
Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue

 , , , and
, , , and
Abstract
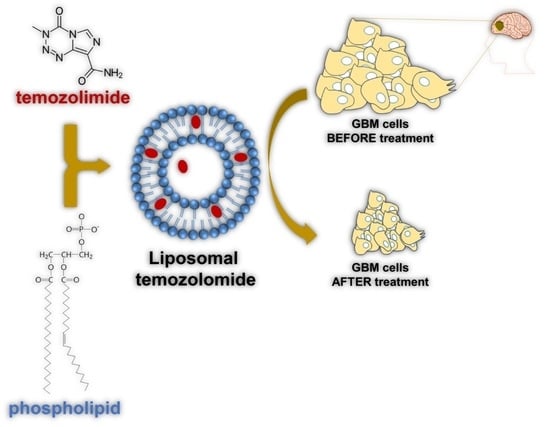
:
1. Introduction
2. Brief History and Overview
2.1. Classical Liposomes
2.2. Engineered Liposomes
2.2.1. Stealth Liposomes
2.2.2. Targeting Liposomes
3. Preparation Techniques
3.1. Overview
3.2. Conventional Methods
3.2.1. Thin-Film Hydration (THF)
3.2.2. Emulsification
3.2.3. Ethanol Injection
3.2.4. Detergent Removal
3.3. Large-Scale Liposome Production Techniques
4. Physico-Chemical Properties
5. Translational Oncology
6. Clinical Trials
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, A.R.; Liu, M.; Khan, M.W.; Zhai, G. Progress in brain targeting drug delivery system by nasal route. J Control. Release 2017, 268, 364–389. [Google Scholar] [CrossRef] [PubMed]
- Vanza, J.; Jani, P.; Pandya, N.; Tandel, H. Formulation and statistical optimization of intravenous temozolomide- loaded PEGylated liposomes to treat glioblastoma multiforme by three-level factorial design. Drug Dev. Ind. Pharm. 2018, 44, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Préat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015, 2, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alphandéry, E. Glioblastoma treatments: An account of recent industrial developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, M.F.G.; Hickman, J.A.; Langdon, S.P.; Chubb, D.; Vickers, L.; Stone, R.; Baig, G.; Goddard, C.; Gibson, N.W.; Slack, J.A.; et al. Antitumor Activity and Pharmacokinetics in Mice of 8-Carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a Novel Drug with Potential as an Alternative to Dacarbazine. Cancer Res. 1987, 47, 5846–5852. [Google Scholar] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanism of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Z.; Liu, H.; Wang, L.; Huang, G. Liposome encapsulated of temozolomide for the treatment of glioma tumor: Preparation, characterization and evaluation. Drug Discov Ther. 2015, 9, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Afzalipour, R.; Khoei, S.; Khoee, S.; Shirvalilou, S.; Raoufi, N.J.; Motevalian, M.; Karimi, M.R. Dual-Targeting Temozolomide Loaded in Folate-Conjugated Magnetic Triblock Copolymer Nanoparticles to Improve the Therapeutic Efficiency of Rat Brain Gliomas. ACS Biomater. Sci. Eng. 2019, 5, 6000–6011. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Temodal, Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/temodal-epar-product-information_en.pdf (accessed on 15 October 2021).
- Ramalho, M.J.; Coelho, M.A.N.; Pereira, M.C. Nanocarriers for the delivery of temozolomide in the treatment of glioblatoma: A review. In Design and Development of New Nanocarriers; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; Chapter 18; pp. 687–722. ISBN 9780128136270. [Google Scholar]
- Glaser, T.; Han, I.; Wu, L.; Zeng, X. Targeted Nanotechnology in Glioblastoma Multiforme. Front. Pharmacol. 2017, 8, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tan, T.; Zhao, L.; Liu, M.; You, Y.; Zeng, Y.; Chen, D.; Xie, T.; Zhang, L.; Fu, C.; et al. Recent Advancements in Liposome-Targeting Strategies for the Treatment of Gliomas: A Systematic Review. ACS Appl. Bio Mater. 2020, 3, 5500–5528. [Google Scholar] [CrossRef] [PubMed]
- Malam, Y.; Loizidou, M.; Seifalian, A.M. Liposomes and nanoparticles: Nanosized vehicles for drug delivery in cancer liposomes and nanoparticles. Trends Pharmacol. 2009, 30, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.B.; Amiji, M.M. A Review of Nanocarrier-Based CNS Delivery Systems. Curr. Drug Deliv. 2006, 3, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangham, A.D.; Horne, R.W. Negative Staining of Phospholipids and their Structural Modification by Surface-active Agents as observed in the Electron Microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Ohama, Y.; Heike, Y.; Sugahara, T.; Sakata, K.; Yoshimura, N.; Hisaeda, Y.; Hosokawa, M.; Takashima, S.; Kato, K. Gene Transfection into HeLa Cells by Vesicle Containing Cationic Peptide Lipid. Biosci. Biotechnol. Biochem. 2005, 69, 1453–1458. [Google Scholar] [CrossRef] [Green Version]
- Omokawaa, Y.; Miyazakib, T.; Waldec, P.; Akiyamad, K.; Sugaharae, T.; Masudaf, S.; Inadag, A.; Ohnishih, Y.; Saekii, T.; Kato, K. In vitro and in vivo anti-tumor effects of novel Span 80 vesicles containing immobilized Eucheuma serra agglutinin. Int. J. Pharm. 2010, 389, 157–167. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Harries, D.; May, S.; Gelbart, W.M.; Ben-Shaul, A. Structure, stability, and thermodynamics of lamellar DNA-lipid complexes. Biophys. J. 1998, 75, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Singh-Moon, R.; Wang, M.; Chaudhuri, D.B.; Ellis, J.A.; Bruce, J.N.; Bigio, I.J.; Straubinger, R.M. Cationic surface charge enhances early regional deposition of liposomes after intracarotid injection. J. Neurooncol. 2014, 120, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood–brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulimondi, F.; Digiacomo, L.; Pozzi, D.; Palchetti, S.; Vulpis, E.; Capriotti, A.L.; Chiozzi, R.Z.; Laganà, A.; Amenitsch, H.; Masuelli, L.; et al. Interplay of protein corona and immune cells controls blood residency of liposomes. Nat. Commun. 2019, 10, 3686. [Google Scholar] [CrossRef] [Green Version]
- Kong, B.; Sun, Y.; Li, Y.; Hu, D. Preparation and the Influencing Factors of Timozolomide Liposomes. Artif. Cells Nanomed. Biotechnol. 2009, 37, 279–282. [Google Scholar] [CrossRef]
- Wauthoz, N.; Deleuze, P.; Hecq, J.; Roland, I.; Saussez, S.; Adanja, I.; Debeir, O.; Decaestecker, C.; Mathieu, V.; Kiss, R.; et al. In vivo assessment of temozolomide local delivery for lung cancer inhalation therapy. Eur. J. Pharm. Sci. 2010, 39, 402–411. [Google Scholar] [CrossRef] [Green Version]
- Hamzawy, M.A.; Abo-youssef, A.M.; Salem, H.F.; Mohammed, S.A. Antitumor activity of intratracheal inhalation of temozolomide (TMZ) loaded into gold nanoparticles and/or liposomes against urethane-induced lung cancer in BALB/c mice. Drug Deliv. 2017, 24, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Temozolomide, No 14163, Product Information, Cayman Chemical. 2017. Available online: https://www.caymanchem.com/product/14163/temozolomide (accessed on 30 October 2021).
- Bregoli, L.; Movia, D.; Gavigan-Imedio, J.D.; Lysaght, J.; Reynolds, J.; Prina-Mello, A. Nanomedicine applied to translational oncology: A future perspective on cancer treatment. Nanomedicine 2016, 12, 81–103. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.A.; de Rosales, R.T.M.; La-Beck, N.M. Translational considerations in nanomedicine: The oncology perspective. Adv. Drug Deliv. Rev. 2020, 158, 140–157. [Google Scholar] [CrossRef]
- Wu, W.; Lu, Y.; Qi, J. Oral delivery of liposomes. Ther. Deliv. 2015, 6, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Mahringer, A.; Ott, M.; Fricker, G. The Blood-Brain Barrier: An Introduction to its Structure and Function. In The Blood-Brain Barrier; Fricker, G., Ott, M., Mahringer, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–20. ISBN 978-3-662-43786-5. [Google Scholar]
- Nieto Montesinos, R. Liposomal Drug Delivery to the Central Nervous System. In Liposomes; Catala, A., Ed.; InTech-Open Science Open Minds: London, UK, 2017; pp. 213–241. ISBN 978-953-51-3580-7. [Google Scholar]
- Haluska, C.K.; Riske, K.A.; Marchi-Artzner, V.; Lehn, J.M.; Lipowsky, R.; Dimova, R. Time scales of membrane fusion revealed by direct imaging of vesicle fusion with high temporal resolution. Proc. Natl. Acad. Sci. USA 2006, 103, 15841–15846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrink, S.; Mark, A.E. The Mechanism of Vesicle Fusion as Revealed by Molecular Dynamics Simulations. J. Am. Chem. Soc. 2003, 125, 11144–11145. [Google Scholar] [CrossRef] [Green Version]
- Silvander, M. Progress in Colloid and Polymer Science; Springer: Berlin/Heidelberg, Germany, 2008; pp. 35–40. ISBN 978-3-540-45291-1. [Google Scholar]
- Storm, G.; Belliot, S.O.; Daemen, T.; Lasic, D.D. Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system. Adv. Drug Deliv. 1995, 17, 31–48. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Storm, G.; Crommelin, D.J.A. Liposomes: Quo vadis? Pharm. Sci. Technol. Today 1998, 1, 19–31. [Google Scholar] [CrossRef]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [Green Version]
- Chow, T.H.; Lin, Y.Y.; Hwang, J.J.; Wang, H.E.; Tseng, Y.L.; Wang, S.J.; Liu, R.S.; Lin, W.J.; Yang, C.S.; Ting, G. Improvement of biodistribution and therapeutic index via increase of polyethylene glycol on drug-carrying liposomes in an HT-29/luc xenografted mouse model. Anticancer. Res. 2009, 29, 2111–2120. [Google Scholar]
- Subhan, M.A.; Yalamarty, S.S.K.; Filipczak, N.; Parveen, F.; Torchilin, V.P. Recent Advances in Tumor Targeting via EPR Effect for Cancer Treatment. J. Pers. Med. 2021, 11, 571. [Google Scholar] [CrossRef]
- Hu, J.; Wang, J.; Wang, G.; Yao, Z.; Dang, X. Pharmacokinetics and antitumor efficacy of DSPE-PEG2000 polymeric liposomes loaded with quercetin and temozolomide: Analysis of their effectiveness in enhancing the chemosensitization of drug-resistant glioma cells. Int. J. Mol. Med. 2016, 37, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Ye, X.; Wu, Y.; Wang, H.; Sheng, C.; Peng, D.; Chen, W. Time Interval of Two Injections and First-Dose Dependent of Accelerated Blood Clearance Phenomenon Induced by PEGylated Liposomal Gambogenic Acid: The Contribution of PEG-Specific IgM. J. Pharm. Sci. 2019, 108, 641–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSweeney, M.D.; Price, L.S.L.; Wessler, T.; Ciociola, E.C.; Herity, L.B.; Piscitelli, J.A.; DeWalle, A.C.; Harris, T.N.; Chan, A.K.P.; Saw, R.S.; et al. Overcoming anti-PEG antibody mediated accelerated blood clearance of PEGylated liposomes by pre-infusion with high molecular weight free PEG. J. Control. Release 2019, 311–312, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Tapeinos, C.; Battaglini, M.; Ciofani, G. Advances in the design of solid lipid nanoparticles and nanostructured lipid carriers for targeting brain diseases. J. Control. Release 2017, 264, 306–332. [Google Scholar] [CrossRef]
- Lin, C.Y.; Li, R.J.; Huang, C.Y.; Wei, K.C.; Chen, P.Y. Controlled release of liposome-encapsulated temozolomide for brain tumour treatment by convection-enhanced delivery. J. Drug Target. 2018, 26, 325–332. [Google Scholar] [CrossRef]
- Nordling-David, M.M.; Yaffe, R.; Guez, D.; Meirow, H.; Last, D.; Grad, E.; Salomon, S.; Sharabi, S.; Levi-Kalisman, Y.; Golomb, G.; et al. Liposomal temozolomide drug delivery using convection enhanced delivery. J. Control. Release 2017, 261, 138–146. [Google Scholar] [CrossRef]
- Brem, S.; Tyler, B.; Li, K.; Pradilla, G.; Legnani, F.; Caplan, J.; Brem, H. Local delivery of temozolomide by biodegradable polymers is superior to oral administration in a rodent glioma model. Cancer Chemother. Pharmacol. 2007, 60, 643–650. [Google Scholar] [CrossRef]
- Huwyler, J.; Wu, D.; Pardridge, W.M. Brain drug delivery of small molecules using immunoliposomes. Proc. Natl. Acad. Sci. USA 1996, 93, 14164–14169. [Google Scholar] [CrossRef] [Green Version]
- Arcella, A.; Palchetti, S.; Digiacomo, L.; Pozzi, D.; Capriotti, A.L.; Frati, L.; Oliva, M.A.; Tsaouli, G.; Rota, R.; Screpanti, I.; et al. Brain Targeting by Liposome–Biomolecular Corona Boosts Anticancer Efficacy of Temozolomide in Glioblastoma Cells. ACS Chem. Neurosci. 2018, 9, 3166–3174. [Google Scholar] [CrossRef]
- Kumar, S.; Dutta, J.; Dutta, P.K.; Koh, J. A systematic study on chitosan-liposome based systems for biomedical applications. Int. J. Biol. Macromol. 2020, 160, 470–481. [Google Scholar] [CrossRef]
- Liu, X.; Ma, L.; Mao, Z.; Gao, C. Chitosan-Based Biomaterials for Tissue Repair and Regeneration. In Chitosan for Biomaterials II; Jayakumar, R., Prabaharan, M., Muzzarelli, R., Eds.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2011; pp. 81–127. ISBN 978-3-642-24061-4. [Google Scholar]
- Ibrahim, H.M.; El-Zairy, E.M.R. Chitosan as a Biomaterial—Structure, Properties, and Electrospun Nanofibers. In Concepts, Compounds and the Alternatives of Antibacterials; Bobbarala, V., Ed.; IntechOpen: London, UK, 2015; pp. 82–83. ISBN 978-953-51-5417-4. [Google Scholar]
- Zhang, H.; Li, Y.; Zhang, X.; Liu, B.; Zhao, H.; Chen, D. Directly determining the molecular weight of chitosan with atomic force microscopy. Front. Nanosci. Nanotechnol. 2016, 2, 123–127. [Google Scholar] [CrossRef]
- Pavinatto, A.; Pavinatto, F.J.; Delezuk, J.A.d.M.; Nobre, T.M.; Souza, A.L.; Campana-Filho, S.P.; Oliveira, O.N. Low molecular-weight chitosans are stronger biomembrane model perturbants. Colloids Surf. 2013, 104, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Alomrani, A.; Badran, M.; Harisa, G.I.; ALshehry, M.; Alhariri, M.; Alshamsan, A.; Alkholief, M. The use of chitosan-coated flexible liposomes as a remarkable carrier to enhance the antitumor efficacy of 5-fluorouracil against colorectal cancer. Saudi Pharm. J. 2019, 27, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Grozdova, I.; Melik-Nubarov, N.; Efimova, A.; Ezhov, A.; Krivtsov, G.; Litmanovich, E.; Yaroslavov, A. Intracellular delivery of drugs by chitosan-based multi-liposomal complexes. Colloids Surf. B. 2020, 193, 111062. [Google Scholar] [CrossRef]
- Yaroslavov, A.A.; Efimova, A.A.; Krasnikov, E.A.; Trosheva, K.S.; Popov, A.S.; Melik-Nubarov, N.S.; Krivtsov, G.G. Chitosan-based multi-liposomal complexes: Synthesis, biodegradability, and cytotoxicity. Int. J. Biol. Macromol. 2021, 177, 455–462. [Google Scholar] [CrossRef]
- Gao, W.; Hu, C.-M.J.; Fang, R.H.; Zhang, L. Liposome-like nanostructures for drug delivery. J. Mater. Chem. B. 2013, 1, 6569–6585. [Google Scholar] [CrossRef]
- Wu, Z.H.; Ping, Q.N.; Wei, Y.; Lai, J.M. Hypoglycemic efficacy of chitosan-coated insulin liposomes after oral administration in mice. Acta Pharmacol. 2004, 25, 966–972. [Google Scholar] [PubMed]
- Salade, L.; Wauthoz, N.; Vermeersch, M.; Amighi, K.; Goole, J. Chitosan-coated liposome dry-powder formulations loaded with ghrelin for nose-to-brain delivery. Eur. J. Pharm. Biopharm. 2018, 129, 257–266. [Google Scholar] [CrossRef]
- Lee, E.-H.; Lim, S.-J.; Lee, M.-K. Chitosan-coated liposomes to stabilize and enhance transdermal delivery of indocyanine green for photodynamic therapy of melanoma. Carbohydr. Polym. 2019, 224, 115143. [Google Scholar] [CrossRef]
- Şalva, E.; Turan, S.Ö.; Eren, F.; Akbuğa, J. The enhancement of gene silencing efficiency with chitosan-coated liposome formulations of siRNAs targeting HIF-1α and VEGF. Int. J. Pharm. 2015, 478, 147–154. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, T.; Liu, Y.; Wang, Q.; Xing, S.; Li, L.; Wang, L.; Liu, L.; Gao, D. Ursolic acid liposomes with chitosan modification: Promising antitumor drug delivery and efficacy. Mater. Sci. Eng. C 2017, 71, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Masarone, M.; Armenia, E.; Giudice, A.; Barbarisi, M.; Caraglia, M.; Barbarisi, A.; Persico, M. Chitosan-coated liposomes loaded with butyric acid demonstrate anticancer and anti-inflammatory activity in human hepatoma HepG2 cells. Oncol. Rep. 2019, 41, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, S.S.; Alshehri, S.; Altamimi, M.A.; Hussain, A.; Qamar, W.; Gilani, S.J.; Zafar, A.; Alruwaili, N.K.; Alanazi, S.; Almutairy, B.K. Formulation of Piperine–Chitosan-Coated Liposomes: Characterization and In Vitro Cytotoxic Evaluation. Molecules 2021, 26, 3281. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.; De Giglio, E.; Cafagna, D.; Denora, N.; Agrimi, G.; Cassano, T.; Gaetani, S.; Cuomo, V.; Trapani, G. Characterization and evaluation of chitosan nanoparticles for dopamine brain delivery. Int. J. Pharm. 2011, 419, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Blanco, J.; Martín-Sabroso, C.; Torres-Suárez, A.-I. In vitro screening of nanomedicines through the blood brain barrier: A critical review. Biomaterials 2016, 103, 229–255. [Google Scholar] [CrossRef]
- Sahin, A.; Yoyen-Ermis, D.; Caban-Toktas, S.; Horzum, U.; Aktas, Y.; Couvreur, P.; Esendagli, G.; Capan, Y. Evaluation of brain-targeted chitosan nanoparticles through blood-brain barrier cerebral microvessel endothelial cells. J. Microencapsul. 2017, 34, 659–666. [Google Scholar] [CrossRef]
- Harris, R. Chitosan-based drug delivery systems for brain cancer therapy. URMSE 2021, 2, 31–35. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-)clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Fang, Z.; Shen, Y.; Gao, D. Stimulus-responsive nanocarriers for targeted drug delivery. New J. Chem. 2021, 45, 4534–4544. [Google Scholar] [CrossRef]
- Kim, S.S.; Rait, A.; Kim, E.; DeMarco, J.; Pirollo, K.F.; Chang, E.H. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 2015, 369, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Schvartz, I.; Seger, D.; Shaltiel, S. Vitronectin. Int. J. Biochem. Cell Biol. 1999, 31, 539–544. [Google Scholar] [CrossRef]
- Kim, J.S.; Shin, D.H.; Kim, J.-S. Dual-targeting immunoliposomes using angiopep-2 and CD133 antibody for glioblastoma stem cells. J. Control. Release 2018, 269, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Han, T.L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Weizman, A.; Zeineh, N.; Kahana, M.; Obeid, F.; Allon, N.; Gavish, M. Liposomal Carrier Conjugated to APP-Derived Peptide for Brain Cancer Treatment. Cell Mol. Neurobiol. 2021, 41, 1019–1029. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, H.; Xue, X. Blood–brain barrier penetrating liposomes with synergistic chemotherapy for glioblastoma treatment. Biomat. Sci. 2022. [Google Scholar] [CrossRef]
- Xin, H.; Jiang, Y.; Lv, W.; Xu, J. Liposome-Based drug Delivery for Brain Tumor Theranostics. In Nanotechnology-Based Targeted Drug Delivery Systems for Brain Tumors; Academic Press: Cambridge, MA, USA, 2018; pp. 245–266. ISBN 9780128122495. [Google Scholar]
- Gallego, L.; Cena, V. Potential advancements in the treatment of difficult-to-treat glioblastoma through nanoparticle drug delivery. Expert Opin. Drug Deliv. 2020, 17, 1541–1554. [Google Scholar] [CrossRef]
- Mojarad-Jabali, S.; Farshbaf, M.; Walker, P.R.; Hemmati, S.; Fatahi, Y.; Zakeri-Milani, P.; Sarfraz, M.; Valizadeh, H. An update on actively targeted liposomes in advanced drug delivery to glioma. Int. J. Pharm. 2021, 602, 120645. [Google Scholar] [CrossRef]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I.; Velázquez-Fernández, J.B.; Vallejo-Cardona, A.A. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Raza Shah, M.R.; Imran, M.; Ullah, S. Lipid-Based Nanocarriers for Drug Delivery and Diagnosis; Elsevier: Amsterdam, The Netherlands, 2017; pp. 68–69. ISBN 978-0-323-52729-3. [Google Scholar]
- Small, D.M. The Physical Chemistry of Lipids, From Alkanes to Phospholipids. In Handbook of Lipid Research; Plenum Press: New York, NY, USA, 1986; Volume 4, p. 672. ISBN 9780306417634. [Google Scholar]
- Crommelin, D.J.A.; Zuidam, N.J. Hydrolysis of Phospholipids in Liposomes and Stability-Indicating Analytical Techniques. In Liposome Technology, 3rd ed.; Gregoriadis, G., Ed.; Volume 1. Liposome Preparation and Related Techniques; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2007; p. 285. ISBN 978-0-8493-9726-4. [Google Scholar]
- Wagner, A.; Vorauer-Uhl, K. Liposome technology for industrial purposes. J. Drug Deliv. 2011, 2011, 591325. [Google Scholar] [CrossRef] [Green Version]
- Maherani, B.; Arab-Tehrany, E.; Mozafari, M.R.; Gaiani, C.; Linder, M. Liposomes: A Review of Manufacturing Techniques and Targeting Strategies. Curr. Nanosci. 2011, 7, 436–452. [Google Scholar] [CrossRef]
- Ahmed, K.S.; Hussein, S.A.; Ali, A.H.; Korma, S.A.; Lipeng, Q.; Jinghua, C. Liposome: Composition, characterisation, preparation, and recent innovation in clinical applications. J. Drug Target. 2019, 27, 742–761. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef] [PubMed]
- Karn, P.R.; Cho, W.; Hwang, S.J. Liposomal drug products and recent advances in the synthesis of supercritical fluid-mediated liposomes. Nanomedicine 2013, 8, 1529–1548. [Google Scholar] [CrossRef]
- Maja, L.; Željko, K.; Mateja, P. Sustainable technologies for liposome preparation. J. Supercrit. Fluids 2020, 165, 104984. [Google Scholar] [CrossRef]
- Crommelin, D.; Metselaar, J.; Storm, G. Liposomes: The Science and the Regulatory Landscape. In Non-Biological Complex Drugs; Crommelin, D., de Vlieger, J., Eds.; AAPS Advances in the Pharmaceutical Sciences Series; Springer: Cham, Switzerland, 2015; Volume 20, pp. 77–106. ISBN 978-3-319-16241-6. [Google Scholar]
- Costa, A.P.; Xu, X.; Khan, M.A.; Burgess, D.J. Liposome Formation Using a Coaxial Turbulent Jet in Co-Flow. Pharm. Res. 2016, 33, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Laouini, A.; Jaafar-Maalej, C.; Limayem-Blouza, I.; Sfar, S.; Charcosset, C.; Fessi, H. Preparation, Characterization and Applications of Liposomes: State of the Art. J. Coll. Sci. Biotechnol. 2012, 1, 147–168. [Google Scholar] [CrossRef]
- Castañeda-Reyes, E.D.; Perea-Flores, M.J.; Davila-Ortiz, G.; Lee, Y.; Gonzalez de Mejia, E. Development, Characterization and Use of Liposomes as Amphipathic Transporters of Bioactive Compounds for Melanoma Treatment and Reduction of Skin Inflammation: A Review. Int. J. Nanomed. 2020, 15, 7627–7650. [Google Scholar] [CrossRef]
- Jash, A.; Ubeyitogullari, A.; Rizvi, S.S.H. Liposomes for oral delivery of protein and peptide-based therapeutics: Challenges, formulation strategies, and advances. J. Mater. Chem. B 2021, 9, 4773–4792. [Google Scholar] [CrossRef]
- Has, C.; Sunthar, P. A comprehensive review on recent preparation techniques of liposomes. J. Liposome Res. 2020, 30, 336–365. [Google Scholar] [CrossRef]
- Wang, Y.; Grainger, D.W. Lyophilized liposome-based parenteral drug development: Reviewing complex product design strategies and current regulatory environments. Adv. Drug Deliv Rev. 2019, 151–152, 56–71. [Google Scholar] [CrossRef]
- Worsham, R.D.; Thomas, V.; Farid, S.S. Potential of Continuous Manufacturing for Liposomal Drug Products. J. Biotechnol. 2019, 14, 1700740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trucillo, P.; Campardelli, R.; Reverchon, E. Liposomes: From Bangham to Supercritical Fluids. Processes 2020, 8, 1022. [Google Scholar] [CrossRef]
- Bangham, A.D. Surrogate cells or Trojan horses. The discovery of liposomes. Bioessays 1995, 17, 1081–1088. [Google Scholar] [CrossRef]
- Shah, S.; Dhawan, V.; Holm, R.; Nagarsenker, M.S.; Perrie, Y. Liposomes: Advancements and innovation in the manufacturing process. Adv. Drug Deliv. Rev. 2020, 154–155, 102–122. [Google Scholar] [CrossRef]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. In Liposomes; D’Souza, G., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1522, pp. 17–22. ISBN 978-1-4939-6591-5. [Google Scholar]
- Xu, H.; He, L.; Nie, S.; Guan, J.; Zhang, X.; Yang, X.; Pan, W. Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J. Control. Release 2009, 140, 61–68. [Google Scholar] [CrossRef]
- Tian, X.H.; Lin, X.N.; Wei, F.; Feng, W.; Huang, Z.C.; Wang, P.; Ren, L.; Diao, Y. Enhanced brain targeting of temozolomide in polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Int. J. Nanomed. 2011, 6, 445–452. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, M.J.; Andrade, S.; Coelho, M.Á.N.; Loureiro, J.A.; Pereira, M.C. Biophysical interaction of temozolomide and its active metabolite with biomembrane models: The relevance of drug-membrane interaction for Glioblastoma Multiforme therapy. Eur. J. Pharm. Biopharm. 2019, 136, 156–163. [Google Scholar] [CrossRef]
- Waghule, T.; Rapalli, V.K.; Singhvi, G.; Gorantla, S.; Khosa, A.; Dubey, S.K.; Saha, R.N. Design of temozolomide-loaded proliposomes and lipid crystal nanoparticles with industrial feasible approaches: Comparative assessment of drug loading, entrapment efficiency, and stability at plasma pH. J. Liposome Res. 2021, 31, 158–168. [Google Scholar] [CrossRef]
- Drolez, A.; Vandenhaute, E.; Julien, S.; Gosselet, F.; Burchell, J.; Cecchelli, R.; Delannoy, P.; Dehouck, M.-P.; Mysiorek, C. Selection of a Relevant In Vitro Blood-Brain Barrier Model to Investigate Pro-Metastatic Features of Human Breast Cancer Cell Lines. PLoS ONE 2016, 11, e0151155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caddeo, C.; Pons, R.; Carbone, C.; Fernàndez-Busquets, X.; Cardia, M.C.; Maccioni, A.M.; Fadda, A.M.; Manconi, M. Physico-chemical characterization of succinyl chitosan-stabilized liposomes for the oral co-delivery of quercetin and resveratrol. Carbohydr. Polym. 2017, 157, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Currie, J.-C.; Bertrand, Y.; Ché, C.; Nguyen, T.; Régina, A.; Gabathuler, R.; Castaigne, J.-P.; Béliveau, R. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector Angiopep-2. J. Neurochem. 2008, 106, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.Q.; Qi, X.R. Preparation of Drug Liposomes by Reverse-Phase Evaporation. In Liposome-Based Drug Delivery Systems; Lu, W.L., Qi, X.R., Eds.; Biomaterial Engineering; Springer: Berlin/Heidelberg, Germany, 2018; pp. 37–46. ISBN 978-3-662-49231-4. [Google Scholar]
- Szoka, F., Jr.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef] [Green Version]
- Meure, L.A.; Foster, N.R.; Dehghani, F. Conventional and Dense Gas Techniques for the Production of Liposomes: A Review. AAPS PharmSciTech 2008, 9, 798. [Google Scholar] [CrossRef] [Green Version]
- Laloy, E.; Vuillemard, J.C.; Ackermann, H.W.; Robin, O. Preparation of liposomes by a simple emulsification technique. Biotechnol. Tech. 1994, 8, 717–722. [Google Scholar] [CrossRef]
- Shum, H.C.; Lee, D.; Yoon, I.; Kodger, T.; Weitz, D.A. Double emulsion templated monodisperse phospholipid vesicles. Langmuir 2008, 24, 7651–7653. [Google Scholar] [CrossRef]
- Batzri, S.; Korn, E.D. Single bilayer liposomes prepared without sonication. Biochim. Biophys. Acta Biomembr. 1973, 298, 1015–1019. [Google Scholar] [CrossRef]
- Jaafar-Maalej, C.; Diab, R.; Andrieu, V.; Elaissari, A.; Fessi, H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J. Liposome Res. 2010, 20, 228–243. [Google Scholar] [CrossRef]
- Wagner, A.; Platzgummer, M.; Kreismayr, G.; Quendler, H.; Stiegler, G.; Ferko, B.; Vecera, G.; Vorauer-Uhl, K.; Katinger, H. GMP Production of Liposomes—A New Industrial Approach. J. Liposome Res. 2006, 16, 311–319. [Google Scholar] [CrossRef]
- Justo, O.R.; Moraes, A.M. Economical Feasibility Evaluation of an Ethanol Injection Liposome Production Plant. Chem. Eng. Technol. 2010, 33, 15–20. [Google Scholar] [CrossRef]
- Chang, E.H.; Sangsoo, K.; Antonina, R. Targeted Liposomes. U.S. Patent 2021205456A1, 8 July 2021. [Google Scholar]
- Xu, L.; Huang, C.C.; Huang, W.; Tang, W.H.; Rait, A.; Yin, Y.Z.; Cruz, I.; Xiang, L.M.; Pirollo, K.F.; Chang, E.H. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol. Cancer Ther. 2002, 1, 337–346. [Google Scholar] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, R. Liposome preparation by detergent removal. Methods Enzymol. 2003, 367, 46–70. [Google Scholar] [CrossRef]
- Available online: https://www.transparencymarketresearch.com/liposome-drug-delivery-market.html (accessed on 30 October 2021).
- Walde, P.; Ichikawa, S. Enzymes inside lipid vesicles: Preparation, reactivity, and applications. Biomol. Eng. 2001, 18, 143–177. [Google Scholar] [CrossRef]
- Perrie, Y.; Kastner, E.; Khadke, S.; Roces, C.; Stone, P. Manufacturing Methods for Liposome Adjuvants. In Vaccine Adjuvants. Methods in Molecular Biology; Fox, C., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1494, pp. 127–144. ISBN 978-1-4939-6445-1. [Google Scholar]
- Philippot, J.R.; Schuber, F. Liposomes as Tools in Basic Research and Industry, 1st ed.; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 1995; p. 286. ISBN 9780203713259. [Google Scholar]
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER). Liposome Drug Products-Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. In Guidance for Industry; FDA: Silver Spring, MD, USA, 2018. [Google Scholar]
- Mozafari, M.; Reed, C.; Rostron, C. Development of Non-Toxic Liposomal Formulations for Gene and Drug Delivery to the Lung; Technology and Health Care; IOS Press: Amsterdam, The Netherlands, 2002; Volume 10, pp. 342–344. [Google Scholar]
- Mortazavi, S.M.; Mohammadabadi, M.R.; Khosravi-Darani, K.; Mozafari, M.R. Preparation of liposomal gene therapy vectors by a scalable method without using volatile solvents or detergents. J. Biotechnol. 2007, 129, 604–613. [Google Scholar] [CrossRef]
- Nkanga, C.I.; Krause, R.W.M. Encapsulation of Isoniazid-conjugated Phthalocyanine-In-Cyclodextrin-In-Liposomes Using Heating Method. Sci. Rep. 2019, 9, 11485. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K.; Katinger, H. Liposomes produced in a pilot scale: Production, purification, and efficiency aspects. Eur. J. Pharm. Biopharm. 2002, 54, 213–219. [Google Scholar] [CrossRef]
- Wagner, A.; Vorauer-Uhl, K.; Kreismayr, G.; Katinger, H. The crossflow injection technique: An improvement of the ethanol injection method. J. Liposome Res. 2002, 12, 259–270. [Google Scholar] [CrossRef]
- Katinger, H.; Vorauer-Uhl, K.; Wagner, A.; Kreismayr, G. Method and Device for Producing Lipid Vesicles. U.S. Patent 6843942B2, 16 October 2000. [Google Scholar]
- Vorauer-Uhl, K.; Wagner, A.; Katinger, H. Long term stability of rh-Cu/Zn-superoxide dismutase (SOD)-liposomes prepared by the cross-flow injection technique following International Conference on Harmonisation (ICH)-guidelines. Eur. J. Pharm. Biopharm. 2002, 54, 83–87. [Google Scholar] [CrossRef]
- Suleiman, E.; Mayer, J.; Lehner, E.; Kohlhauser, B.; Katholnig, A.; Batzoni, M.; Damm, D.; Temchura, V.; Wagner, A.; Überla, K.; et al. Conjugation of Native-Like HIV-1 Envelope Trimers onto Liposomes Using EDC/Sulfo-NHS Chemistry: Requirements and Limitations. Pharmaceutics 2020, 12, 979. [Google Scholar] [CrossRef] [PubMed]
- Carugo, D.; Bottaro, E.; Owen, J.; Stride, E.; Nastruzzi, C. Liposome production by microfluidics: Potential and limiting factors. Sci. Rep. 2016, 6, 25876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, S.; Caspi, Y.; Meijering, A.E.; Dekker, C. Octanol-assisted liposome assembly on chip. Nat. Commun. 2016, 7, 10447. [Google Scholar] [CrossRef]
- Ganesan, P.; Karthivashan, G.; Park, S.; Kim, J.; Choi, D.K. Microfluidization trends in the development of nanodelivery systems and applications in chronic disease treatments. Int. J. Nanomed. 2018, 13, 6109–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suleiman, E.; Damm, D.; Batzoni, M.; Temchura, V.; Wagner, A.; Überla, K.; Vorauer-Uhl, K. Electrostatically Driven Encapsulation of Hydrophilic, Non-Conformational Peptide Epitopes into Liposomes. Pharmaceutics 2019, 11, 619. [Google Scholar] [CrossRef] [Green Version]
- Kankala, R.K.; Zhang, Y.S.; Wang, S.B.; Lee, C.H.; Chen, A.Z. Supercritical Fluid Technology: An Emphasis on Drug Delivery and Related Biomedical Applications. Adv. Healthc. Mater. 2017, 6, 1700433. [Google Scholar] [CrossRef] [Green Version]
- Bigazzi, W.; Penoy, N.; Evrard, B.; Piel, G. Supercritical fluid methods: An alternative to conventional methods to prepare liposomes. Chem. Eng. J. 2020, 383, 123106. [Google Scholar] [CrossRef]
- Otake, K.; Imura, T.; Sakai, H.; Abe, M. Development of a New Preparation Method of Liposomes Using Supercritical Carbon Dioxide. Langmuir 2001, 17, 3898–3901. [Google Scholar] [CrossRef]
- Aburai, K.; Yagi, N.; Yokoyama, Y.; Okuno, H.; Sakai, K.; Sakai, H.; Sakamoto, K.; Abe, M. Preparation of liposomes modified with lipopeptides using a supercritical carbon dioxide reverse-phase evaporation method. J. Oleo Sci. 2011, 60, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ou, C.; Ye, S.; Song, X.; Luo, S. Construction of nanoscale liposomes loaded with melatonin via supercritical fluid technology. J. Microencapsul. 2017, 34, 687–698. [Google Scholar] [CrossRef]
- Tomsen-Melero, J.; Passemard, S.; García-Aranda, N.; Díaz-Riascos, Z.V.; González-Rioja, R.; Nedergaard Pedersen, J.; Lyngsø, J.; Merlo-Mas, J.; Cristóbal-Lecina, E.; Corchero, J.L.; et al. Impact of Chemical Composition on the Nanostructure and Biological Activity of α-Galactosidase-Loaded Nanovesicles for Fabry Disease Treatment. ACS Appl. Mater. Interfaces 2021, 13, 7825–7838. [Google Scholar] [CrossRef] [PubMed]
- Zabihi, S.; Khoshmaram, A.; Pishnamazi, M.; Borousan, F.; Hezave, A.Z.; Marjani, A.; Pelalak, R.; Kurniawan, T.A.; Shirazian, S. Thermodynamic study on solubility of brain tumor drug in supercritical solvent: Temozolomide case study. J. Mol. Liq. 2021, 321, 114926. [Google Scholar] [CrossRef]
- Li, C.; Deng, Y. A novel method for the preparation of liposomes: Freeze drying of monophase solutions. J. Pharm. Sci. 2004, 93, 1403–1414. [Google Scholar] [CrossRef]
- Sylvester, B.; Porfire, A.; Van Bockstal, P.J.; Porav, S.; Achim, M.; Beer, T.; Tomuţă, I. Formulation Optimization of Freeze-Dried Long-Circulating Liposomes and In-Line Monitoring of the Freeze-Drying Process Using an NIR Spectroscopy Tool. J. Pharm. Sci. 2018, 107, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Deng, Y.; Geng, Y.; Gao, Z.; Zou, J.; Wang, Z. Preparation of submicron unilamellar liposomes by freeze-drying double emulsions. Biochim. Biophys. Acta Biomembr. 2006, 1758, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, N.; Wang, T.; Sun, W.; Li, T. Preparation of submicron liposomes exhibiting efficient entrapment of drugs by freeze-drying water-in-oil emulsions. Chem. Phys. Lipids 2011, 164, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kukuchi, H.; Yamauchi, H.; Hirota, S. A Spray-Drying Method for Mass Production of Liposomes. Chem. Pharm. Bull. 1991, 39, 1522–1527. [Google Scholar] [CrossRef] [Green Version]
- Charnvanich, D.; Vardhanabhuti, N.; Kulvanich, P. Effect of Cholesterol on the Properties of Spray-Dried Lysozyme-Loaded Liposomal Powders. AAPS PharmSciTech 2010, 11, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Omer, H.K.; Hussein, N.R.; Ferraz, A.; Najlah, M.; Ahmed, W.; Taylor, K.M.G.; Elhissi, A.M.A. Spray-Dried Proliposome Microparticles for High-Performance Aerosol Delivery Using a Monodose Powder Inhaler. AAPS PharmSciTech 2018, 19, 2434–2448. [Google Scholar] [CrossRef]
- Skalko-Basnet, N.; Pavelic, Z.; Becirevic-Lacan, M. Liposomes containing drug and cyclodextrin prepared by the one-step spray-drying method. Drug Dev. Ind. Pharm. 2000, 26, 1279–1284. [Google Scholar] [CrossRef]
- Alves, A.; Correia-da-Silva, M.; Nunes, C.; Campos, J.; Sousa, E.; Silva, P.M.A.; Bousbaa, H.; Rodrigues, F.; Ferreira, D.; Costa, P.C.; et al. Discovery of a New Xanthone against Glioma: Synthesis and Development of (Pro)liposome Formulations. Molecules 2019, 24, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, M.I.; Dimitrov, D.S. Liposome electroformation. Faraday Discuss. Chem. Soc. 1986, 81, 303–311. [Google Scholar] [CrossRef]
- Mertins, O.; da Silveira, N.P.; Pohlmann, A.R.; Schröder, A.P.; Marques, C.M. Electroformation of giant vesicles from an inverse phase precursor. Biophys. J. 2009, 96, 2719–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertins, O.; Dimova, R. Insights on the Interactions of Chitosan with Phospholipid Vesicles. Part II: Membrane Stiffening and Pore Formation. Langmuir 2013, 29, 14552–14559. [Google Scholar] [CrossRef]
- Le Berre, M.; Yamada, A.; Reck, L.; Chen, Y.; Baigl, D. Electroformation of Giant Phospholipid Vesicles on a Silicon Substrate: Advantages of Controllable Surface Properties. Langmuir 2008, 24, 2643–2649. [Google Scholar] [CrossRef]
- Ye, H.; Shen, Z.; Yu, L.; Wei, M.; Li, Y. Manipulating nanoparticle transport within blood flow through external forces: An exemplar of mechanics in nanomedicine. Proc. Math. Phys. Eng. Sci. 2018, 474, 20170845. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Li, Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small 2013, 9, 1521–1532. [Google Scholar] [CrossRef] [PubMed]
- Kanásová, M.; Nesměrák, K. Systematic review of liposomes’ characterization methods. Monatsh. Chem. 2017, 148, 1581–1593. [Google Scholar] [CrossRef]
- Robson, A.L.; Dastoor, P.C.; Flynn, J.; Palmer, W.; Martin, A.; Smith, D.W.; Woldu, A.; Hua, S. Advantages and Limitations of Current Imaging Techniques for Characterizing Liposome Morphology. Front. Pharmacol. 2018, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.Ø.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro-Oncology 2012, 14, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Stylli, S.S.; Luwor, R.B.; Ware, T.M.; Tan, F.; Kaye, A.H. Mouse models of glioma. J. Clin. Neurosci. 2015, 22, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.D.; Miller, C.R.; Rossmeisl, J.H. Canine Primary Intracranial Cancer: A Clinicopathologic and Comparative Review of Glioma, Meningioma, and Choroid Plexus Tumors. Front. Oncol. 2019, 9, 1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lainetti, P.d.F.; Zuliani, F.; Leis-Filho, A.F.; Fonseca Alves, R.H.; Fonseca-Alves, C.E. Controlled Drug Delivery Vehicles in Veterinary Oncology: State-of-the-Art and Future Directions. Processes 2020, 8, 541. [Google Scholar] [CrossRef]
- Platt, S.; Nduom, E.; Kent, M.; Freeman, C.; Machaidze, R.; Kaluzova, M.; Wang, L.; Mao, H.; Hadjipanayis, C.G. Canine model of convection-enhanced delivery of cetuximab-conjugated iron-oxide nanoparticles monitored with magnetic resonance imaging. Clin. Neurosurg. 2012, 59, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.C.; Platt, S.R.; Holmes, S.; Kent, M.; Robinson, K.; Howerth, E.; Eagleson, J.; Bouras, A.; Kaluzova, M.; Hadjipanayis, C.G. Convection-enhanced delivery of cetuximab conjugated iron-oxide nanoparticles for treatment of spontaneous canine intracranial gliomas. J. Neurooncol. 2018, 137, 653–663. [Google Scholar] [CrossRef]
- Arami, H.; Patel, C.B.; Madsen, S.J.; Dickinson, P.J.; Davis, R.M.; Zeng, Y.; Sturges, B.K.; Woolard, K.D.; Habte, F.G.; Akin, D.; et al. Nanomedicine for Spontaneous Brain Tumors: A Companion Clinical Trial. ACS Nano 2019, 13, 2858–2869. [Google Scholar] [CrossRef]
- Dickinson, P.J.; LeCouteur, R.A.; Higgins, R.J.; Bringas, J.R.; Larson, R.F.; Yamashita, Y.; Krauze, M.T.; Forsayeth, J.; Noble, C.O.; Drummond, D.C.; et al. Canine spontaneous glioma: A translational model system for convection-enhanced delivery. Neuro-Oncology 2010, 12, 928–940. [Google Scholar] [CrossRef]
- Bredlau, A.L.; Motamarry, A.; Chen, C.; McCrackin, M.A.; Helke, K.; Armeson, K.E.; Bynum, K.; Broome, A.-M.; Haemmerich, D. Localized delivery of therapeutic doxorubicin dose across the canine blood–brain barrier with hyperthermia and temperature sensitive liposomes. Drug Deliv. 2018, 25, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Young, J.S.; Bernal, G.; Polster, S.P.; Nunez, L.; Larsen, G.F.; Mansour, N.; Podell, M.; Yamini, B. Convection-Enhanced Delivery of Polymeric Nanoparticles Encapsulating Chemotherapy in Canines with Spontaneous Supratentorial Tumors. World Neurosurg. 2018, 117, e698–e704. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrials.gov (accessed on 6 September 2021).
- Mahan, V. Clinical Trial Phases. J. Clin. Med. 2014, 5, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- LeBlanc, A.K.; Mazcko, C.N. Improving human cancer therapy through the evaluation of pet dogs. Nat. Rev. Cancer 2020, 20, 727–742. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
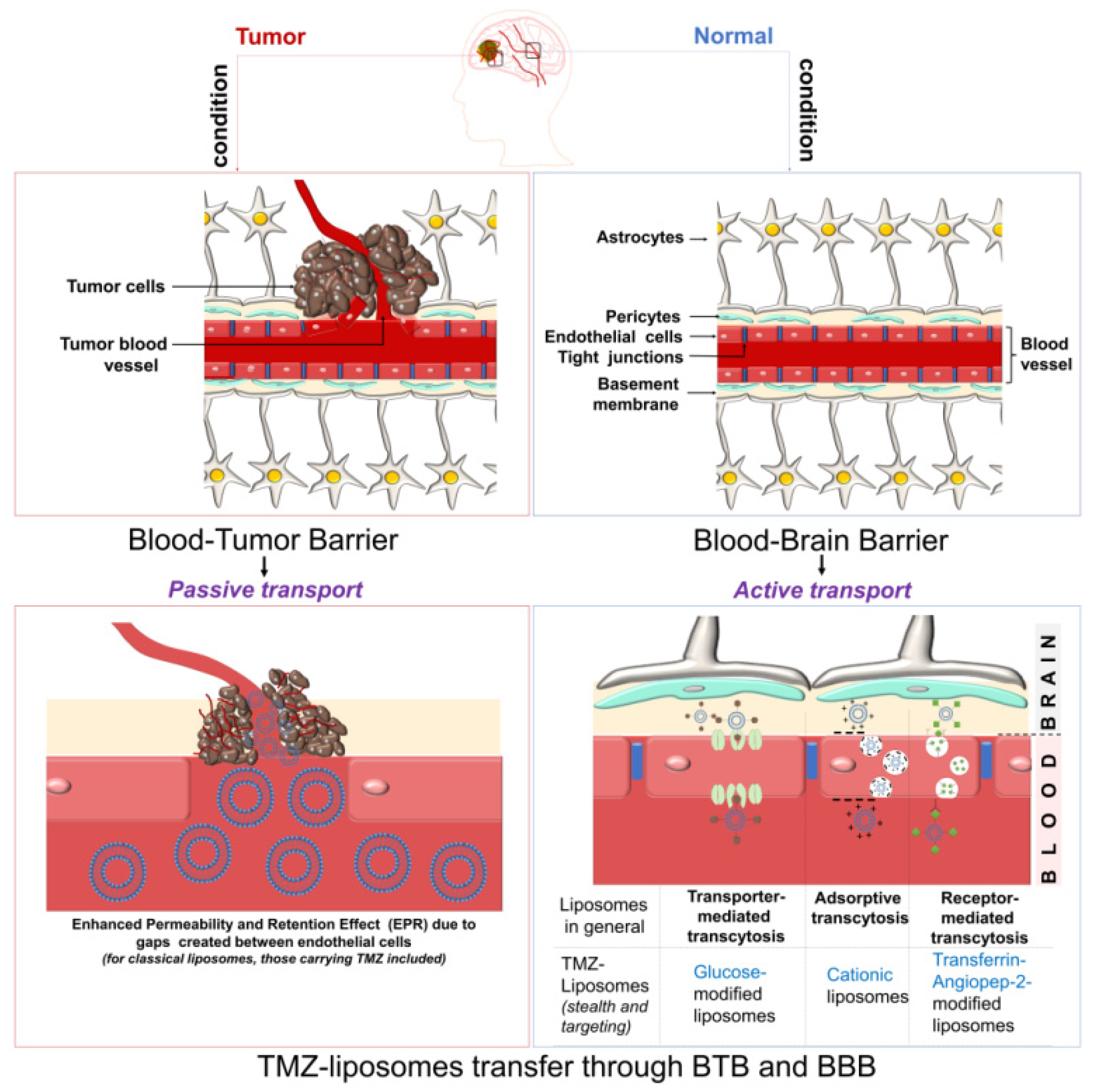{kind=link}
{kind=link}
{kind=link}
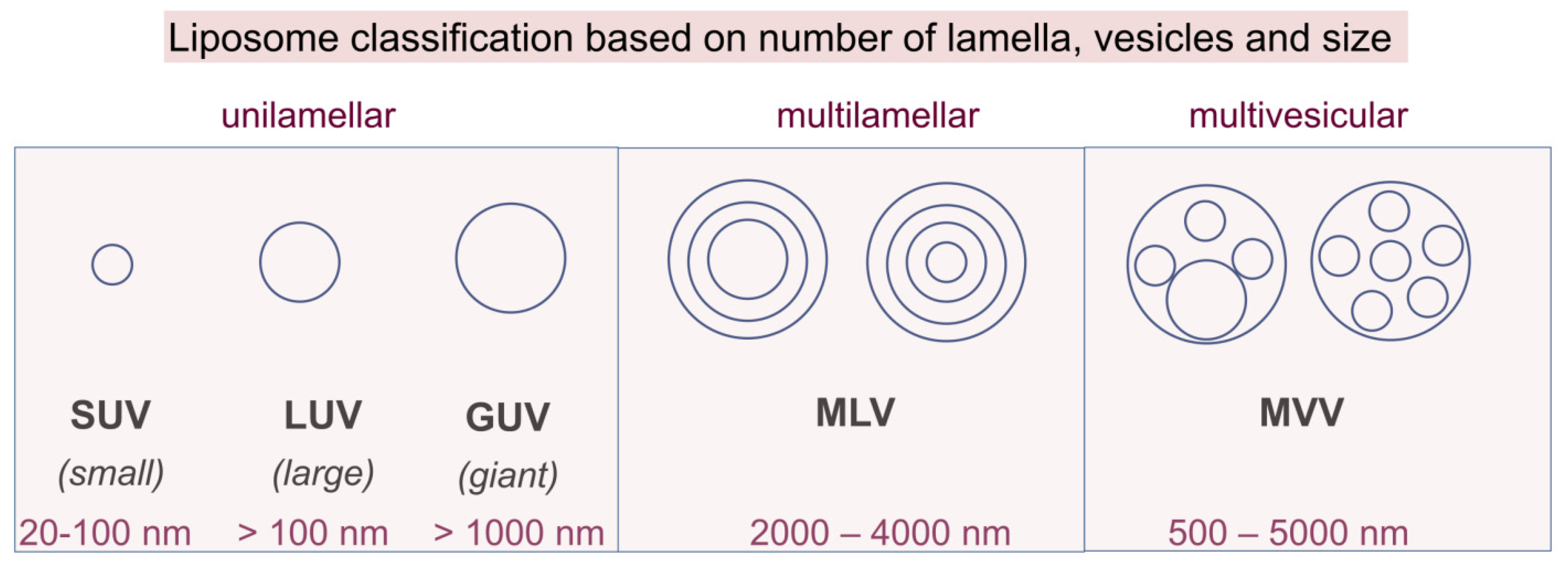{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Head Group Substituent | (Phospho)lipid | Abbreviation | Molecular Weight (g·mol−1) | C/U | TC (°C) | Charge (pH = 7) |
|---|---|---|---|---|---|---|
| Choline | Dilauroyl phosphatidylcholine | DLPC | 621.83 | 12/0 | −2 | Zwitteriionic/Neutral |
| Dimyristoyl phosphatidylcholine | DMPC | 677.93 | 14/0 | 24 | ||
| Dipalmitoyl phosphatidylcholine | DPPC | 805.48 | 16/0 | 41 | ||
| Distearoyl phosphatidylcholine | DSPC | 790.15 | 18/0 | 55 | ||
| Dioleoyl phosphatidylcholine | DOPC | 786.11 | 18/1 | −17 | ||
| Ethanolamine | Dilauroyl phosphatidylethanolamine | DLPE | 579.75 | 12/0 | 29 | |
| Dimyristoyl phosphatidylethanolamine | DMPE | 635.85 | 14/0 | 50 | ||
| Dipalmitoyl phosphatidylethanolamine | DPPE | 691.96 | 16/0 | 60 | ||
| Distearoyl phosphatidylethanolamine | DSPE | 748.07 | 18/0 | 74 | ||
| Dioleoyl phosphatidylethanolamine | DOPE | 744.03 | 18/1 | −16 | ||
| Glycerol | Dilauroyl phosphatidylglycerol | DLPG | 610.8 | 12/0 | −3 | Anionic/Negative |
| Dimyristoyl phosphatidylglycerol | DMPG | 666.9 | 14/0 | 23 | ||
| Dipalmitoyl phosphatidylglycerol | DPPG | 744.95 | 16/0 | 41 | ||
| Distearoyl phosphatidylglycerol | DSPG | 779.1 | 18/0 | 55 | ||
| Dioleoyl phosphatidylglycerol | DOPG | 775.0 | 18/1 | −18 | ||
| Serine | Dilauroyl phosphatidylserine | DLPS | 645.74 | 12/0 | ||
| Dimyristoyl phosphatidylserine | DMPS | 679.9 | 14/0 | 35 | ||
| Dipalmitoyl phosphatidylserine | DPPS | 736.0 | 16/0 | 51 | ||
| Distearoyl phosphatidylserine | DSPS | 792.1 | 18/0 | 68 | ||
| Dioleoyl phosphatidylserine | DOPS | 810.03 | 18/1 | −11 | ||
| - | Dioleoyl trimethylammonium-propane | DOTAP | 698.54 | 18/1 | <5 | Cationic/Positive |
| Size (nm) | Surface Charge (mV) | pH | Morphology /Structure | PL Composition (mg/mL) | EE (%) | Stability | Cell Line | In Vivo | Delivery Way | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 203.4 | −1.60 | - | - | - | 99.20 | - | - | - | IV | [80] |
| 160.0 | ~0 | - | Sphere (TEM) | - | 87.00 | - | - | - | CED | [51] |
| 185.0 | - | - | Sphere (SEM) | - | 90.30 | - | - | - | - | [28] |
| 157.0 | - | 6.46 | Sphere (TEM) | - | 35.45 | - | - | Rabbit mouse | IV | [9] |
| 120.0 | −0.20 | - | Sphere/ unilamellar (cryoTEM) | 28 (HPLC) | (HPLC) | - | CNS-1 rat glioma cell | rat | CED | [52] |
| 137 | −12.10 | - | - | - | 45.10 (HPLC) | - | U87, GL261 | Mice | [81] | |
| 137.4 | −49.90 | - | - | - | - | - | - | [82] | ||
| 135.6 | −26.26 | - | Sphere/ unilamellar (cryoTEM) | - | 56.11 (UV-Vis) | Size, EE% 93 months | - | Rats | IV | [2] |
| 41.4 | 30.10 | - | - | - | 45.23 | - | U87-luc2 U251 U87R | Mice | IV | [78] |
| 196.5 | 30.50 | Sphere (TEM) | HPLC UV-Vis FT-IR | 53.58–66.25 (HPLC) | - | U87/TR | Rats | Intraperi-toneal | [47] | |
| 133.0 | −5.68 | Sphere (TEM) | Size, CMC | GL261 | Mice | IV | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amarandi, R.-M.; Ibanescu, A.; Carasevici, E.; Marin, L.; Dragoi, B. Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue. Pharmaceutics 2022, 14, 308. https://doi.org/10.3390/pharmaceutics14020308
Amarandi R-M, Ibanescu A, Carasevici E, Marin L, Dragoi B. Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue. Pharmaceutics. 2022; 14(2):308. https://doi.org/10.3390/pharmaceutics14020308
Chicago/Turabian StyleAmarandi, Roxana-Maria, Alina Ibanescu, Eugen Carasevici, Luminita Marin, and Brindusa Dragoi. 2022. "Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue" Pharmaceutics 14, no. 2: 308. https://doi.org/10.3390/pharmaceutics14020308