | → | 44.29 | [M + H]+ | 20 | 25 | 5.48 (5.45–5.62) | 5.2 | 6 | 0.05 |
| D-ethambutol | 208.78 | → | 48.29 | [M + H]+ | 20 | 25 | 5.50 (5.45–5.67) | 5.2 | 6 | 0.05 | |
| delamanid | 534.37 | → | 352.07 | [M + H]+ | 35 | 25 | 1.87 (1.70–2.51) | 1.2 | 3.3 | 0.05 | |
| D-delamanid | 538.30 | → | 356.07 | [M + H]+ | 35 | 25 | 1.95 (1.73–2.62) | 1.2 | 3.3 | 0.05 | |
| pyrazinamide | 123.68 | → | 78.88 | [M + H]+ | 20 | 20 | 1.19 (1.16–1.21) | 0 | 2 | 0.05 | |
| D-pyrazinamide | 127.21 | → | 82.85 | [M + H]+ | 20 | 20 | 1.19 (1.09–1.21) | 0 | 2 | 0.05 | |
| meropenem | 383.95 | → | 68.04 | [M + H]+ | 25 | 30 | 4.58 (4.43–4.68) | 4 | 5 | 0.05 | |
| D-meropenem | 389.96 | → | 68.06 | [M + H]+ | 25 | 30 | 4.59 (4.44–4.68) | 4 | 5 | 0.05 | |
| clavulanic acid * | 198.00 | → | 136.00 | [M − H]− | 20 | 10 | 1.17 * | 0 | 4 | 0.10 | |
| amikacin | 586.06 | → | 163.07 | [M + H]+ | 30 | 30 | 6.94 (6.62–7.29) | 6 | 8 | 0.50 | |
| D-amikacin | 590.11 | → | 162.52 | [M + H]+ | 30 | 30 | 6.93 (6.60–7.28) | 6 | 8 | 0.50 | |
| streptomycin | 582.03 | → | 263.10 | [M + H]+ | 65 | 35 | 6.12 (6.07–6.35) | 5.7 | 6.8 | 0.50 | |
| prothionamide | 180.57 | → | 120.94 | [M + H]+ | 35 | 25 | 1.76 (1.65–2.25) | 1.2 | 3 | 0.05 | |
| D-prothionamide | 187.58 | → | 127.38 | [M + H]+ | 35 | 25 | 1.79 (1.70–2.31) | 1.2 | 3 | 0.05 | |
| PAS | 153.64 | → | 91.17 | [M + H]+ | 25 | 25 | 1.09 (0.98–1.21) | 0.7 | 1.4 | 0.05 | |
| D-PAS | 159.94 | → | 96.09 | [M + H]+ | 25 | 25 | 1.07 (1.04–1.12) | 0.7 | 1.4 | 0.05 | |
| Excluded | capreomycin IB § | 326.21 | → | 70.18 | [M + 2H]2+ | 25 | 20 | 7.60 (7.45–8.70) | 6.5 | 10 | 0.50 |
| capreomycin IA § | 334.27 | → | 70.24 | [M + 2H]2+ | 25 | 20 | 7.79 (7.61–8.97) | 6.5 | 10 | 0.50 | |
| kanamycin | 484.46 | → | 162.48 | [M + H]+ | 35 | 25 | 7.55 (7.46–8.27) | 5 | 9 | 0.50 | |
| D-kanamycin | 490.63 | → | 162.52 | [M + H]+ | 35 | 25 | 7.31 (7.24–7.95) | 5 | 9 | 0.50 | |
| gentamicin | 477.43 | → | 157.15 | [M + H]+ | 30 | 20 | 12.07 (10.21–13.81) | 9 | 22 | 0.50 |
| WHO Group | Analyte | Calibration Range [µg/mL] | R2 | Carry-Over [% LLOQ] | Sample Sets nlow; nmed; nhigh | Recovery [%] (%CV) | Matrix Effect [%] (%CV) |
|---|---|---|---|---|---|---|---|
| First-line | rifampicin | 0.1–10 | 0.9935 | 1.3 | 1; 1; 1 | 102.6 (7.5) | 135.4 (13.1) |
| rifabutin | 0.005–0.5 | 0.9952 | 2.4 | 2; 2; 2 | 96.9 (6.1) | 106.3 (6.4) | |
| isoniazid | 0.05–5 | 0.9951 | 0.0 | 1; 1; 1 | 101.2 (6.7) | 101.3 (3.9) | |
| Group A | levofloxacin | 0.025–2.5 | 0.9947 | 6.3 | 2; 2; 2 | 98.0 (4.2) | 98.2 (7.8) |
| moxifloxacin | 0.01–1 | 0.9916 | 0.0 | 3; 3; 4 | 96.2 (7.6) | 107.3 (5.1) | |
| bedaquiline | 0.01–1 | 0.9969 | 0.6 | 1; 1; 1 | 86.2 (5.5) | 96.6 (4.6) | |
| linezolid | 0.05–5 | 0.9968 | 4.1 | 1; 1; 1 | 93.3 (6.0) | 96.9 (4.9) | |
| Unclassified | pretomanid * | 0.015–1 | 0.9902 | 0.0 | 2; 2; 2 | 97.2 (13.4) | 106.7 (35.5) |
| Group B | clofazimine | 0.005–0.5 | 0.9974 | 0.0 | 3; 3; 4 | 96.1 (7.7) | 102.1 (3.9) |
| cycloserine | 0.1–10 | 0.9914 | 0.0 | 3; 3; 4 | 94.6 (10.3) | 116.2 (4.8) | |
| Group C | ethambutol | 0.025–2.5 | 0.9919 | 8.4 | 1; 1; 1 | 91.8 (9.2) | 103.6 (6.6) |
| delamanid | 0.005–0.5 | 0.9959 | 0.0 | 3; 3; 3 | 83.0 (9.8) | 101.1 (16.4) | |
| pyrazinamide | 0.1–10 | 0.9953 | 19.0 | 1; 1; 1 | 105.6 (6.7) | 97.2 (3.3) | |
| meropenem | 0.1–10 | 0.9916 | 6.7 | 1; 1; 1 | 76.1 (10.3) | 105.9 (6.1) | |
| clavulanic acid | – | - | - | - | - | - | |
| amikacin | 0.1–10 | 0.9835 | 0.0 | 1; 1; 1 | 47.8 (13.3) | 139.8 (11.3) | |
| Streptomycin $ | 0.2–10 | 0.9913 | 0.0 | 1; 1; 1 | 56.3 (28.1) | 183.6 (35.8) | |
| prothionamide | 0.01–1 | 0.9974 | 1.0 | 3; 3; 4 | 97.0 (7.8) | 98.9 (12.7) | |
| PAS | 0.1–10 | 0.9957 | 1.2 | 2; 2; 2 | 95.8 (4.0) | 100.7 (7.3) | |
| excluded | capreomycin IB $ | 0.2–10 | 0.9903 | 0.0 | 3; 3; 4 | 49.9 (24.1) | 231.7 (32.6) |
| capreomycin IA $ | 0.1–10 | 0.9879 | 0.0 | 3; 3; 4 | 47.7 (23.6) | 206.5 (30.2) | |
| kanamycin | 0.25–10 | 0.9910 | 16.7 | 2; 2; 2 | 63.9 (15.3) | 153.5 (12.2) |
| WHO Group | Analyte | Sample Sets nlow; nmed; nhigh | Val. | Accuracy [%] (wthn−Day Prec. [%]; btw−Day prec. [%]) | 80 % Beta−Expectation Tolerance Interval [%] | Accuracy [%] (Precision [%CV]) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| QClow | QCmed | QChigh | QClow | QCmed | QChigh | Patient Samples | ||||
| First−line | rifampicin | 2; 2; 2 | ext | 122.2 (1.9; 9.7) | 113.0 (3.4; 5.6) | 114.5 (2.4; 1.5) | −14.9; +59.4 | −10.6; +36.7 | +9.9; +19.2 | - |
| rifabutin | 3; 3; 2 | int | 103.8 (2.1; 20.8) | 103.0 (3.5; 14.4) | 102.1 (3.3; 3.4) | −41.7; +49.3 | −29.0; +35.1 | −8.1; +12.3 | - | |
| isoniazid | 2; 2; 2 | ext | 160.3 (2.2; 14.8) | 141.2 (1.0; 0.7) | 146.9 (4.6; 7.6) | +4.2; +116.4 | +39.1; +43.3 | +14.8; +78.9 | - | |
| Group A | levofloxacin | 3; 3; 3 | ext | 95.7 (5.8; 17.8) | 97.1 (6.8; 14.4) | 105.0 (4.5; 13.4) | −44.7; +36.1 | −36.9; +31.1 | −25.5; +35.5 | - |
| moxifloxacin | 5; 6; 6 | int | 113.8 (9.0; 12.9) | 106.7 (4.9; 20.6) | 114.9 (4.6; 11.0) | −10.0; +37.6 | −26.9; +40.3 | −3.5; +33.2 | 113.6 (7.5) | |
| bedaquiline | 3; 3; 3 | ext | 97.2 (6.2; 24.4) | 94.3 (3.5; 3.9) | 102.0 (4.9; 10.1) | −57.2; +51.7 | −14.5; +3.1 | −21.9; +25.9 | 106.5 (11.2) | |
| linezolid | 3; 3; 3 | ext | 110.5 (5.3; 12.9) | 108.3 (3.5; 12.9) | 106.3 (3.6; 14.2) | −19.3; +40.4 | −20.5; +37.1 | −25.5; +38.1 | 102.6 (8.4) | |
| Unclassified | pretomanid * | 3; 3; 3 | int | 91.5 (20.0; 37.6) | 96.1 (19.3; 29.2) | 96.7 (13.3; 38.3) | −99.1; +82.1 | −67.9; +60.0 | −90.6; +84.1 | - |
| Group B | clofazimine | 5; 6; 6 | int | 108.6 (8.2; 13.1) | 106.2 (3.5; 15.8) | 113.2 (3.4; 11.7) | −15.3; +32.5 | −19.6; +32.0 | −6.1; +32.5 | 90.1 (9.6) |
| cycloserine | 4; 5; 5 | int | 103.3 (14.7; 11.9) | 97.7 (6.7; 17.3) | 95.9 (8.4; 17.6) | −24.2; +30.8 | −32.0; +27.4 | −35.2; +27.1 | 105.7 (11.7) | |
| Group C | ethambutol | 2; 5; 5 | int | 115.6 (7.7; 8.8) | 123.4 (5.0; 8.1) | 114.1 (4.8; 10.3) | −9.7; +41.0 | +8.7; +38.2 | −4.0; +32.3 | - |
| delamanid | 3; 4; 3 | int | 114.0 (5.0; 9.4) | 113.0 (4.4; 13.5) | 107.4 (6.1; 13.2) | −8.6; +36.6 | −12.8; +38.7 | −23.6; +38.4 | 113.6 (16.4) | |
| pyrazinamide | 2; 2; 2 | ext | 116.1 (2.0; 9.5) | 108.1 (1.1; 4.1) | 113.6 (2.8; 2.2) | −20.5; +52.7 | −7.9; +24.0 | +7.0; +20.1 | − | |
| meropenem | 2; 2; 2 | int | 101.5 (6.7; 1.7) | 95.4 (7.1; 4.7) | 101.1 (8.5; 0.0) | −8.6; +11.6 | −18.9; +9.8 | −11.1; +13.4 | 92.3 (10.8) | |
| clavulanic acid | - | - | - | - | - | - | - | - | ||
| amikacin | 1; 1; 1 | int | 93.5 (6.3; 6.3) § | −15.2; +2.2 § | - | |||||
| streptomycin $ | 3; 4; 4 | int | 131.8 (19.0; 13.0) | 106.1 (16.3; 60.2) | 82.5 (23.6; 37.3) | −2.5; +66.1 | −107.5; +119.7 | −88.5; +53.5 | - | |
| prothionamide | 3; 4; 3 | int | 113.6 (10.2; 6.1) | 111.1 (5.5; 11.5) | 111.2 (10.4; 6.2) | −3.7; +31.0 | −10.3; +32.5 | −6.5; +28.9 | - | |
| PAS | 3; 3; 3 | int | 109.0 (2.5; 16.4) | 109.8 (1.9; 15.1) | 90.8 (2.5; 14.1) | −27.1; +45.2 | −23.3; +42.9 | −40.3; +21.9 | - | |
| Excluded | capreomycin IB $ | 3; 4; 4 | int | 71.0 (13.5; 43.9) | 46.2 (12.7; 29.4) | 33.5 (19.5; 32.5) | −128.2; +70.2 | −111.8; +4.2 | −130.0; −3.0 | - |
| capreomycin IA $ | 3; 4; 4 | int | 61.5 (12.6; 47.0) | 38.6 (12.4; 29.5) | 27.7 (20.0; 34.7) | −143.8; +66.8 | −119.4; −3.4 | −139.5; −5.0 | - | |
| kanamycin | 3; 3; 3 | int | 103.7 (5.6; 6.8) | 114.7 (3.7; 10.4) | 95.8 (10.6; 30.0) | −11.2; +18.6 | −9.1; +38.5 | −72.8; +64.4 | - | |
| TDM Target | Patient 1 T1 | Patient 2 T3 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Parameter | Value | DST § | MIC § | Dose [mg] § | TDM1 | DST | MIC | Dose [mg] | TDM3 | |
| Group A | levofloxacin | AUC/MIC | >146 | R | 4 | - | - | R | 7.5 | - | - |
| moxifloxacin | fAUC/MIC | >53 | r | 2 | 1200 | 21.26 | r | 2 | 1600 | 18.49 | |
| bedaquiline | AUC/MIC | >74.6 | S | 0.015 | 200 | 2703.33 | r | 2 | 300 | 54.03 | |
| linezolid | fAUC/MIC | >125 | S | 0.25 | 600 | 273.53 | R | >1 | - | - | |
| Unclassified | pretomanid | - | - | - | - | - | - | - | - | - | - |
| Group B | clofazimine | Cmin; Cmax * [µg/mL] | 0.52; 0.79 | S | 0.12 | 100 | 0.25; 0.52 | r | 2 | 200 | 0.54; 0.68 |
| cycloserine $ | % T > MIC [%] | >30 | - | 4 | 750 | 100 | - | 30 | 1000 | 80.67 | |
| Group C | ethambutol | AUC/MIC | >119 | R | 10 | - | - | R | 7.5 | - | - |
| delamanid | Cmin; Cmax * [µg/mL] | 0.17; 0.31 | R | >0.48 | - | - | S | <0.06 | 2 × 100 | 0.32; 0.58 | |
| pyrazinamide | AUC/MIC | >8.42 | R | >100 | - | - | R | >100 | - | - | |
| meropenem | % T > MIC [%] | >60 % | - | - | 3 × 2000/2 h | 62.3 (MIC:8) § | R | 16 | 3 × 2000/3 h | 46.30 | |
| clavulanic acid | - | - | - | - | 3 × 125 | - | - | - | 3 × 125 | - | |
| amikacin | Cmax/MIC | >75 | S | <0.25 | - | - | R | >1 | - | - | |
| streptomycin | fCmax/MIC | >20 | S | - | - | - | R | - | - | - | |
| prothionamide | AUC/MIC | >56.2 | R | 2 | - | - | R | >5 | - | - | |
| PAS | Cmax * [µg/mL] | 300–500 | S | <4 | - | - | R | >16 | 11820 | 440.38 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köhler, N.; Karaköse, H.; Grobbel, H.-P.; Hillemann, D.; Andres, S.; König, C.; Kalsdorf, B.; Brehm, T.T.; Böttcher, L.; Friesen, I.; et al. A Single-Run HPLC–MS Multiplex Assay for Therapeutic Drug Monitoring of Relevant First- and Second-Line Antibiotics in the Treatment of Drug-Resistant Tuberculosis. Pharmaceutics 2023, 15, 2543. https://doi.org/10.3390/pharmaceutics15112543
Köhler N, Karaköse H, Grobbel H-P, Hillemann D, Andres S, König C, Kalsdorf B, Brehm TT, Böttcher L, Friesen I, et al. A Single-Run HPLC–MS Multiplex Assay for Therapeutic Drug Monitoring of Relevant First- and Second-Line Antibiotics in the Treatment of Drug-Resistant Tuberculosis. Pharmaceutics. 2023; 15(11):2543. https://doi.org/10.3390/pharmaceutics15112543
Chicago/Turabian StyleKöhler, Niklas, Hande Karaköse, Hans-Peter Grobbel, Doris Hillemann, Sönke Andres, Christina König, Barbara Kalsdorf, Thomas Theo Brehm, Laura Böttcher, Inna Friesen, and et al. 2023. "A Single-Run HPLC–MS Multiplex Assay for Therapeutic Drug Monitoring of Relevant First- and Second-Line Antibiotics in the Treatment of Drug-Resistant Tuberculosis" Pharmaceutics 15, no. 11: 2543. https://doi.org/10.3390/pharmaceutics15112543