

Excipient Impact on Fenofibrate Equilibrium Solubility in Fasted and Fed Simulated Intestinal Fluids Assessed Using a Design of Experiment Protocol

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dual Level Design of Experiment (DoE) and Data Analysis
2.3. Equilibrium Solubility Measurement
2.3.1. Preparation of Stock Systems
- Preparation of Lipid Suspension
- Preparation of Sodium Oleate Solution
- Preparation Buffer Solution
- Preparation of excipient stock solutions
2.3.2. Preparation of Experimental Measurement Solutions
- Preparation of Individual Design of Experiment Solutions
2.4. HPLC Analysis
3. Results
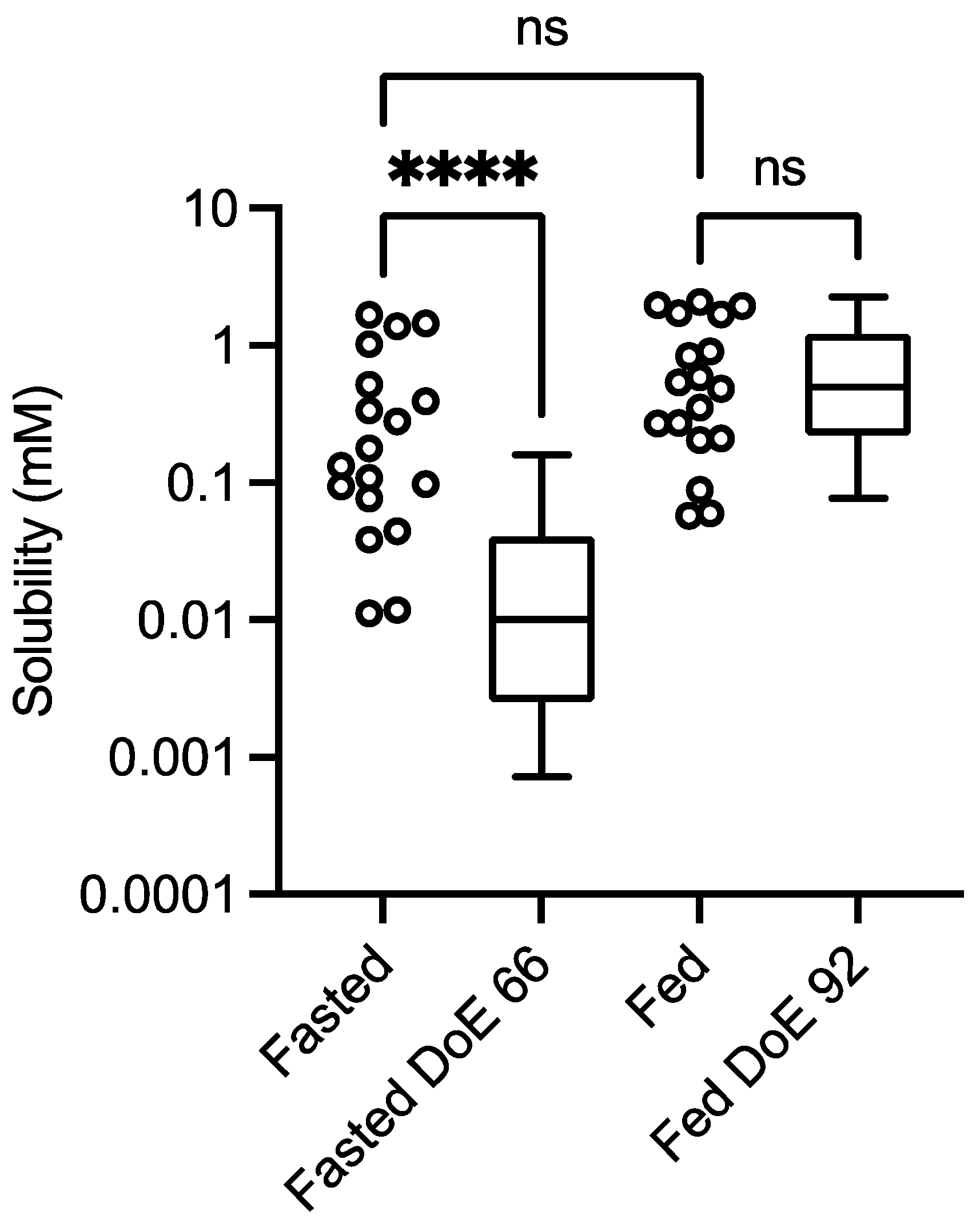
3.1. Control Solubility Measurement
3.1.1. Solubility Distribution

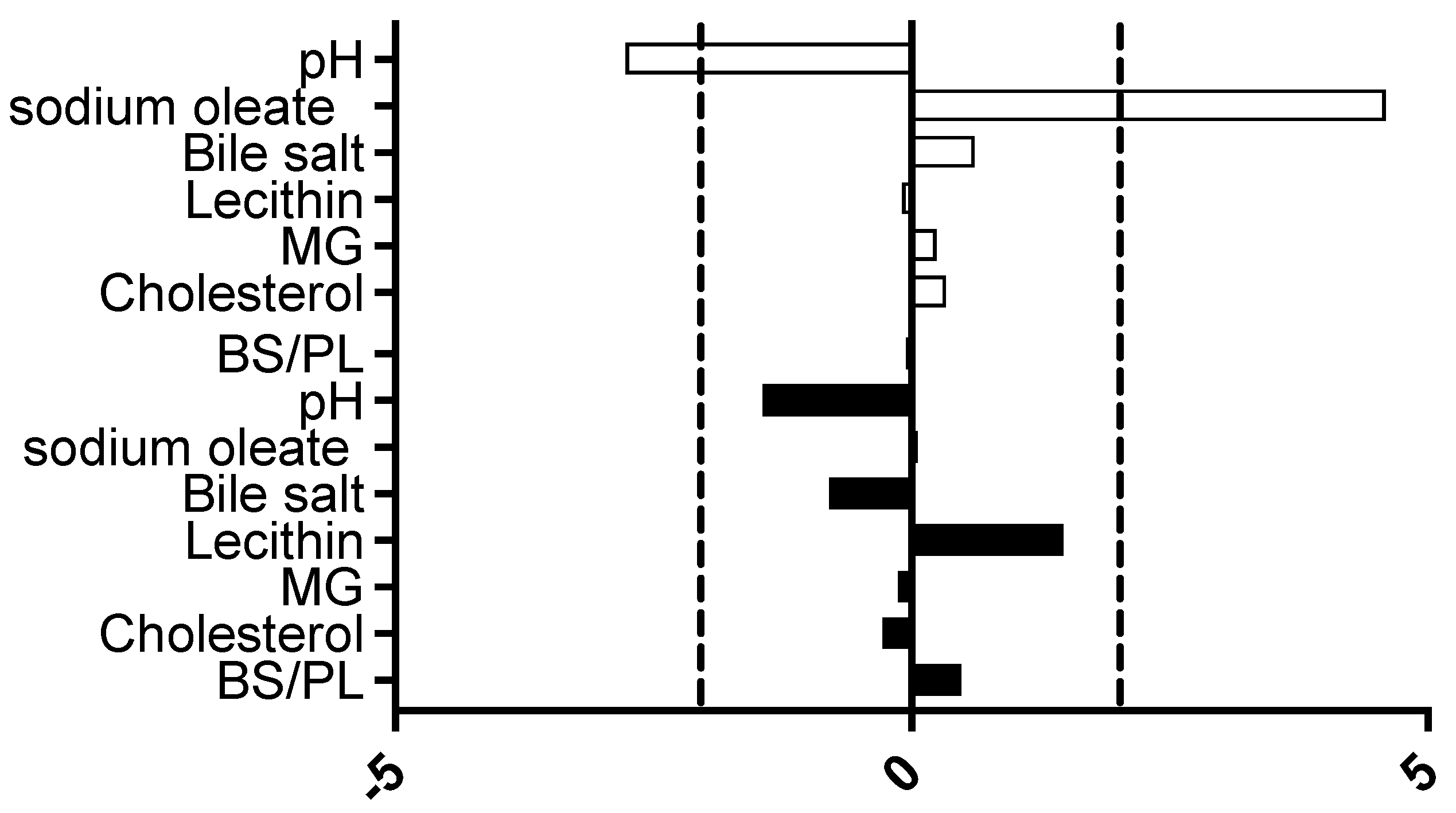
3.1.2. Solubility Influence of DoE Factors
 represents fasted study arm;
represents fasted study arm;  represents fed study arm. Bar to the right of 0 on x axis is positive effect on solubility, bar length indicates the magnitude of the effect, vertical dashed black lines indicate statistical significance threshold (p < 0.05).
represents fasted study arm; represents fed study arm. Bar to the right of 0 on x axis is positive effect on solubility, bar length indicates the magnitude of the effect, vertical dashed black lines indicate statistical significance threshold (p < 0.05).
represents fed study arm. Bar to the right of 0 on x axis is positive effect on solubility, bar length indicates the magnitude of the effect, vertical dashed black lines indicate statistical significance threshold (p < 0.05).
represents fasted study arm; represents fed study arm. Bar to the right of 0 on x axis is positive effect on solubility, bar length indicates the magnitude of the effect, vertical dashed black lines indicate statistical significance threshold (p < 0.05).
3.1.3. Statistical Considerations
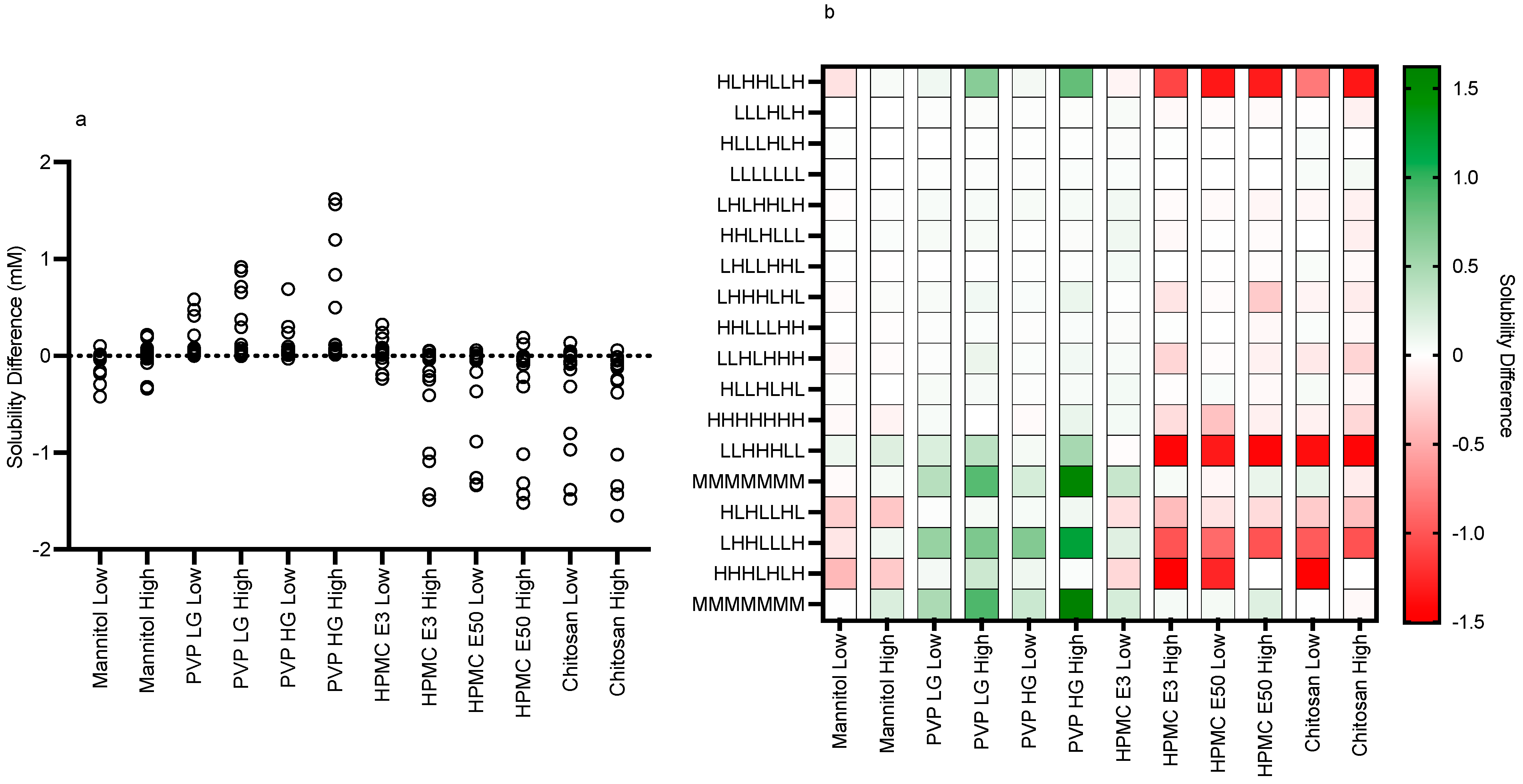
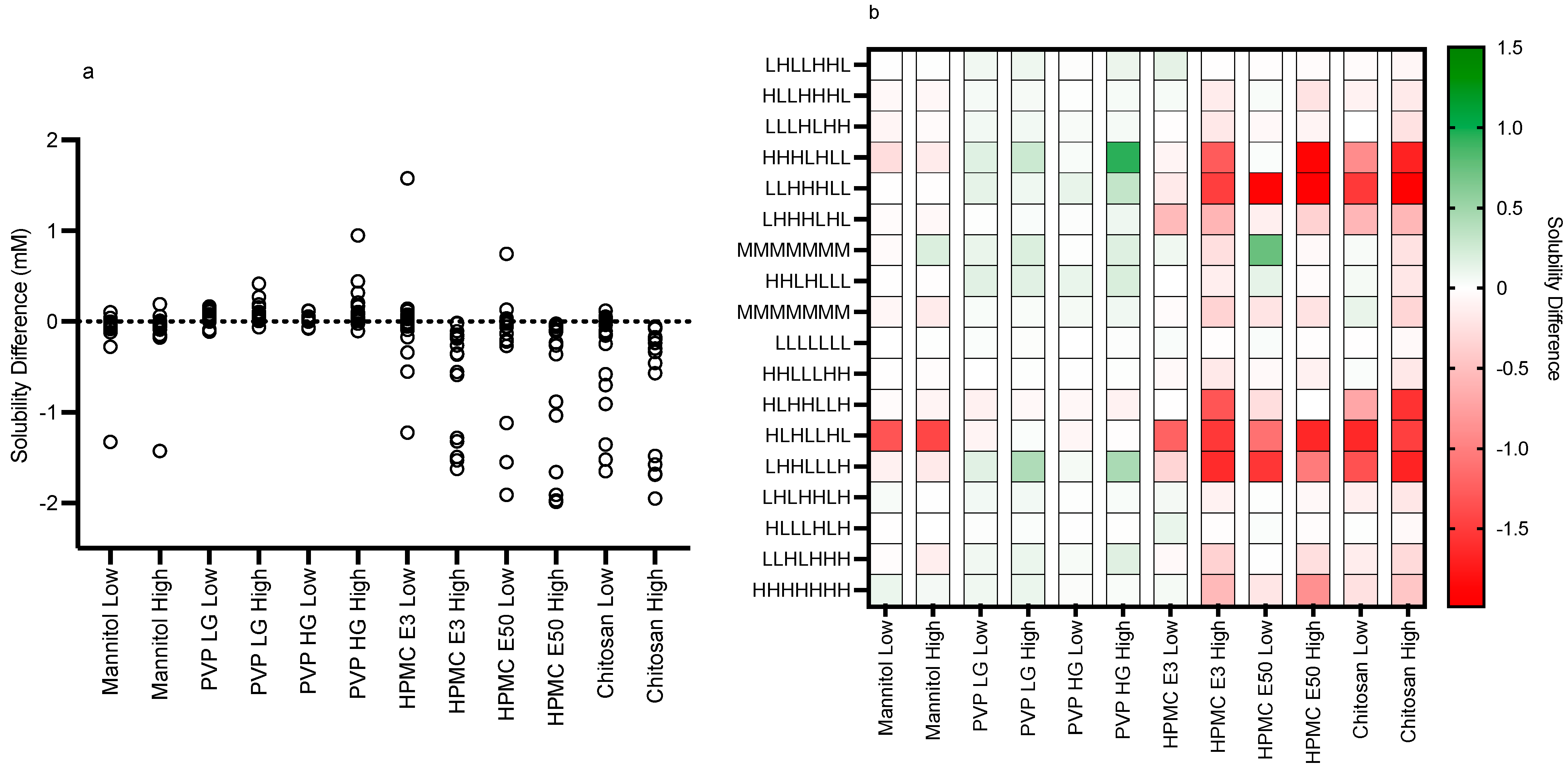
3.2. Impact of Excipients on Equilibrium Solubility
3.2.1. Excipient Concentrations
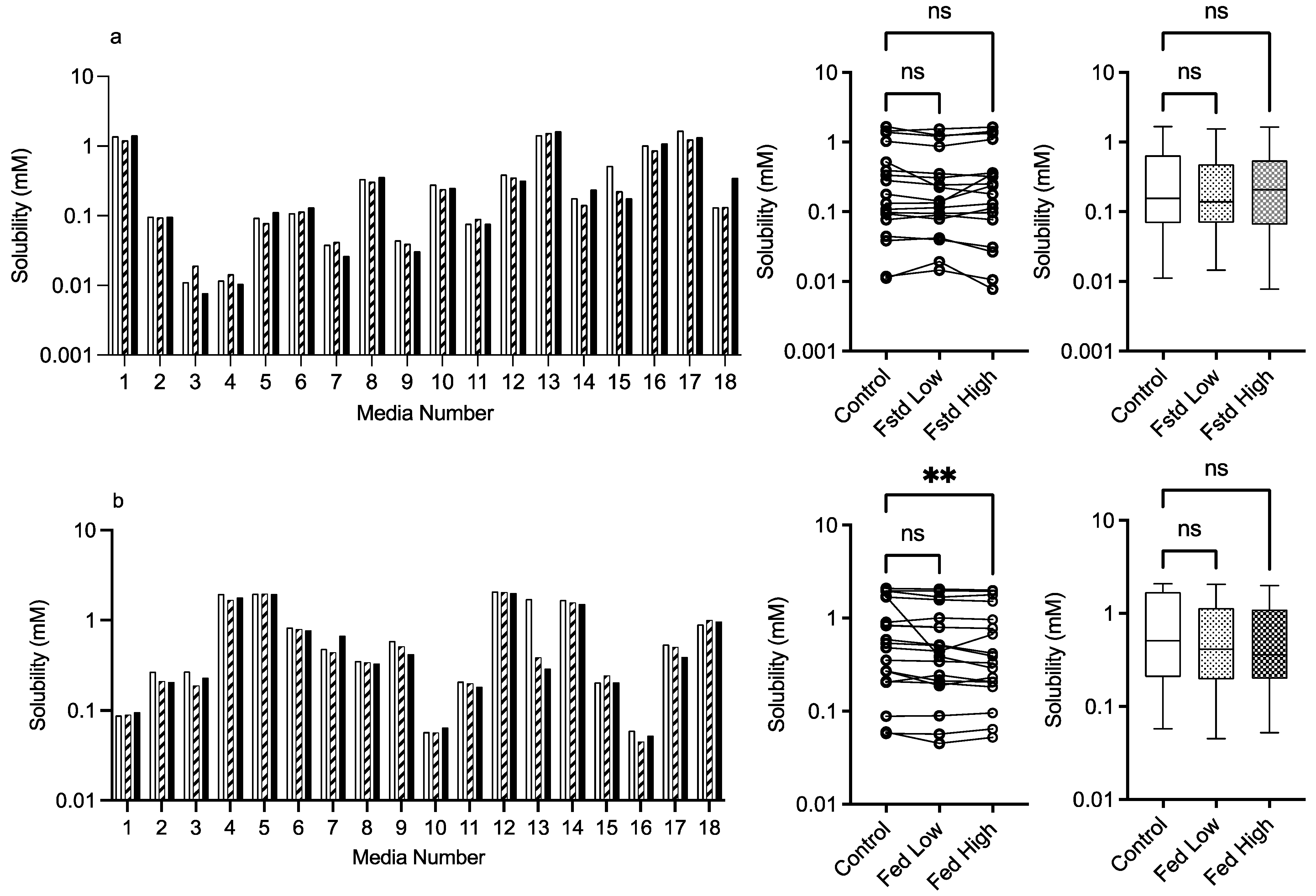
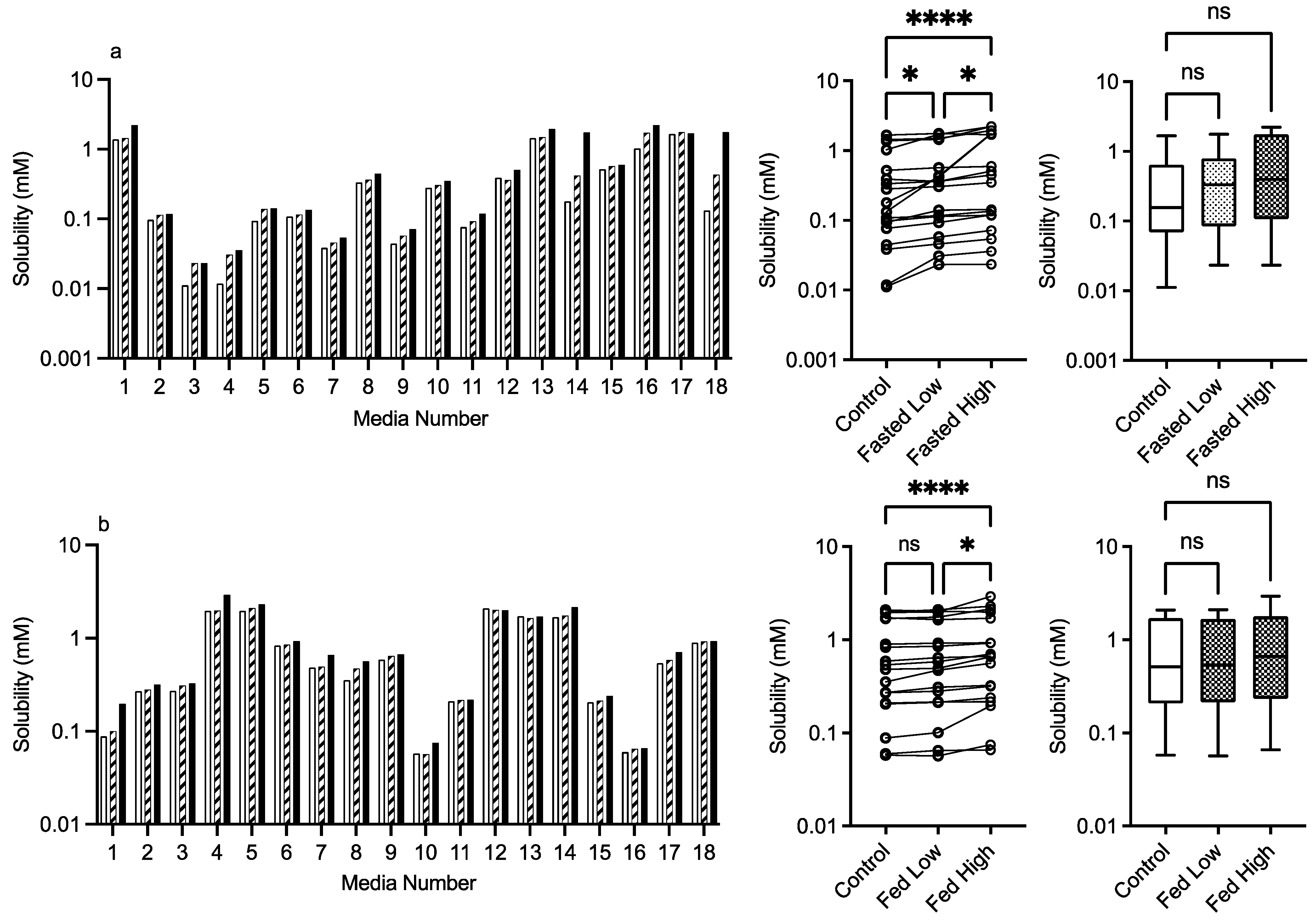
3.2.2. Mannitol
control (no excipient),  low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient,
low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient,  low excipient concentration,
low excipient concentration,  high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05. (b). ** p = 0.0124.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05. (b). ** p = 0.0124.
high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05. (b). ** p = 0.0124.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05. (b). ** p = 0.0124.
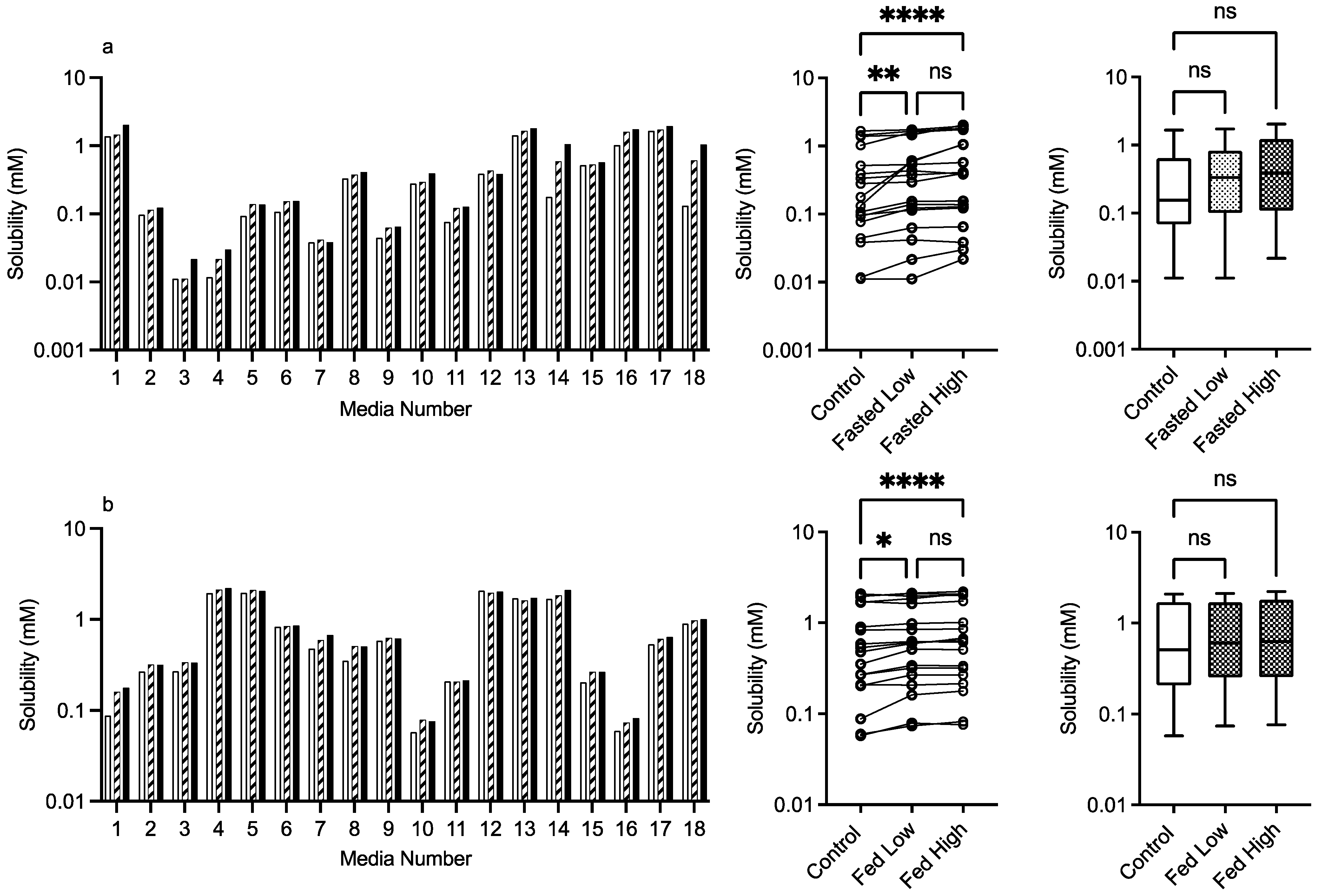
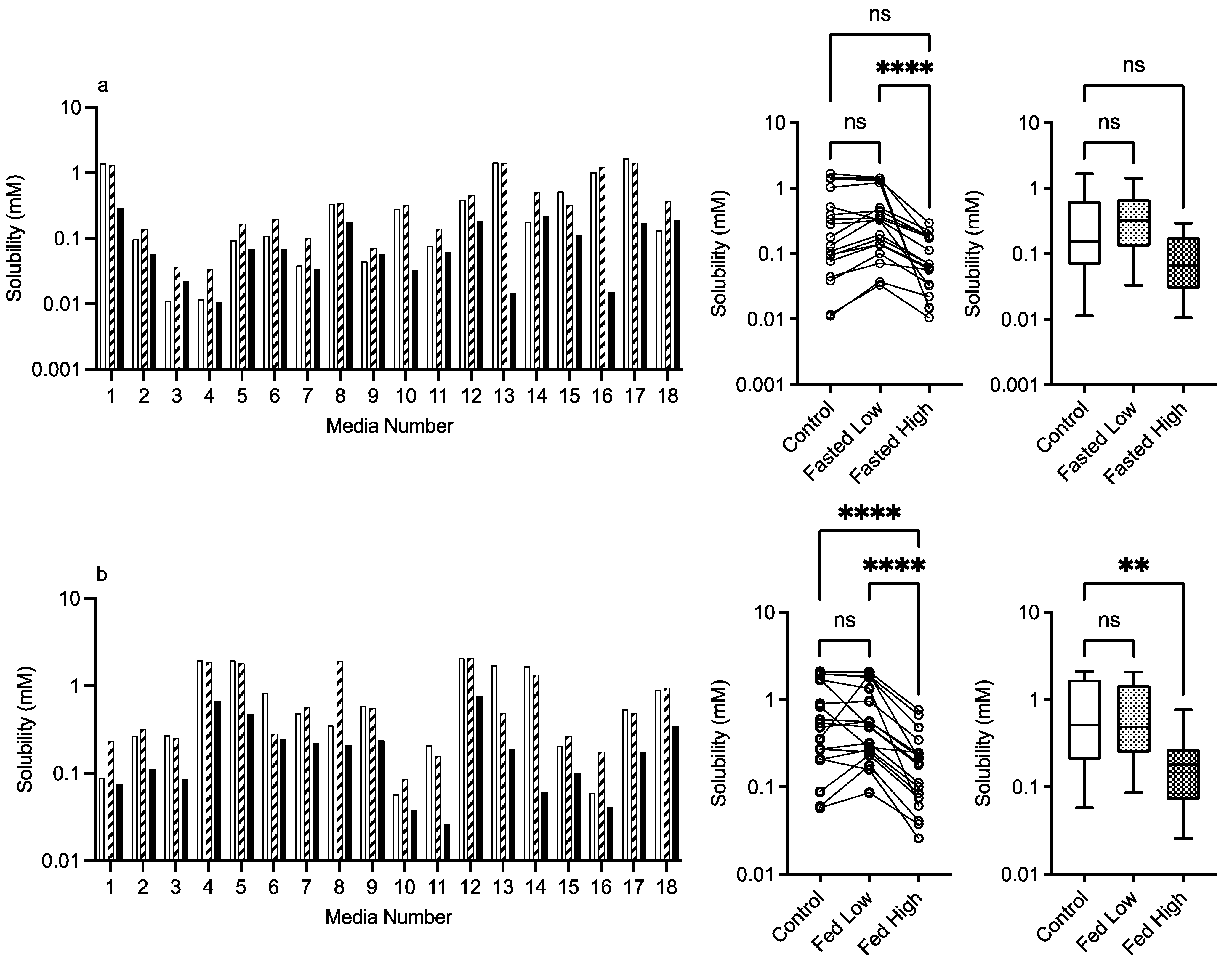
3.2.3. PVP K12 and K29/32
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1 (a) ns = no significant difference, p > 0.05; ** p = 0.0081; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1 (a) ns = no significant difference, p > 0.05; ** p = 0.0081; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001. control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; * p = 0.0138; **** p < 0.0001.
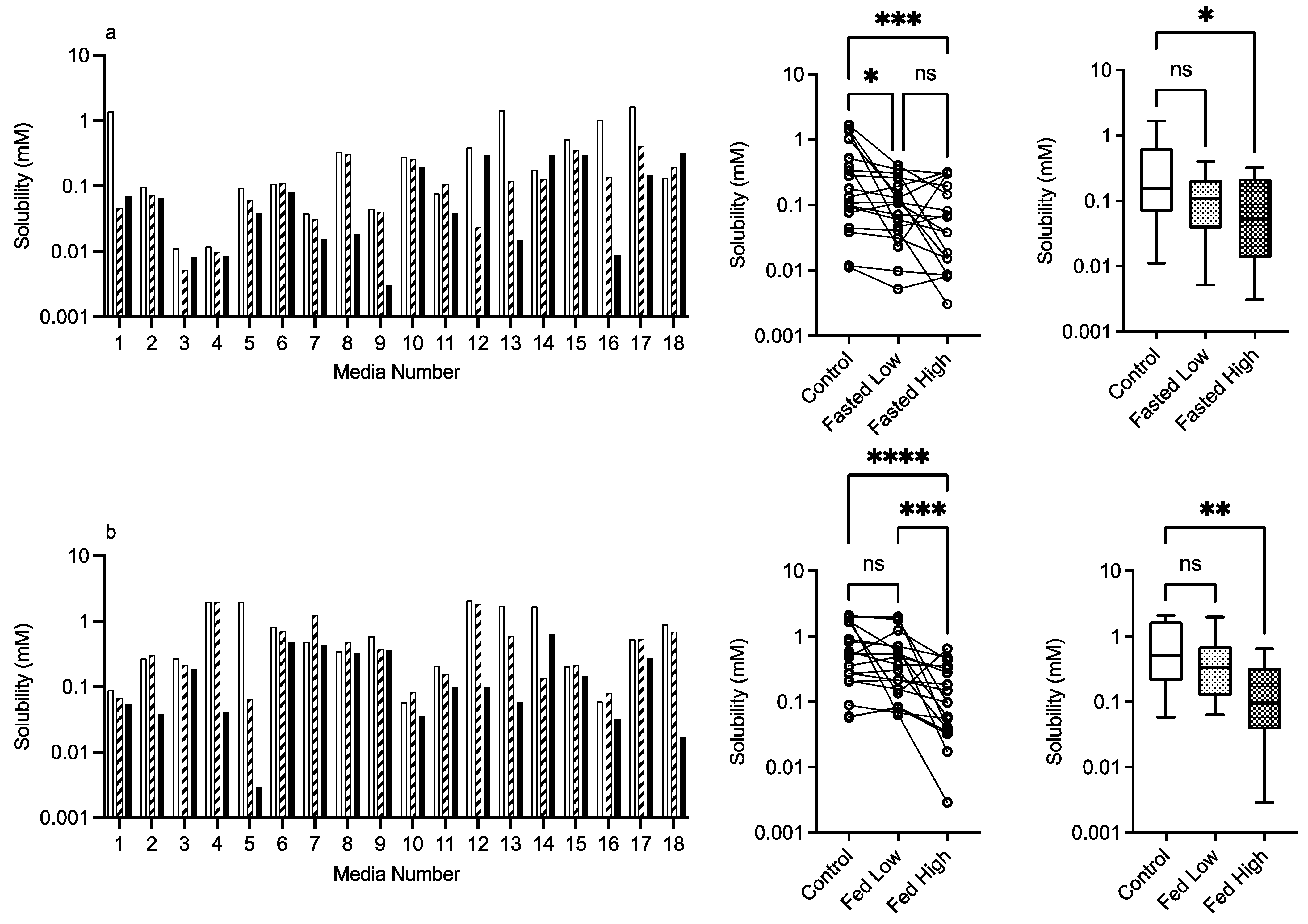
3.2.4. HPMC E3 and E50
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; ** p = 0.0077; **** p < 0.0001.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) ns = no significant difference, p > 0.05; **** p < 0.0001. (b) ns = no significant difference, p > 0.05; ** p = 0.0077; **** p < 0.0001. control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; * p < 0.0373; *** p < 0.0002; Kruskal–Wallis comparison, * p < 0.0263. (b) ns = no significant difference, p > 0.05; *** p = 0.0002; **** p < 0.0001; ** p = 0.0020.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; * p < 0.0373; *** p < 0.0002; Kruskal–Wallis comparison, * p < 0.0263. (b) ns = no significant difference, p > 0.05; *** p = 0.0002; **** p < 0.0001; ** p = 0.0020.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; * p < 0.0373; *** p < 0.0002; Kruskal–Wallis comparison, * p < 0.0263. (b) ns = no significant difference, p > 0.05; *** p = 0.0002; **** p < 0.0001; ** p = 0.0020.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; * p < 0.0373; *** p < 0.0002; Kruskal–Wallis comparison, * p < 0.0263. (b) ns = no significant difference, p > 0.05; *** p = 0.0002; **** p < 0.0001; ** p = 0.0020.
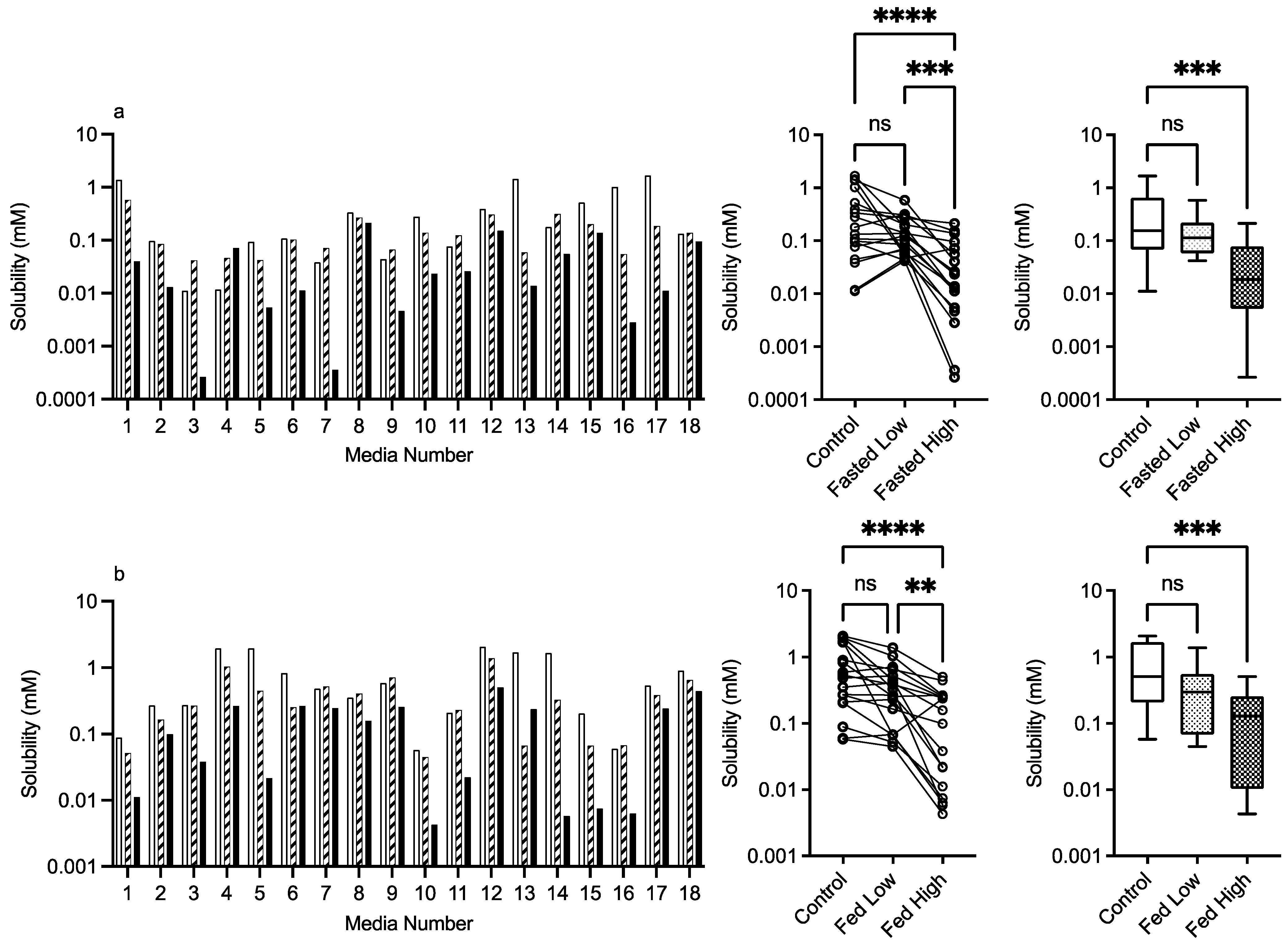
3.2.5. Chitosan
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; *** p < 0.0007; **** p < 0.0001; Kruskal–Wallis comparison, *** p < 0.0004. (b) ns = no significant difference, p > 0.05; ** p = 0.0042; **** p < 0.0001; Kruskal–Wallis comparison, *** p < 0.0004.
control (no excipient), low excipient concentration, high excipient concentration. Matched Friedman non-parametric comparison of groups (control, low excipient and high excipient concentration). Non-matched Kruskal–Wallis comparison of groups, control no excipient, low excipient concentration, high excipient concentration. Box and whisker plots—see legend in Figure 1. (a) Friedman comparison: ns = no significant difference, p > 0.05; *** p < 0.0007; **** p < 0.0001; Kruskal–Wallis comparison, *** p < 0.0004. (b) ns = no significant difference, p > 0.05; ** p = 0.0042; **** p < 0.0001; Kruskal–Wallis comparison, *** p < 0.0004.


4. Discussion
4.1. Control Excipient-Free Solubility Measurements
4.2. Effect of Excipients on Fenofibrate Equilibrium Solubility
4.2.1. Mannitol
4.2.2. PVP K12 and K29/32
4.2.3. HPMC E3 and E50
4.2.4. Chitosan
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis For a Biopharmaceutic Drug Classification—The Correlation Of In-Vitro Drug Product Dissolution and In-Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, J.; Butler, J.; Dressman, J. A Refined Developability Classification System. J. Pharm. Sci. 2018, 107, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J. 2010, 12, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Chae, S.I.; Noh, Y.S.; Lee, S.J.; Shim, W.S.; Yoon, J.M.; Hwang, S.J.; Lee, K.T.; Chung, E.K. Pharmacokinetics and bioequivalence of two fenofibrate choline formulations in healthy subjects under fed and fasted condition. Int. J. Clin. Pharmacol. Ther. 2019, 57, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, S.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Intestinal drug solubility estimation based on simulated intestinal fluids: Comparison with solubility in human intestinal fluids. Eur. J. Pharm. Sci. 2011, 43, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Roos, C.; Johansson, P.; Tannergren, C.; Lundqvist, A.; Langguth, P.; Sjoblom, M.; Sjogren, E.; Lennernas, H. The effects of three absorption-modifying critical excipients on the in vivo intestinal absorption of six model compounds in rats and dogs. Int. J. Pharm. 2018, 547, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Augustijns, P.; Wuyts, B.; Hens, B.; Annaert, P.; Butler, J.; Brouwers, J. A review of drug solubility in human intestinal fluids: Implications for the prediction of oral absorption. Eur. J. Pharm. Sci. 2014, 57, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Bou-Chacra, N.; Melo, K.J.C.; Morales, I.A.C.; Stippler, E.S.; Kesisoglou, F.; Yazdanian, M.; Lobenberg, R. Evolution of Choice of Solubility and Dissolution Media After Two Decades of Biopharmaceutical Classification System. AAPS J. 2017, 19, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Khadra, I.; Zhou, Z.; Dunn, C.; Wilson, C.G.; Halbert, G. Statistical investigation of simulated intestinal fluid composition on the equilibrium solubility of biopharmaceutics classification system class II drugs. Eur. J. Pharm. Sci. 2015, 67, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G.W. Statistical investigation of simulated fed intestinal media composition on the equilibrium solubility of oral drugs. Eur. J. Pharm. Sci. 2017, 99, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Ainousah, B.E.; Perrier, J.; Dunn, C.; Khadra, I.; Wilson, C.G.; Halbert, G. Dual Level Statistical Investigation of Equilibrium Solubility in Simulated Fasted and Fed Intestinal Fluid. Mol. Pharm. 2017, 14, 4170–4180. [Google Scholar] [CrossRef] [PubMed]
- Abuhassan, Q.; Khadra, I.; Pyper, K.; Halbert, G.W. Small scale in vitro method to determine a bioequivalent equilibrium solubility range for fasted human intestinal fluid. Eur. J. Pharm. Biopharm. 2021, 168, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Ines Silva, M.; Khadra, I.; Pyper, K.; Halbert, G.W. Small scale in vitro method to determine a potential bioequivalent equilibrium solubility range for fed human intestinal fluid. Eur. J. Pharm. Biopharm. 2022, 177, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T. Solubility and dissolution profile assessment in drug discovery. Drug Metab. Pharmacokinet. 2007, 22, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Rask, M.B.; Knopp, M.M.; Olesen, N.E.; Holm, R.; Rades, T. Influence of PVP/VA copolymer composition on drug-polymer solubility. Eur. J. Pharm. Sci. 2016, 85, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Tajber, L.; Tian, Y.; Olesen, N.E.; Jones, D.S.; Kozyra, A.; Lobmann, K.; Paluch, K.; Brennan, C.M.; Holm, R.; et al. Comparative Study of Different Methods for the Prediction of Drug-Polymer Solubility. Mol. Pharm. 2015, 12, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.C. Handbook of Pharmaceutical Excipients, 7th ed.; Pharmaceutical Press: London, UK, 2013; Volume 18, p. 544. [Google Scholar] [CrossRef]
- McPherson, S.; Perrier, J.; Dunn, C.; Khadra, I.; Davidson, S.; Ainousah, B.E.; Wilson, C.G.; Halbert, G. Small scale design of experiment investigation of equilibrium solubility in simulated fasted and fed intestinal fluid. Eur. J. Pharm. Biopharm. 2020, 150, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.S.; Kumar, V.; Singh, U.P.; Bhat, H.R.; Mazumder, B. Physicochemical characterization and in vitro dissolution studies of solid dispersions of ketoprofen with PVP K30 and d-mannitol. Saudi Pharm. J. 2013, 21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.A.; Ford, S.J.; Walker, A.A.; Hargreaves, R.H.J.; Halbert, G.W. Development of a lyophilised RH1 formulation: A novel DT diaphorase activated alkylating agent. J. Pharm. Pharmacol. 2002, 54, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Yalkowsky, S.H.; Rubino, J.T. Solubilization by Cosolvents. 1. Organic Solutes in Propylene-Glycol Water Mixtures. J. Pharm. Sci. 1985, 74, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Kleberg, K.; Jacobsen, J.; Mullertz, A. Characterising the behaviour of poorly water soluble drugs in the intestine: Application of biorelevant media for solubility, dissolution and transport studies. J. Pharm. Pharmacol. 2010, 62, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Bevernage, J.; Forier, T.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Excipient-mediated supersaturation stabilization in human intestinal fluids. Mol. Pharm. 2011, 8, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Pigliacelli, C.; Belton, P.; Wilde, P.; Qi, S. Probing the molecular interactions between pharmaceutical polymeric carriers and bile salts in simulated gastrointestinal fluids using NMR spectroscopy. J. Colloid Interface Sci. 2019, 551, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Martini, L.G.; Ford, J.L.; Roberts, M. The use of hypromellose in oral drug delivery. J. Pharm. Pharmacol. 2005, 57, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Z.; Chen, X.G.; Liu, N.; Wang, S.X.; Liu, C.S.; Meng, X.H.; Liu, C.G. Protonation constants of chitosan with different molecular weight and degree of deacetylation. Carbohydr. Polym. 2006, 65, 194–201. [Google Scholar] [CrossRef]
- Kubbinga, M.; Augustijns, P.; Garcia, M.A.; Heinen, C.; Wortelboer, H.M.; Verwei, M.; Langguth, P. The effect of chitosan on the bioaccessibility and intestinal permeability of acyclovir. Eur. J. Pharm. Biopharm. 2019, 136, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Nadai, M.; Tajiri, C.; Yoshizumi, H.; Suzuki, Y.; Zhao, Y.L.; Kimura, M.; Tsunekawa, Y.; Hasegawa, T. Effect of chitosan on gastrointestinal absorption of water-insoluble drugs following oral administration in rats. Biol. Pharm. Bull. 2006, 29, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Cano-Cebrian, M.J.; Olander, T.; Hedeland, M.; Sjoblom, M.; Lennernas, H. Regional Intestinal Drug Permeability and Effects of Permeation Enhancers in Rat. Pharmaceutics 2020, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Riethorst, D.; Mols, R.; Duchateau, G.; Tack, J.; Brouwers, J.; Augustijns, P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J. Pharm. Sci. 2016, 105, 673–681. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | MWt (g/mol) | Substance | Fasted State | Fed State | ||
|---|---|---|---|---|---|---|
| Lower | Upper | Lower | Upper | |||
| Bile salt | 515.70 | Sodium taurocholate | 1.5 mM | 5.9 mM | 3.6 mM | 15 mM |
| Lecithin | 750.00 | Phosphatidylcholine | 0.2 mM | 0.75 mM | 0.5 mM | 3.75 mM |
| Fatty acid | 304.44 | Sodium oleate | 0.5 mM | 15 mM | 0.8 mM | 25 mM |
| Mono-glyceride | 358.57 | Glyceryl monooleate | 0.1 mM | 2.8 mM | 1 mM | 9 mM |
| Cholesterol | 386.65 | Cholesterol | 0.1 mM | 0.26 mM | 0.13 mM | 1 mM |
| pH | Sodium hydroxide/hydrochloric acid qs | 5 | 7 | 5 | 7 | |
| BS:PL ratio | 7.5 | 7.9 | 7.2 | 4 | ||
| Excipient/Concentration | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mannitol | PVP LG | PVP K29/32 | HPMC E3 | HPMC E50 | Chitosan | |||||||
| Media | Low | High | Low | High | Low | High | Low | High | Low | High | Low | High |
| Factor | Fasted | |||||||||||
| pH | −S | −S | −S | −S | −S | −S | −S | NS | NS | NS | NS | NS |
| Na Oleate | +S | +S | +S | +S | +S | +S | +S | +S | +S | +S | +S | NS |
| Bile Salt | NS | NS | NS | NS | NS | NS | NS | +S | NS | NS | NS | NS |
| Lecithin | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | +S |
| MG | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Cholesterol | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| BS/PL ratio | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Fed | ||||||||||||
| pH | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | −S |
| Na Oleate | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Bile Salt | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Lecithin | NS | NS | NS | NS | NS | NS | NS | +S | NS | NS | NS | +S |
| MG | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Cholesterol | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| BS/PL ratio | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ainousah, B.E.; Khadra, I.; Halbert, G.W. Excipient Impact on Fenofibrate Equilibrium Solubility in Fasted and Fed Simulated Intestinal Fluids Assessed Using a Design of Experiment Protocol. Pharmaceutics 2023, 15, 2484. https://doi.org/10.3390/pharmaceutics15102484
Ainousah BE, Khadra I, Halbert GW. Excipient Impact on Fenofibrate Equilibrium Solubility in Fasted and Fed Simulated Intestinal Fluids Assessed Using a Design of Experiment Protocol. Pharmaceutics. 2023; 15(10):2484. https://doi.org/10.3390/pharmaceutics15102484
Chicago/Turabian StyleAinousah, Bayan E., Ibrahim Khadra, and Gavin W. Halbert. 2023. "Excipient Impact on Fenofibrate Equilibrium Solubility in Fasted and Fed Simulated Intestinal Fluids Assessed Using a Design of Experiment Protocol" Pharmaceutics 15, no. 10: 2484. https://doi.org/10.3390/pharmaceutics15102484