
Anticancer Tungstenocenes with a Diverse Set of (O,O–), (O,S–) and (O,N–) Chelates—A Detailed Biological Study Using an Improved Evaluation via 3D Spheroid Models
 , , , , and
, , , , and
Abstract
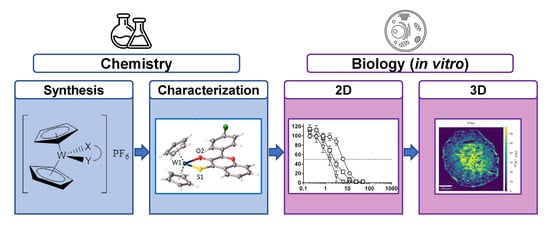
:
1. Introduction
2. Materials and Methods
2.1. Experimental Part
2.2. Synthesis
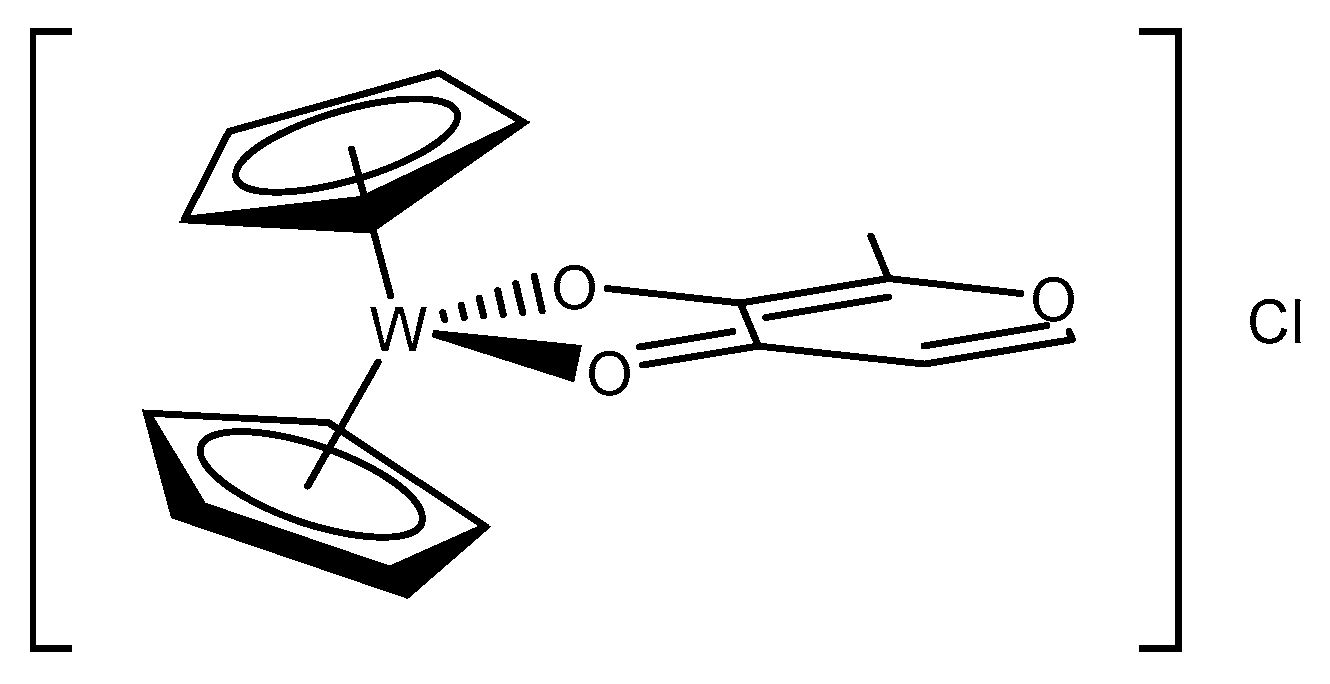
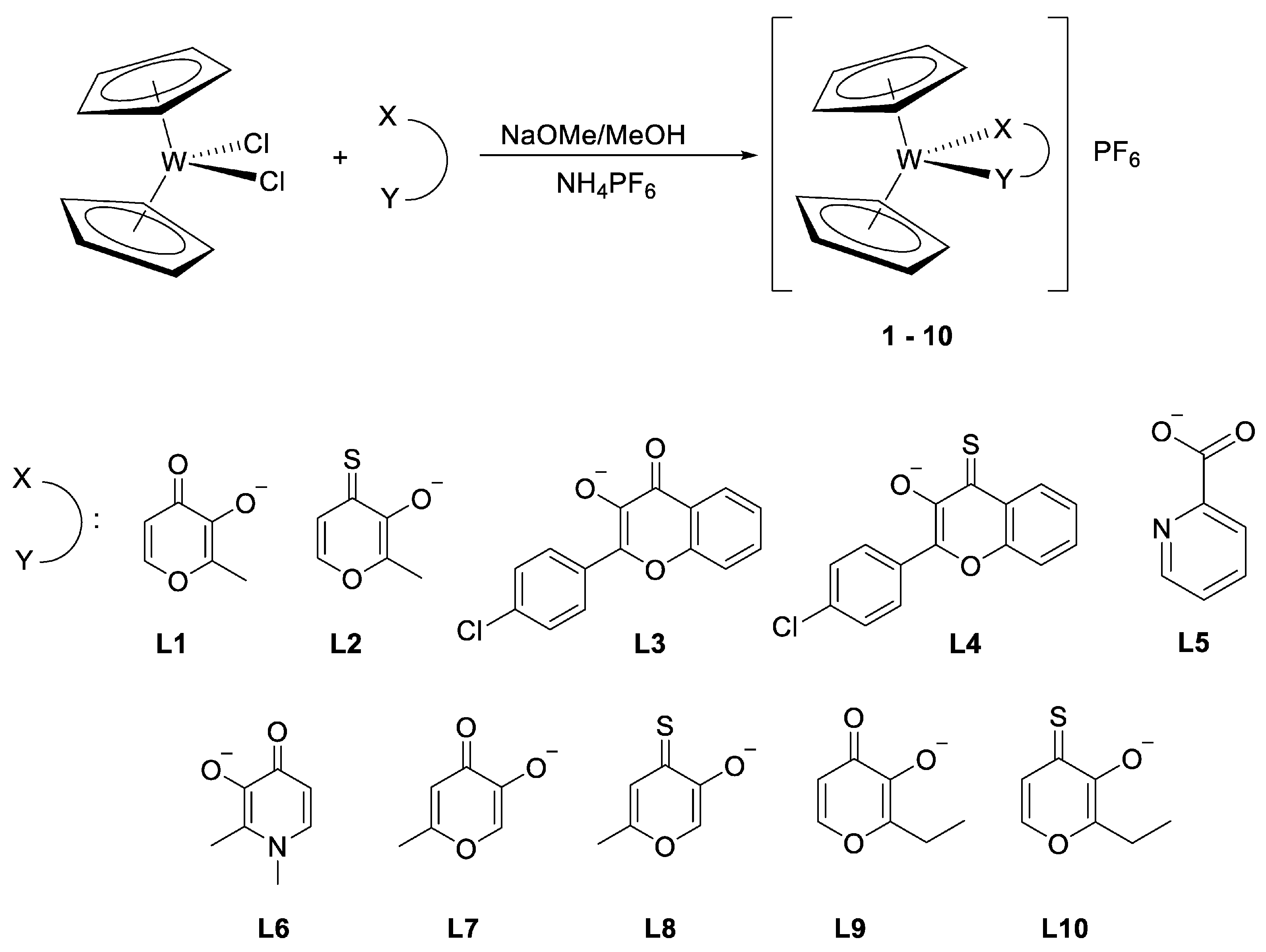
2.2.1. General Complexation Synthesis A
2.2.2. General Complexation Synthesis B
Bis(η5-cyclopentadienyl)[2-methyl-3-(oxo-κO)-4-(1H)-pyran-4-ato-κO]tungsten(IV) hexafluorophosphate (1)
Bis(η5-cyclopentadienyl)[2-methyl-3-(oxo-κO)-4-(1H)-pyran-4-ato-κO]tungsten(IV) chloride (1A)
Bis(η5-cyclopentadienyl)[2-methyl-3-(oxo-κO)-4-(1H)-pyran-4-thionato-κS]tungsten(IV) hexafluorophosphate (2)
Bis(η5-cyclopentadienyl)[2-(4-chlorophenyl)-3-(oxo-κO)-4(H)-chromen-4-oato-κO]tungsten(IV) hexafluorophosphate (3)
Bis(η5-cyclopentadienyl)[2-(4-chlorophenyl)-3-(oxo-κO)-4(H)-chromen-4-thionato-κS]tungsten(IV) hexafluorophosphate (4)
Bis(η5-cyclopentadienyl)[2-(carboxylato-κO)-pyridine-κN]tungsten(IV) hexafluorophosphate (5)
Bis(η5-cyclopentadienyl)[1,2-dimethyl-3-(oxo-κO)-4-(1H)-pyridonato-κO]tungsten(IV) hexafluorophosphate (6)
Bis(η5-cyclopentadienyl)[2-methyl-5-(oxo-κO)-4-(1H)-pyran-4-ato-κO] tungsten(IV) hexafluorophosphate (7)
Bis(η5-cyclopentadienyl)[2-methyl-5-(oxo-κO)-pyran-4-(1H)-thionato-κS] tungsten(IV) hexafluorophosphate (8)
Bis(η5-cyclopentadienyl)[2-ethyl-3-(oxo-κO)-4-(1H)-pyran-4-ato-κO] tungsten(IV) hexafluorophosphate (9)
Bis(η5-cyclopentadienyl)[2-ethyl-3-(oxo-κO)-4-(1H)-pyran-4-thionato-κS] tungsten(IV) hexafluorophosphate (10)
2.3. Cell Lines and Culture Conditions
2.4. Spheroid Formation and Growth
2.5. Cell Viability (MTT Assay)
2.6. Cell Viability (Resazurin Assay)
2.7. Apoptosis Studies
2.8. Electrophoretic dsDNA Plasmid Assay
2.9. Reactive Oxygen Species (ROS) Detection
2.10. LA-ICP-TOF-MS Analysis
2.11. Immunofluorescence Staining
3. Results and Discussion
3.1. Synthesis and Characterisation
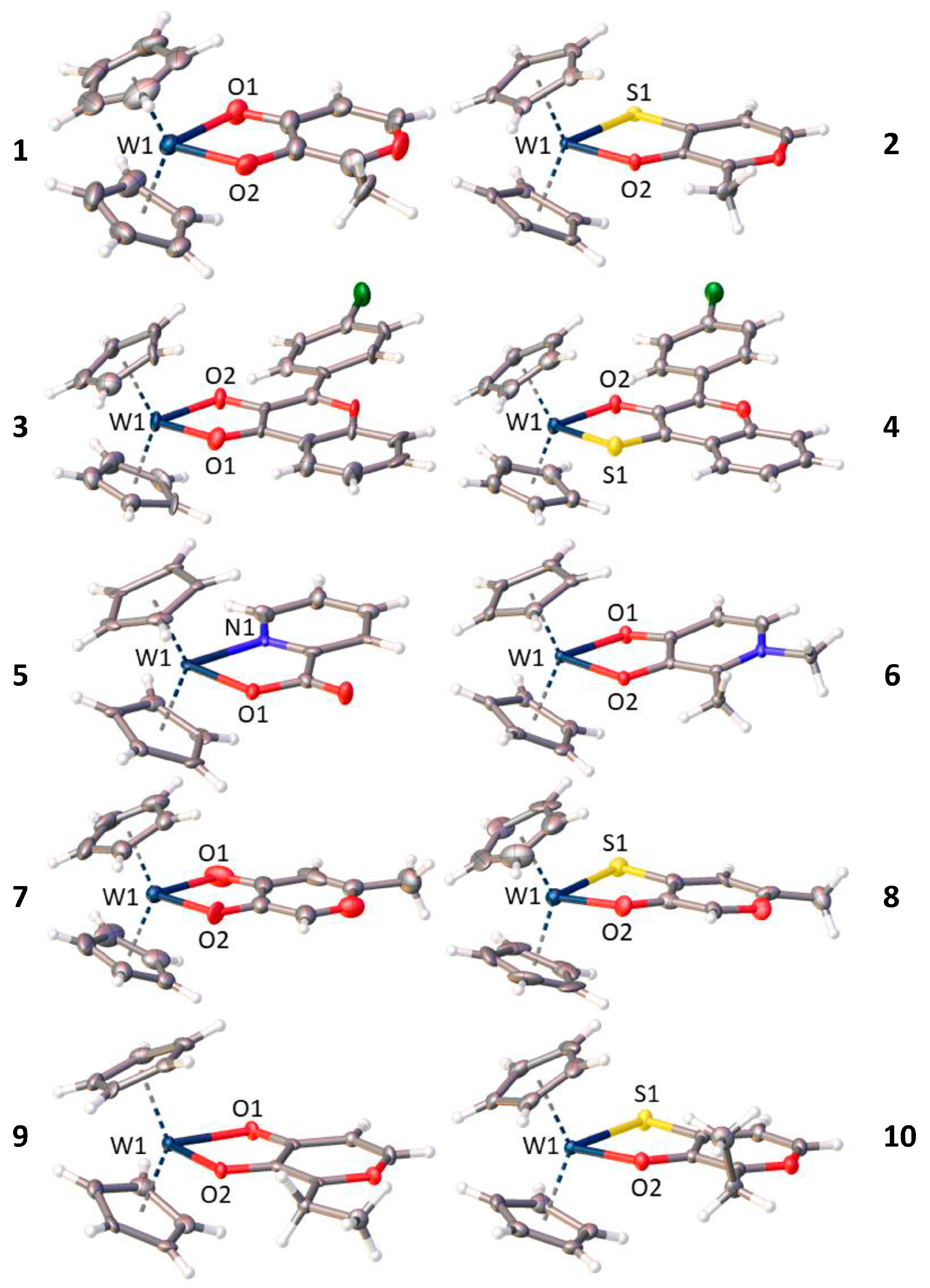
3.2. X-ray Diffractometry
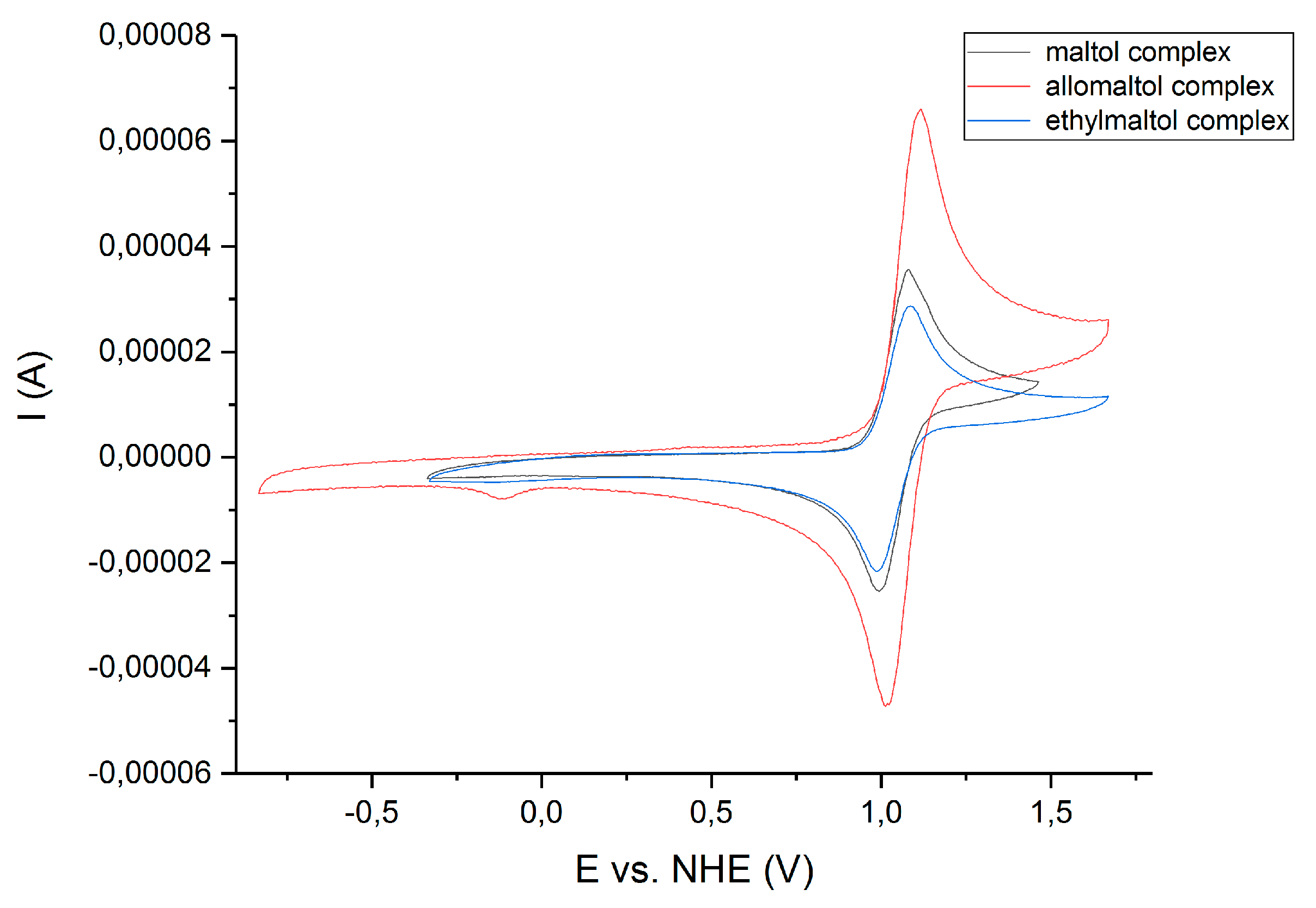
3.3. Cyclic Voltammetry
3.4. Aqueous Solubility and Stability Measurements
3.5. Cytotoxic Activity In Vitro (MTT Assay)
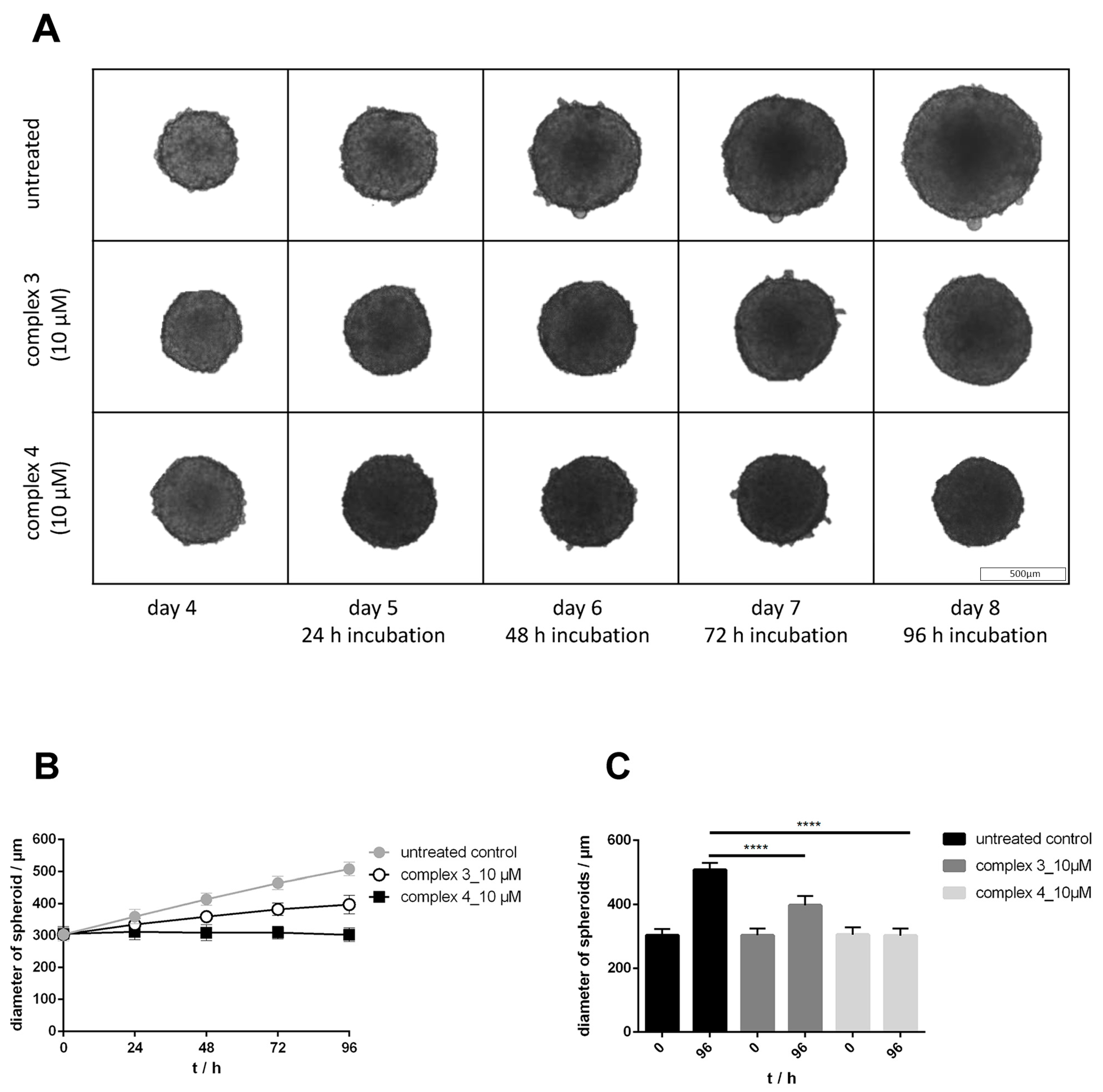
3.6. Drug Sensitivity in 2D and 3D Cultures, Effects on Growth and Morphology
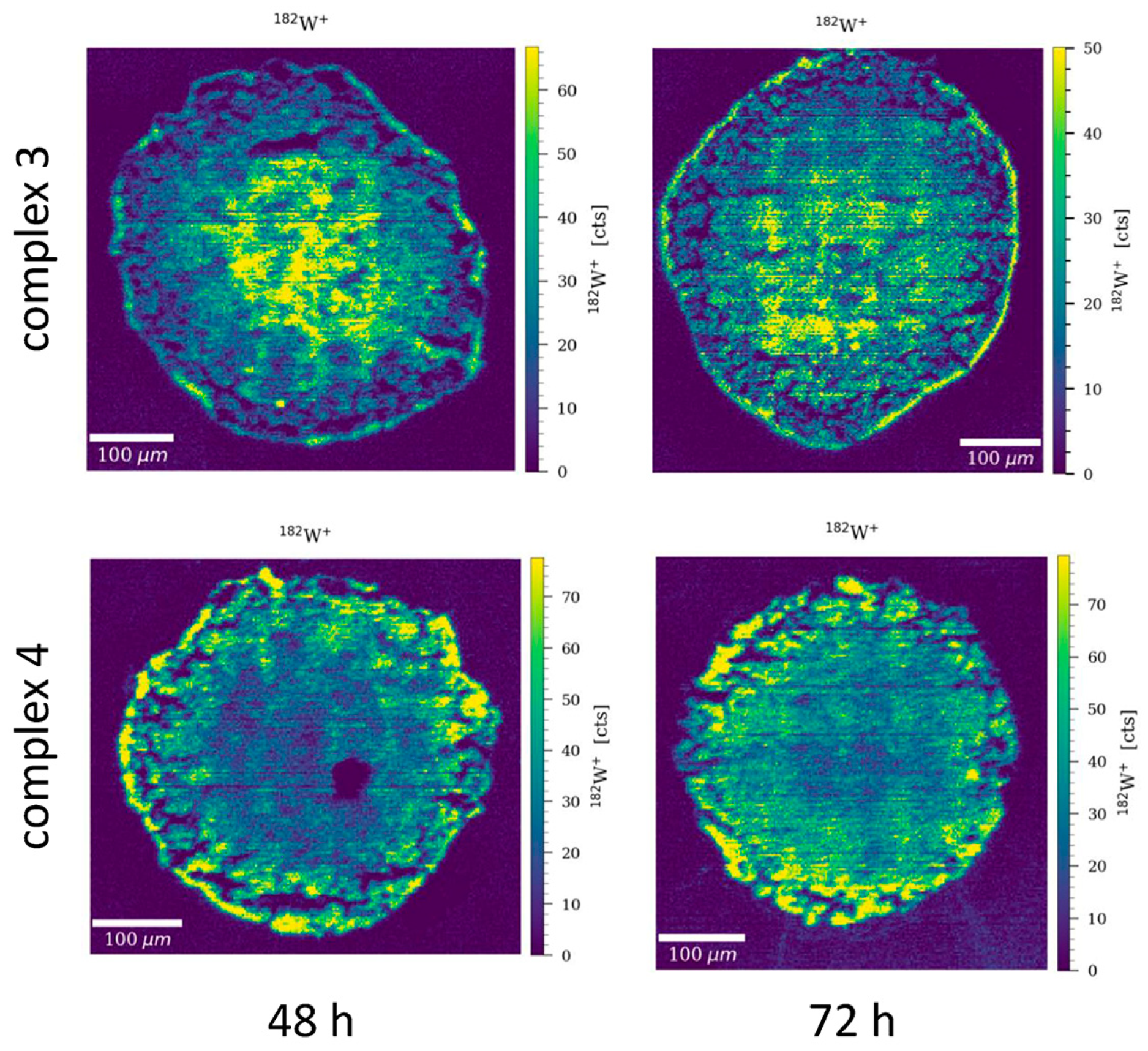
3.7. W Distribution in HCT116 Tumor Spheroids
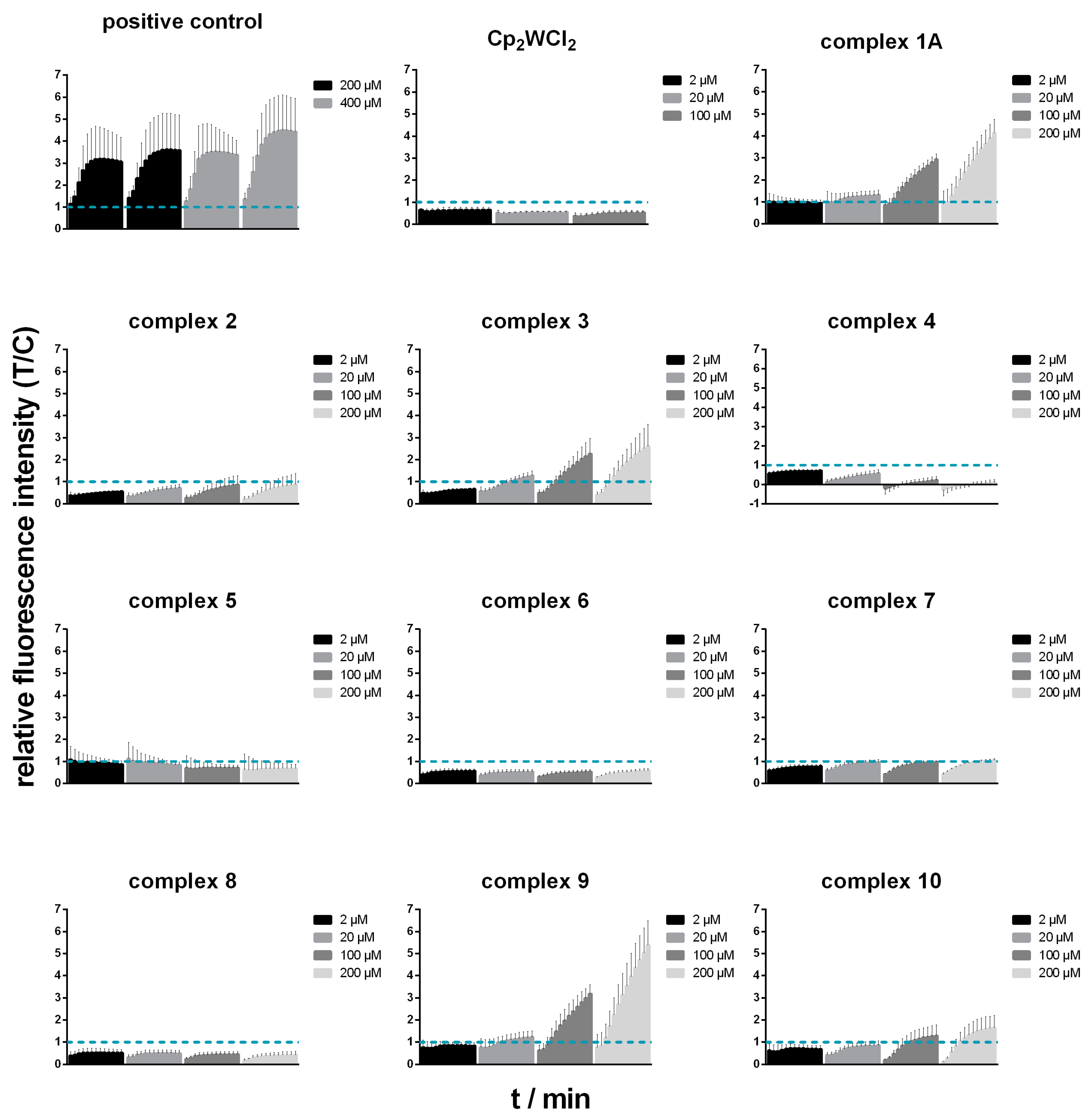
3.8. Oxidative Stress Studies
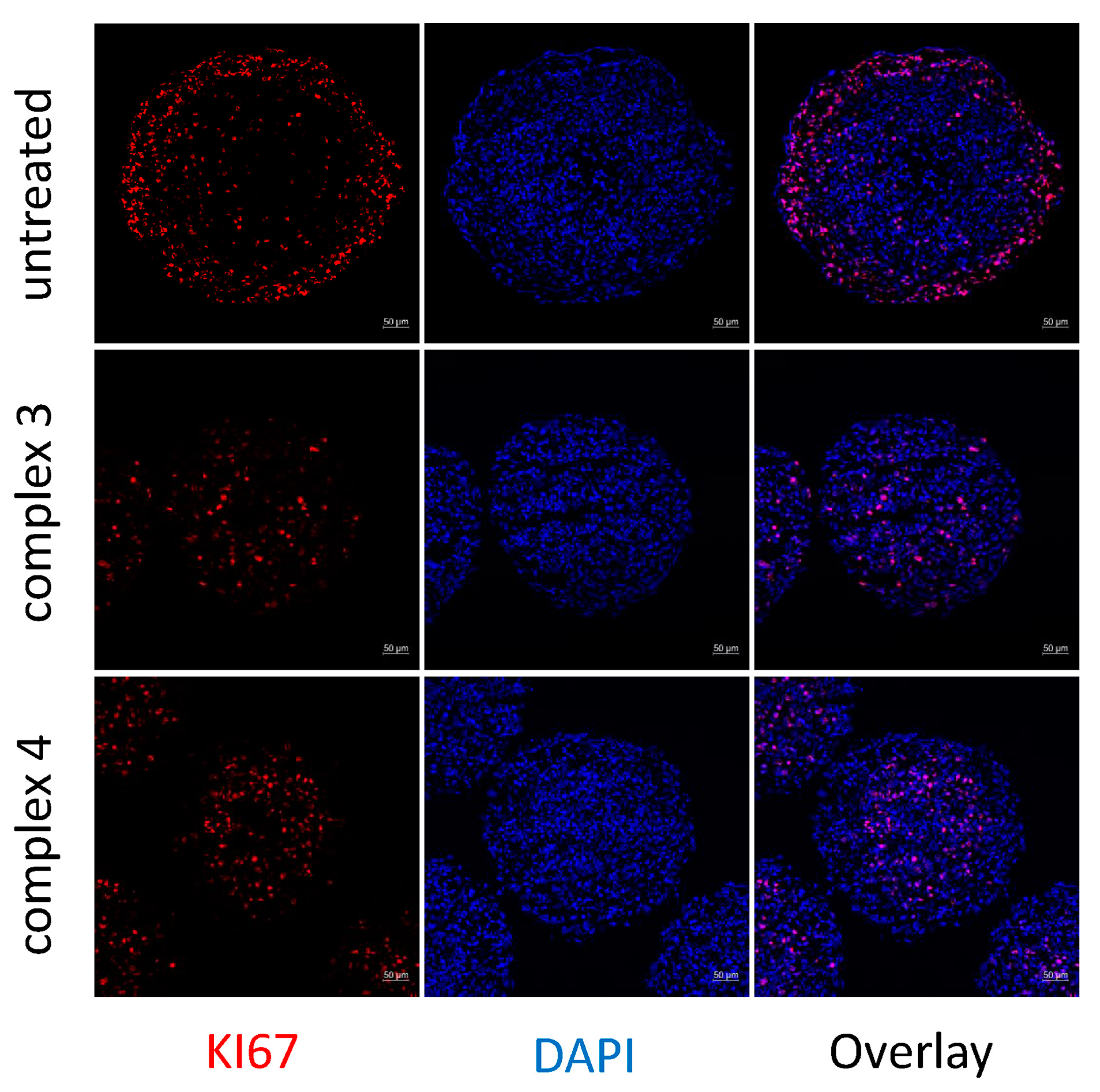
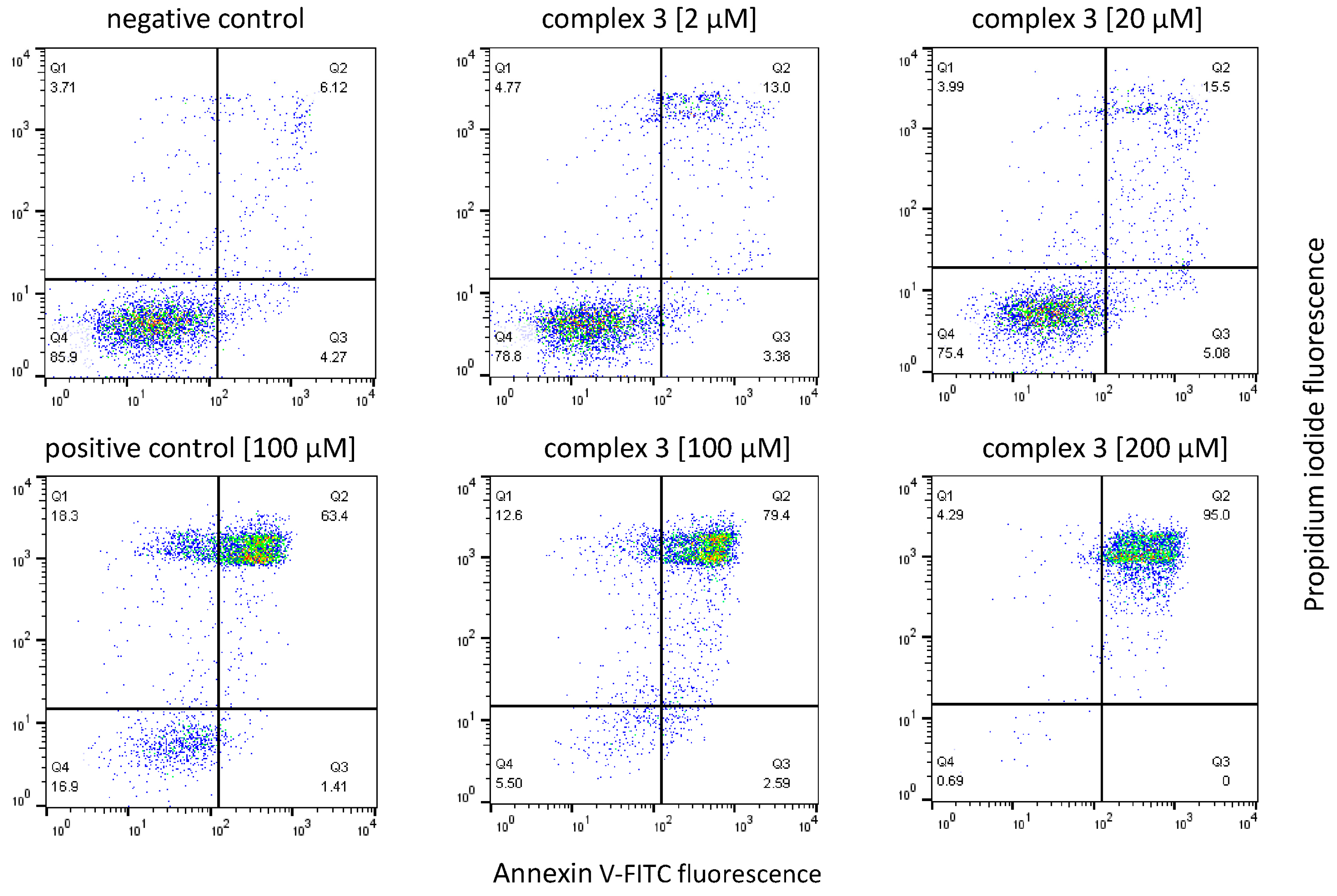
3.9. Apoptosis Studies
3.10. Cell-Free DNA Interaction Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gasser, G.; Ott, I.; Metzler-Nolte, N. Organometallic anticancer compounds. J. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef]
- Jaouen, G.; Top, S.; Vessières, A. Organometallics Targeted to Specific Biological Sites: The Development of New Therapies. In Bioorganometallics: Biomolecules, Labeling, Medicine; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2005. [Google Scholar]
- Schirrmacher, V. Quo Vadis Cancer Therapy? LAP Lambert Academic Publishing: London, UK, 2017. [Google Scholar]
- Keith, L.S.; Moffett, D.B.; Rosemond, Z.A.; Wohlers, D.W. ATSDR evaluation of health effects of tungsten and relevance to public health. Toxicol. Ind. Health 2007, 23, 347–387. [Google Scholar] [CrossRef]
- Vyskočil, A.; Viau, C. Assessment of molybdenum toxicity in humans. J. Appl. Toxicol. 1999, 19, 185–192. [Google Scholar] [CrossRef]
- Köpf, H.; Köpf-Maier, P. Titanocene Dichloride—The First Metallocene with Cancerostatic Activity. Angew. Chem. Int. Ed. Engl. 1979, 18, 477–478. [Google Scholar] [CrossRef]
- Köpf-Maier, P.; Leitner, M.; Voigtländer, R.; Köpf, H. Molybdocene Dichloride as an Antitumor Agent. Z. fur Nat. CA J. Biosci. 1979, 34, 1174–1176. [Google Scholar]
- Köpf-Maier, P.; Leitner, M.; Köpf, H. Tumor Inhibition by Metallocenes: Antitumor Activity of Niobocene and Tungstocene Dichlorides. J. Inorg. Nucl. Chem. 1980, 42, 1789–1791. [Google Scholar] [CrossRef]
- Köpf-Maier, P.; Köpf, H. Vanadocen-dichlorid—Ein weiteres Antitumor-Agens aus der Metallocenreihe/Vanadocene Dichloride—Another Antitumor Agent from the Metallocene Series. Z. Nat. B 1979, 34, 805–807. [Google Scholar] [CrossRef]
- Ghosh, P.; D’Cruz, O.J.; Narla, R.K.; Uckun, F.M. Apoptosis-Inducing Vanadocene Compounds against Human Testicular Cancer. Clin. Cancer Res. 2000, 6, 1536–1545. [Google Scholar]
- Harding, M.M.; Mokdsi, G. Antitumour Metallocenes: Structure-Activity Studies and Interactions with Biomolecules. Curr. Med. Chem. 2000, 7, 1289–1303. [Google Scholar] [CrossRef]
- Köpf-Maier, P. Complexes of Metals Other than Platinum as Antitumour Agents. Eur. J. Clin. Pharmacol. 1994, 47, 1–16. [Google Scholar] [CrossRef]
- Strohfeldt, K.; Tacke, M. Bioorganometallic fulvene-derived titanocene anti-cancer drugs. Chem. Soc. Rev. 2008, 37, 1174–1187. [Google Scholar] [CrossRef]
- Waern, J.B.; Harding, M.M. Bioorganometallic chemistry of molybdocene dichloride. J. Organomet. Chem. 2004, 689, 4655–4668. [Google Scholar] [CrossRef]
- Gao, L.M.; Vera, J.L.; Matta, J.; Meléndez, E. Synthesis and cytotoxicity studies of steroid-functionalized titanocenes as potential anticancer drugs: Sex steroids as potential vectors for titanocenes. J. Biol. Inorg. Chem. 2010, 15, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Garcia, M.; Ortega-Zuniga, C.; Meléndez, E. New tungstenocenes containing 3-hydroxy-4-pyrone ligands: Antiproliferative activity on HT-29 and MCF-7 cell lines and binding to human serum albumin studied by fluorescence spectroscopy and molecular modeling methods. J. Biol. Inorg. Chem. 2013, 18, 195–209. [Google Scholar] [CrossRef]
- Gomez-Ruiz, S.; Maksimovic-Ivanic, D.; Mijatovic, S.; Kaluderovic, G.N. On the discovery, biological effects, and use of Cisplatin and metallocenes in anticancer chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, E. Metallocenes as Target Specific Drugs for Cancer Treatment. Inorg. Chim. Acta 2012, 393, 36–52. [Google Scholar] [CrossRef] [Green Version]
- Kuo, L.Y.; Kanatzidis, M.G.; Sabat, M.; Tipton, A.L.; Marks, T.J. Metallocene Antitumor Agents. Solution and Solid-State Molybdenocene Coordination Chemistry of DNA Constituents. J. Am. Chem. Soc. 1991, 113, 9027–9045. [Google Scholar] [CrossRef]
- Balzarek, C.; Weakley, T.J.R.; Kuo, L.Y.; Tyler, D.R. Investigation of the Monomer−Dimer Equilibria of Molybdocenes in Water. Organometallics 2000, 19, 2927–2931. [Google Scholar] [CrossRef]
- Kandioller, W.; Reikersdorfer, M.; Theiner, S.; Roller, A.; Hejl, M.; Jakupec, M.A.; Malarek, M.S.; Keppler, B.K. The Impact of Leaving Group Variation on the Anticancer Activity of Molybdenocenes. Organometallics 2018, 37, 3909–3916. [Google Scholar] [CrossRef]
- Feliciano, I.; Matta, J.; Meléndez, E. Water-soluble molybdenocene complexes with both proliferative and antiproliferative effects on cancer cell lines and their binding interactions with human serum albumin. J. Biol. Inorg. Chem. 2009, 14, 1109–1117. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, E. Bioorganometallic Chemistry of Molybdenocene Dichloride and Its Derivatives. J. Organomet. Chem. 2012, 706–707, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, V.; Cseh, K.; Hejl, M.; Vician, P.; Neuditschko, B.; Meier-Menches, S.M.; Janker, L.; Bileck, A.; Gajic, N.; Kronberger, J.; et al. Highly Cytotoxic Molybdenocenes with Strong Metabolic Effects Inhibit Tumour Growth in Mice. Chem. Eur. J. 2022, 29, e202202648. [Google Scholar] [CrossRef]
- Kandioller, W.; Kurzwernhart, A.; Hanif, M.; Meier, S.M.; Henke, H.; Keppler, B.K.; Hartinger, C.G. Pyrone derivatives and metals: From natural products to metal-based drugs. J. Organomet. Chem. 2011, 696, 999–1010. [Google Scholar] [CrossRef]
- Kandioller, W.; Hartinger, C.G.; Nazarov, A.A.; Bartel, C.; Skocic, M.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Maltol-derived ruthenium-cymene complexes with tumor inhibiting properties: The impact of ligand-metal bond stability on anticancer activity in vitro. Chemistry 2009, 15, 12283–12291. [Google Scholar] [CrossRef]
- Kandioller, W.; Hartinger, C.G.; Nazarov, A.A.; Kuznetsov, M.L.; John, R.O.; Bartel, C.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. From Pyrone to Thiopyrone Ligands−Rendering Maltol-Derived Ruthenium(II)−Arene Complexes That Are Anticancer Active in Vitro. Organometallics 2009, 28, 4249–4251. [Google Scholar] [CrossRef]
- Reddy, V.D.; Dayal, D.; Szalda, D.J.; Cosenza, S.C.; Ramana Reddy, M.V. Syntheses, structures, and anticancer activity of novel organometallic ruthenium–maltol complexes. J. Organomet. Chem. 2012, 700, 180–187. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Seth, N.; Gupta, V.D.; Nöth, H.; Polborn, K.; Thomann, M.; Schwenk, H. Synthesis and Structure of Organotin(IV) Complexes of Maltol. Chem. Ber. 1994, 127, 1895–1900. [Google Scholar] [CrossRef]
- Lamboy, J.L.; Pasquale, A.; Rheingold, A.L.; Meléndez, E. Synthesis, Solution and Solid State Structure of Titanium-Maltol Complex. Inorg. Chim. Acta 2007, 360, 2115–2120. [Google Scholar] [CrossRef] [Green Version]
- Berasaluce, I.; Cseh, K.; Roller, A.; Hejl, M.; Heffeter, P.; Berger, W.; Jakupec, M.A.; Kandioller, W.; Malarek, M.S.; Keppler, B.K. The First Anticancer Tris(pyrazolyl)borate Molybdenum(IV) Complexes: Tested In Vitro and In Vivo-A Comparison of O,O-, S,O-, and N,N-Chelate Effects. Chem. Eur. J. 2020, 26, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The Effects of Plant Flavonoids on Mammalian Cells: Implications for Inflammation, Heart Disease, and Cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Grant, R.S.; Coggan, S.E.; Smythe, G.A. The Physiological Action of Picolinic Acid in the Human Brain. Int. J. Tryptophan Res. IJTR 2009, 2, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasumoto, E.; Nakano, K.; Nakayachi, T.; Morshed, S.R.M.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Kawase, M.; Sakagami, H. Cytotoxic Activity of Deferiprone, Maltol and Related Hydroxyketones against Human Tumor Cell Lines. Anticancer Res. 2004, 24, 755–762. [Google Scholar] [PubMed]
- Williams, D.B.; Lawton, M. Drying of organic solvents: Quantitative evaluation of the efficiency of several desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Chaves, S.; Gil, M.; Canario, S.; Jelic, R.; Romao, M.J.; Trincao, J.; Herdtweck, E.; Sousa, J.; Diniz, C.; Fresco, P.; et al. Biologically relevant O,S-donor compounds. Synthesis, molybdenum complexation and xanthine oxidase inhibition. Dalton Trans. 2008, 13, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Kurzwernhart, A.; Kandioller, W.; Bachler, S.; Bartel, C.; Martic, S.; Buczkowska, M.; Mühlgassner, G.; Jakupec, M.A.; Kraatz, H.-B.; Bednarski, P.J.; et al. Structure−Activity Relationships of Targeted RuII(η6-p-Cymene) Anticancer Complexes with Flavonol-Derived Ligands. J. Med. Chem. 2012, 55, 10512–10522. [Google Scholar] [CrossRef]
- Enyedy, E.A.; Sija, E.; Jakusch, T.; Hartinger, C.G.; Kandioller, W.; Keppler, B.K.; Kiss, T. Solution equilibria of anticancer ruthenium(II)-(eta(6)-p-cymene)-hydroxy(thio)pyr(id)one complexes: Impact of sulfur vs. oxygen donor systems on the speciation and bioactivity. J. Inorg. Biochem. 2013, 127, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Johnson, S.L.; Jacobsen, J.A.; Miller, M.T.; Chen, L.H.; Pellecchia, M.; Cohen, S.M. Chelator fragment libraries for targeting metalloproteinases. ChemMedChem 2010, 5, 195–199. [Google Scholar] [CrossRef]
- Pavlishchuk, V.V.; Addison, A.W. Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar] [CrossRef]
- Cseh, K.; Geisler, H.; Stanojkovska, K.; Westermayr, J.; Brunmayr, P.; Wenisch, D.; Gajic, N.; Hejl, M.; Schaier, M.; Koellensperger, G.; et al. Arene Variation of Highly Cytotoxic Tridentate Naphthoquinone-Based Ruthenium(II) Complexes and In-Depth In Vitro Studies. Pharmaceutics 2022, 14, 2466. [Google Scholar] [CrossRef]
- Scaffidi-Domianello, Y.Y.; Legin, A.; Jakupec, M.A.; Arion, V.B.; Kukushkin, V.Y.; Galanski, M.S.; Keppler, B.K. Synthesis, characterization, and cytotoxic activity of novel potentially pH-sensitive nonclassical platinum(II) complexes featuring 1,3-dihydroxyacetone oxime ligands. Inorg. Chem. 2011, 50, 10673–10681. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd ed.; The University of West London: Middlesex, UK, 2002; Volume 124, p. 1830. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Berger, W.; Elbling, L.; Micksche, M. Expression of the major vault protein LRP in human non-small-cell lung cancer cells: Activation by short-term exposure to antineoplastic drugs. Int. J. Cancer 2000, 88, 293–300. [Google Scholar] [CrossRef]
- Kubanik, M.; Tu, J.K.Y.; Söhnel, T.; Hejl, M.; Jakupec, M.A.; Kandioller, W.; Keppler, B.K.; Hartinger, C.G. Expanding on the Structural Diversity of Flavone- Derived RutheniumII(ƞ6-arene) Anticancer Agents. Metallodrugs 2015, 1, 24–35. [Google Scholar] [CrossRef]
- Khater, M.; Ravishankar, D.; Greco, F.; Osborn, H.M. Metal complexes of flavonoids: Their synthesis, characterization and enhanced antioxidant and anticancer activities. Future Med. Chem 2019, 11, 2845–2867. [Google Scholar] [CrossRef] [PubMed]
- Duval, K.; Grover, H.; Han, L.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontoura, J.C.; Viezzer, C.; Dos Santos, F.G.; Ligabue, R.A.; Weinlich, R.; Puga, R.D.; Antonow, D.; Severino, P.; Bonorino, C. Comparison of 2D and 3D cell culture models for cell growth, gene expression and drug resistance. Mater. Sci. Eng. C 2020, 107, 110264. [Google Scholar] [CrossRef]
- Nirmalanandhan, V.S.; Duren, A.; Hendricks, P.; Vielhauer, G.; Sittampalam, G.S. Activity of anticancer agents in a three-dimensional cell culture model. Drug Dev. Technol. 2010, 8, 581–590. [Google Scholar] [CrossRef]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Paul Solomon, F.D. 3D Cell Culture Systems Advantages and Applications. J. Cell. Physiol. 2014, 230, 16–26. [Google Scholar] [CrossRef]
- Wang, H.; Brown, P.C.; Chow, E.C.Y.; Ewart, L.S.; Ferguson, S.S.; Fitzpatrick, S.; Freedman, B.S.; Guo, G.L.; Hedrich, W.; Heyward, S.; et al. 3D cell culture models: Drug pharmacokinetics, safety assessment, and regulatory consideration. Clin. Transl. Sci. 2021, 14, 1659–1680. [Google Scholar] [CrossRef]
- Bonnier, F.; Keating, M.E.; Wróbel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Uxa, S.; Castillo-Binder, P.; Kohler, R.; Stangner, K.; Müller, G.A.; Engeland, K. Ki-67 gene expression. Cell Death Differ. 2021, 28, 3357–3370. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer (Review). Mol. Med. Rep 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Kanwar, S.S. Phosphatidylserine: A cancer cell targeting biomarker. Semin. Cancer Biol. 2018, 52, 17–25. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Kupcho, K.; Shultz, J.; Hurst, R.; Hartnett, J.; Zhou, W.; Machleidt, T.; Grailer, J.; Worzella, T.; Riss, T.; Lazar, D.; et al. A real-time, bioluminescent annexin V assay for the assessment of apoptosis. Apoptosis 2019, 24, 184–197. [Google Scholar] [CrossRef] [Green Version]
- Eray, M.; Mättö, M.; Kaartinen, M.; Andersson, L.C.; Pelkonen, J. Flow Cytometric Analysis of Apoptotic Subpopulations with a Combination of Annexin V-FITC, Propidium Iodide, and SYTO 17. Cytometry 2001, 43, 134–142. [Google Scholar] [CrossRef]
- Göschl, S.; Schreiber-Brynzak, E.; Pichler, V.; Cseh, K.; Heffeter, P.; Jungwirth, U.; Jakupec, M.A.; Berger, W.; Keppler, B.K. Comparative studies of oxaliplatin-based platinum(IV) complexes in different in vitro and in vivo tumor models. Metallomics 2017, 9, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valiahdi, S.M.; Heffeter, P.; Jakupec, M.A.; Marculescu, R.; Berger, W.; Rappersberger, K.; Keppler, B.K. The gallium complex KP46 exerts strong activity against primary explanted melanoma cells and induces apoptosis in melanoma cell lines. Melanoma Res. 2009, 19, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, H.; Westermayr, J.; Cseh, K.; Wenisch, D.; Fuchs, V.; Harringer, S.; Plutzar, S.; Gajic, N.; Hejl, M.; Jakupec, M.A.; et al. Tridentate 3-Substituted Naphthoquinone Ruthenium Arene Complexes: Synthesis, Characterization, Aqueous Behavior, and Theoretical and Biological Studies. Inorg. Chem. 2021, 60, 9805–9819. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths [Å] | |||||
|---|---|---|---|---|---|
| Compound | W–O2 | W–O/S/N | W–Cpcentroid | Ring Slippage | C1=O/S1 |
| 2 | 2.067 (2) | 2.441 (1) | 1.9821 (8) | 0.151 | 1.710 (2) |
| 3 | 2.072 (7) | 2.127 (8) | 1.965 (6)–1.979 (6) | 0.106–0.147 | 1.309 (12) |
| 4 | 2.0812 (15) | 2.4298 (5) | 1.9776 (11)–1.9824 (12) | 0.130–0.173 | 1.699 (2) |
| 5 | 2.097 (9) | 2.157 (11) | 1.966 (3)–1.972 (3) | 0.107–0.128 | 1.35 (2) |
| 6 | 2.0940 (19) | 2.097 (2) | 1.9690 (14)–1.9745 (14) | 0.143–0.161 | 1.312 (3) |
| 7 | 2.102 (8) | 2.114 (13) | 1.964 (5) | 0.143 | 1.32 (2) |
| 8 | 2.091 (11) | 2.454 (4) | 1.974 (10)–1.975 (9) | 0.117–0.153 | 1.709 (15) |
| 9 | 2.074 (5) | 2.118 (4) | 1.970 (2) | 0.156 | 1.287 (8) |
| 10 | 2.082 (1) | 2.454 (1) | 1.9822 (11)–1.9832 (12) | 0.125–0.145 | 1.706 (2) |
| Compound | E1/2 (V) |
|---|---|
| 1 | 1.037 |
| 2 | 1.047 |
| 3 | 1.059 |
| 4 | 1.082 |
| 5 | 1.330 |
| 6 | 0.885 |
| 7 | 1.071 |
| 8 | 1.080 |
| 9 | 1.038 |
| 10 | 1.064 |
| Cp2WCl2 | 0.648 |
| IC50 Values [µM] | ||||
|---|---|---|---|---|
| Compound | A549 | SW480 | CH1/PA-1 | IMR-90 |
| [Cp2W(L1)]PF6 (1) | >200 | >200 | 85 ± 41 | n. d. |
| [Cp2W(L1)]Cl (1A) | >200 | >200 | 147 ± 18 | n. d. |
| [Cp2Mo(L1)]PF6 * | >200 | >200 | >200 | n. d. |
| [Cp2W(L2)]PF6 (2) | 36 ± 6 | 36 ± 7 | 24 ± 3 | 107 ± 30 |
| [Cp2Mo(L2)]PF6 * | 106 ± 15 | 55 ± 9 | 29 ± 10 | n. d. |
| [Cp2W(L3)]PF6 (3) | 19 ± 3 | 5.4 ± 1.1 | 2.7 ± 0.4 | 38.2 ± 0.4 |
| [Cp2Mo(L3)]PF6 * | 24 ± 4 | 5.6 ± 0.2 | 1.7 ± 0.4 | n. d. |
| [Cp2W(L4)]PF6 (4) | 6.3 ± 0.5 | 2.4 ± 0.5 | 1.6 ± 0.5 | 11 ± 2 |
| [Cp2Mo(L4)]PF6 * | 5.7 ± 1.0 | 1.6 ± 0.2 | 0.72 ± 0.13 | n. d. |
| [Cp2W(L5)]PF6 (5) | >200 | 147 ± 21 | >200 | n. d. |
| [Cp2Mo(L5)]PF6 * | >200 | >200 | >200 | n. d. |
| 6 | 141 ± 36 | 154 ± 29 | >200 | n. d. |
| 7 | >200 | >200 | 152 ± 16 | n. d. |
| 8 | >200 | 129 ± 14 | 93 ± 15 | n. d. |
| 9 | >200 | 101 ± 15 | >200 | n. d. |
| 10 | 53 ± 8 | 13 ± 1 | 21 ± 1 | 114 ± 33 |
| Cp2WCl2 | >200 | 38 ± 21 | 2.4 ± 0.5 | n. d. |
| Cp2MoCl2 | >200 * | >200 * | >200 * | n. d. |
| Cisplatin | 3.8 ± 1.0 ** | 2.3 ± 0.2 ** | 0.073 ± 0.001 ** | 14 ± 1 |
| IC50 Values [µM] | ||||||
|---|---|---|---|---|---|---|
| Compound | HCT116 | HT29 | MCF-7 | |||
| 2D | 3D | 2D | 3D | 2D | 3D | |
| 1A | >200 | >200 | >200 | >200 | >200 | >200 |
| 2 | 98 ± 12 | 158 ± 28 | 53 ± 9 | >200 | 66 ± 12 | >200 |
| 3 | 9.2 ± 1.4 | 21 ± 4 | 17 ± 4 | 86 ± 2 | 6.5 ± 1.8 | 44 ± 1 |
| 4 | 6.0 ± 0.9 | 15 ± 4 | 5.5 ± 1.0 | 31 ± 6 | 2.7 ± 0.7 | 19 ± 2 |
| 5 | >200 | >200 | >200 | >200 | >200 | >200 |
| 6 | >200 | >200 | >200 | >200 | 172 ± 45 | >200 |
| 7 | >200 | >200 | >200 | >200 | >200 | >200 |
| 8 | >200 | 178 ± 14 | >200 | >200 | 163 ± 32 | >200 |
| 9 | >200 | >200 | >200 | >200 | 147 ± 15 | >200 |
| 10 | 42 ± 3 | 69 ± 12 | 54 ± 2 | >200 | 29 ± 4 | >200 |
| Cp2WCl2 | >100 | >100 | >100 | >100 | >100 | >100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cseh, K.; Berasaluce, I.; Fuchs, V.; Banc, A.; Schweikert, A.; Prado-Roller, A.; Hejl, M.; Wernitznig, D.; Koellensperger, G.; Jakupec, M.A.; et al. Anticancer Tungstenocenes with a Diverse Set of (O,O–), (O,S–) and (O,N–) Chelates—A Detailed Biological Study Using an Improved Evaluation via 3D Spheroid Models. Pharmaceutics 2023, 15, 1875. https://doi.org/10.3390/pharmaceutics15071875
Cseh K, Berasaluce I, Fuchs V, Banc A, Schweikert A, Prado-Roller A, Hejl M, Wernitznig D, Koellensperger G, Jakupec MA, et al. Anticancer Tungstenocenes with a Diverse Set of (O,O–), (O,S–) and (O,N–) Chelates—A Detailed Biological Study Using an Improved Evaluation via 3D Spheroid Models. Pharmaceutics. 2023; 15(7):1875. https://doi.org/10.3390/pharmaceutics15071875
Chicago/Turabian StyleCseh, Klaudia, Iker Berasaluce, Valentin Fuchs, Alexandra Banc, Andreas Schweikert, Alexander Prado-Roller, Michaela Hejl, Debora Wernitznig, Gunda Koellensperger, Michael A. Jakupec, and et al. 2023. "Anticancer Tungstenocenes with a Diverse Set of (O,O–), (O,S–) and (O,N–) Chelates—A Detailed Biological Study Using an Improved Evaluation via 3D Spheroid Models" Pharmaceutics 15, no. 7: 1875. https://doi.org/10.3390/pharmaceutics15071875