Novel Approaches for the Treatment of Pulmonary Tuberculosis

 , , , , and
, , , , and
Abstract
:1. Introduction
2. History of Treatments of TB and Associated Limitations
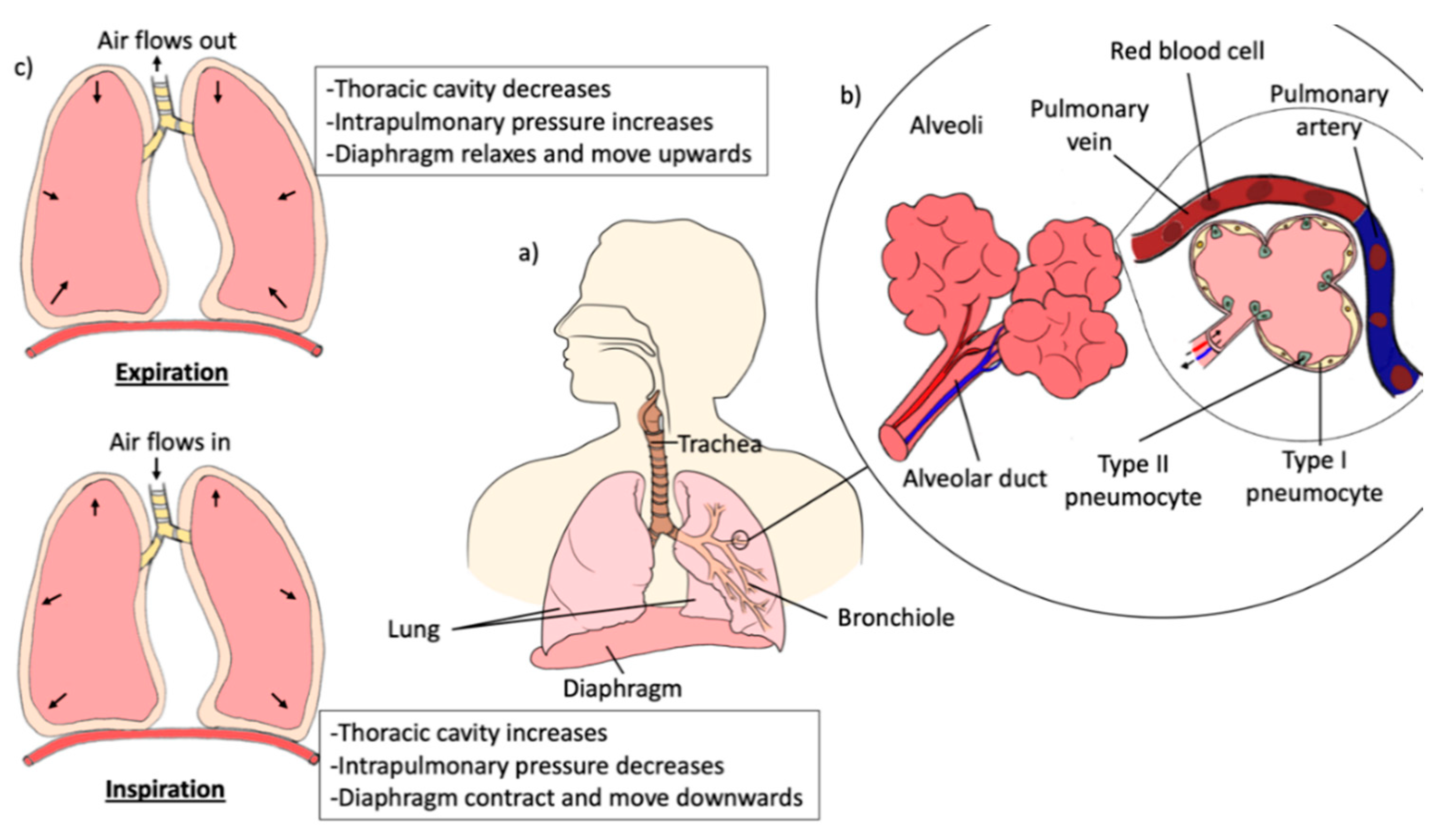
3. Anatomy and Physiology of the Lungs
4. Different Biological Barriers and Factors Affecting Lung Drug Delivery
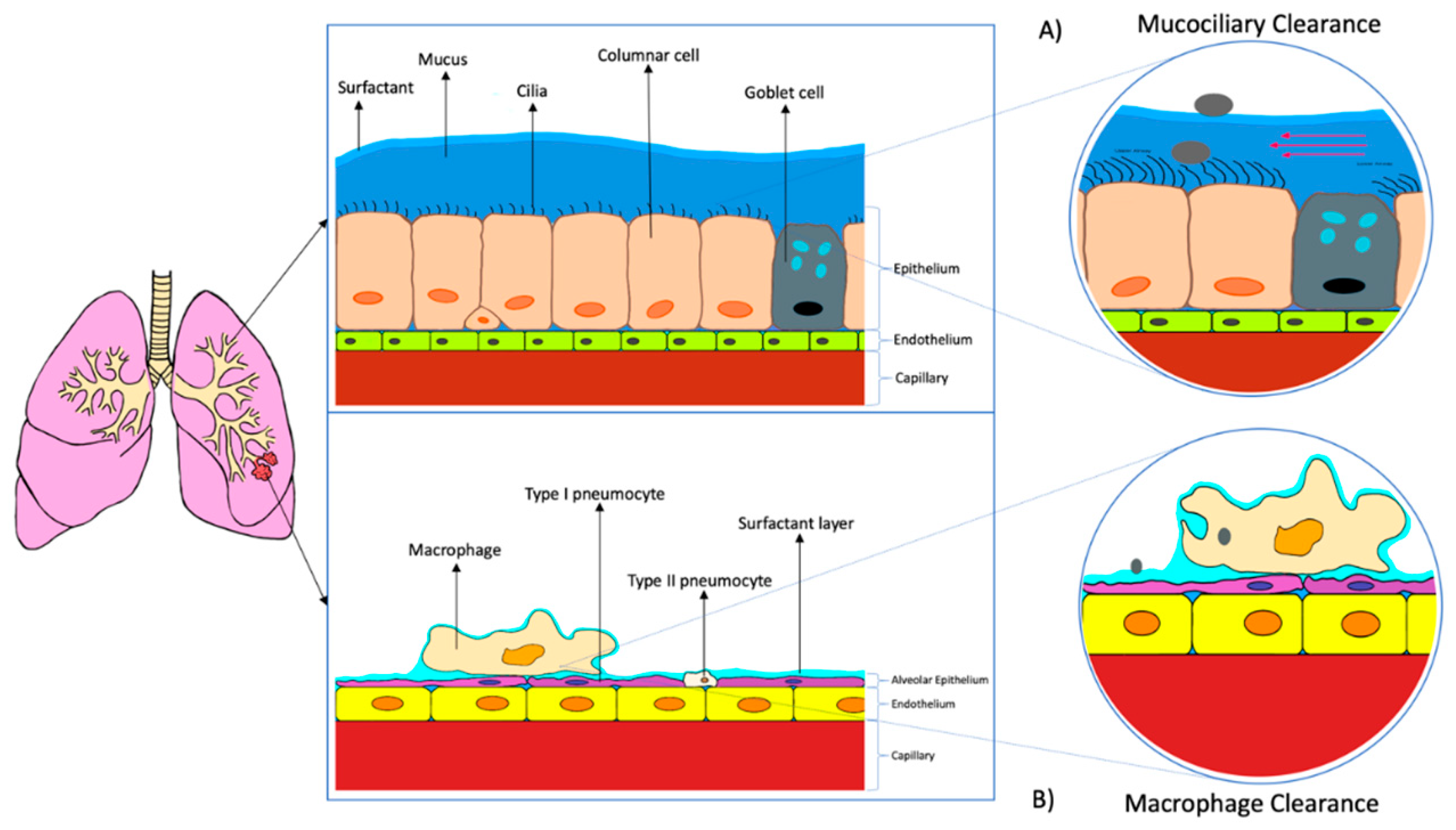
4.1. Biological Barriers of Lungs
4.2. Factors Affecting Lung Drugs Delivery
4.2.1. Physiological Factors
- (a)
- Lung morphology: The architecture of the lungs’ airways can affect the efficiency of pulmonary drug delivery as the diameter of the airways decrease from the trachea to bronchioles. The risk of impaction increases for every bifurcation/branching which will decrease the efficiency in pulmonary drug delivery into deep lungs [42,43]. Furthermore, the airflow in the airways will be influenced and results in the generation of turbulent (due to the sudden decrease in the diameter of airways). The formation of turbulent will increase the deposition of the drug particles on the upper airways [44].
- (b)
- Inspiration flow rate: When the airflow rate is fast, the drug particles are more likely to deposit on the upper airways and oropharynx. In contrast, a slow/moderate airflow will reduce the momentum and the possibility of impaction. Therefore, more particles can be travel to the lower respiratory tract. Research showed that there are fewer drug particles deposited in deep lungs for the inspiratory flow rate at 60 L/min compared to at 15 L/min due to the strong turbulent dispersion [45,46].
- (c)
- Breath-holding: Breath-holding can prolong the residence time of the drug particles in the respiratory tract to allow the occurrence of sedimentation and increase the lung dose [42]. For instance, Horváth et al., demonstrated that the lung dose increased by a mean value of 21.4 percent (5 s breath-hold) and 42.4 percent (25 s breath-hold) compared to no breath-holding [47].
- (d)
- Disease state: Most of the respiratory diseases such as cystic fibrosis, asthma and chronic bronchitis will cause the narrowing of airways and the bronchial obstruction due to the inflammation and excessive production of mucus [44,48] The change in the diameter of airways will result in the change in airflow velocities, turbulent, and air resistance. As a result, a higher amount of drug particles will deposit in the upper respiratory tract rather than deep lungs [44].
4.2.2. Pharmaceutical Factors
- (a)
- Aerosol performance: The aerosol performance of a dispersion device is significant to lung drug delivery. For example, the aerodynamic diameter of 1–5 µm, fine particle fraction > 35%, and emitted dose of > 90% are the 3 important criteria for good aerosol performance for dry powder inhaler [49,50]. In addition, the airflow velocities of a dispersion device can also influence the lung drug delivery as the greater the velocities of the airflow, the drug particles are more likely to impact in the oropharyngeal area [42,46].
- (b)
- Particle shape/morphology: The shape of the drug particles can influence the particle adhesion in lungs. For instance, researchers showed that the particles with pollen or needles shape enhanced the drug deposition in smaller airways which increased the effectiveness of lung drug deposition [51,52]. Hassan et al., also reported that the pollen shape particles possess the benefits of higher fine particle fraction and minimized drug loss [51].
- (c)
- Physical stability: The physical stability of drug particles is extremely important because the drug particles need to go through several environments with high humidity before reaching the targeted site. Premature deposition may occur if the drug particles are unstable [42]. The physical instability of the drug particles may also lead to loss of encapsulated active pharmaceutical ingredients as well as the aggregation or sedimentation of drugs during storage [50]. Moreover, the physical stability of inhalers is also significant in ensuring the delivery of an equivalent and sufficient dose over the shelf life [53].
5. Novel Approaches for Dry Powder Inhalable Lung Drug Delivery
5.1. Approaches for Pyrazinamide (PZA) Delivery’
5.2. Delivery Approaches of Isoniazid
5.3. Delivery Approaches of Ethambutol
5.4. Delivery Approaches of Rifamycins
5.4.1. Rifampicin Formulation Approaches
5.4.2. Rifabutin Formulation Approaches
5.4.3. Rifapentine Formulation Approaches
5.5. Delivery Approaches of Formulations Containing Therapeutic Combinations
5.6. Delivery Approaches for Vaccines
6. Beneficial Aspects of Proliposomal Dry Powder Inhaler over Other Nanocarriers in Treatment of Tuberculosis
7. Safety Concern of Dry Powder Inhaler Formulation
7.1. Safety of Lipid-Based Carriers Intended to Lung Delivery
7.2. Safety Concern of Polymeric-Based Nano/Micro Carriers for the Treatment of Tuberculosis
8. Clinical Aspect of Dry Powder Inhaler Formulation against Tuberculosis
9. Expert Opinion on Drug Delivery on Tuberculosis Treatment
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO|Global Tuberculosis Report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 29 November 2019).
- Kiazyk, S.; Ball, T. Latent tuberculosis infection: An overview. Can. Commun. Dis. Rep. 2017, 43, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Sulis, G.; Roggi, A.; Matteelli, A.; Raviglione, M.C. Tuberculosis: Epidemiology and control. Mediterr. J. Hematol. Infect. Dis. 2014, 6, 2014070. [Google Scholar] [CrossRef] [PubMed]
- Seung, K.J.; Keshavjee, S.; Rich, M.L. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5, a017863. [Google Scholar] [CrossRef] [Green Version]
- Demile, B.; Zenebu, A.; Shewaye, H.; Xia, S.; Guadie, A. Risk factors associated with multidrug-resistant tuberculosis (MDR-TB) in a tertiary armed force referral and teaching hospital, Ethiopia. BMC Infect. Dis. 2018, 18, 249. [Google Scholar] [CrossRef] [PubMed]
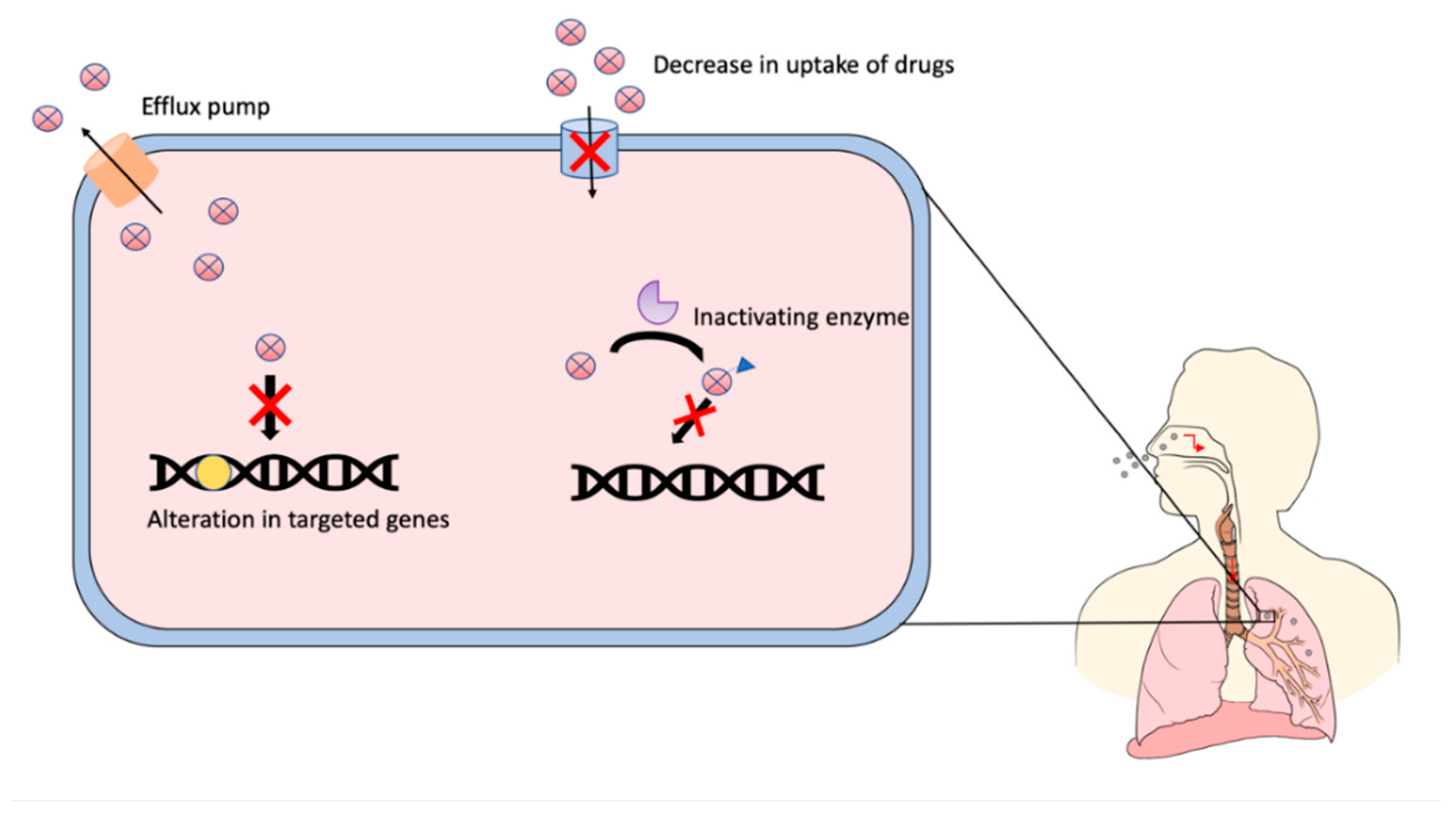
- Hameed, H.M.A.; Islam, M.M.; Chhotaray, C.; Wang, C.; Liu, Y.; Tan, Y.; Li, X.; Tan, S.; Delorme, V.; Yew, W.W.; et al. Molecular targets related drug resistance mechanisms in MDR-, XDR-, and TDR-Mycobacterium tuberculosis strains. Front. Cell. Infect. Microbiol. 2018, 8, 114. [Google Scholar] [CrossRef]
- Almeida, P.E.; Silva, D.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef]
- Ghajavand, H.; Kamakoli, M.K.; Khanipour, S.; Dizaji, S.P.; Masoumi, M.; Jamnani, F.R.; Fateh, A.; Yaseri, M.; Siadat, S.D.; Vaziri, F. Scrutinizing the drug resistance mechanism of multi- and extensively-drug resistant Mycobacterium tuberculosis: Mutations versus efflux pumps. Antimicrob. Resist. Infect. Control. 2019, 8, 70. [Google Scholar] [CrossRef]
- Abraham, A.O.; Nasiru, A.U.; Abdulazeez, A.K.; Seun, O.O.; Ogonna, D.W. Mechanism of Drug Resistance in Mycobacterium Tuberculosis. Am. J. Biomed. Sci. Res. 2020, 5, 378–383. [Google Scholar] [CrossRef]
- Gandhi, N.R.; Brust, J.C.M.; Sarita Shah, N. A new era for treatment of drug-resistant tuberculosis. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention History|World TB Day|TB|CDC. Available online: https://www.cdc.gov/tb/worldtbday/default.htm (accessed on 16 September 2020).
- John Frith History of Tuberculosis. Part 1—Phthisis, Consumption and the White Plague 2014, 22 No.2. Available online: https://jmvh.org/article/history-of-tuberculosis-part-1-phthisis-consumption-and-the-white-plague/ (accessed on 16 September 2020.).
- Cambau, E.; Drancourt, M. Steps towards the discovery of Mycobacterium tuberculosis by Robert Koch, 1882. Eur. Soc. Clin. Infect. Dis. 2014, 20, 196–201. [Google Scholar] [CrossRef] [Green Version]
- John, F.M.; Dean, E.S.; Philip, C.H. Treatment of Tuberculosis: A Historical Perspective. Ann. Am. Thorac. Soc. 2015, 12, 1749–1759. [Google Scholar]
- Steenken, W.; Wolinsky, E. Streptomycin therapy in experimental tuberculosis of guinea pigs infected intracerebrally with virulent tubercle bacilli. Science 1947, 106, 638–639. [Google Scholar] [CrossRef] [PubMed]
- Feldman, W.H.; Hinshaw, H.C.; Mann, F.C. Streptomycin in Experimental Tuberculosis. Am. Rev. Tuberc. 1945, 52, 269–298. [Google Scholar]
- Pfuetze, K.H.; Pyle, M.M.; Hinshaw, H.C.; Feldman, W.H. The first clinical trial of streptomycin in human tuberculosis. Am. Rev. Tuberc. 1955, 71, 752–754. [Google Scholar] [PubMed]
- Pandey, B.; Grover, S.; Kaur, J.; Grover, A. Analysis of mutations leading to para-aminosalicylic acid resistance in Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 13617. [Google Scholar] [CrossRef] [Green Version]
- Kerantzas, C.A.; Jacobs, W.R. Origins of combination therapy for tuberculosis: Lessons for future antimicrobial development and application. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Nachega, J.B.; Chaisson, R.E. Tuberculosis Drug Resistance: A Global Threat. Clin. Infect. Dis. 2003, 36, S24–S30. [Google Scholar] [CrossRef] [Green Version]
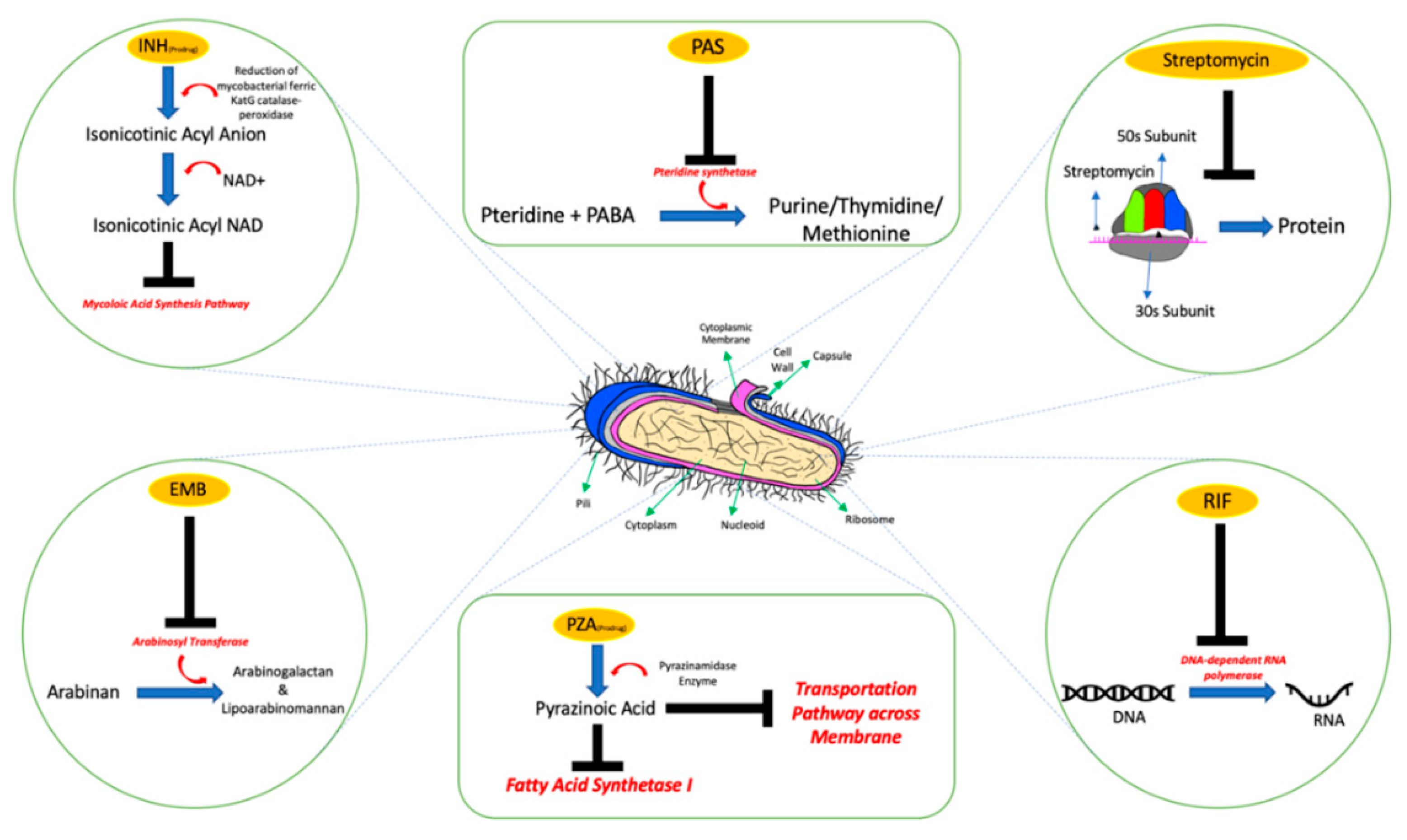
- Chakraborty, S.; Rhee, K.Y. Tuberculosis drug development: History and evolution of the mechanism-based paradigm. Cold Spring Harb. Perspect. Med. 2015, 5, a021147. [Google Scholar] [CrossRef] [Green Version]
- Stagg, H.R.; Lipman, M.C.; McHugh, T.D.; Jenkins, H.E. Isoniazid-resistant tuberculosis: A cause for concern? Int. J. Tuberc. Lung Dis. 2017, 21, 129–139. [Google Scholar] [CrossRef]
- Chang, K.C.; Leung, C.C.; Yew, W.W.; Leung, E.C.C.; Leung, W.M.; Tam, C.M.; Zhang, Y. Pyrazinamide may improve fluoroquinolone-based treatment of multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5465–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol. Spectr. 2014, 2, 1–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ethambutol|C10H24N2O2—PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ethambutol (accessed on 16 September 2020).
- Aung, H.H.; Sivakumar, A.; Gholami, S.K.; Venkateswaran, S.P.; Gorain, B.; Md, S. An Overview of the Anatomy and Physiology of the Lung. In Nanotechnology-Based Targeted Drug Delivery Systems for Lung Cancer; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–20. [Google Scholar]
- Chaudhry, R.; Bordoni, B. Anatomy, Thorax, Lungs; StatPearls: Treasure Island, FL, USA. Available online: https://www.ncbi.nlm.nih.gov/books/NBK470197/ (accessed on 20 September 2020).
- Peate, I. Anatomy and physiology, 10. The Respiratory System. Br. J. Healthc. Assist. 2018, 12, 178–181. [Google Scholar] [CrossRef]
- Patwa, A.; Shah, A. Anatomy and physiology of respiratory system relevant to anaesthesia. Indian J. Anaesth. 2015, 59, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Tomashefski, J.F.; Farver, C.F. Anatomy and histology of the lung. In Dail and Hammar’s Pulmonary Pathology; Springer: New York, NY, USA, 2008; Volume 1, pp. 20–48. ISBN 9780387983950. [Google Scholar]
- Murgia, X.; De Souza Carvalho, C.; Lehr, C.M. Overcoming the pulmonary barrier: New insights to improve the efficiency of inhaled therapeutics. Eur. J. Nanomed. 2014, 6, 157–169. [Google Scholar] [CrossRef]
- Knudsen, L.; Ochs, M. The micromechanics of lung alveoli: Structure and function of surfactant and tissue components. Histochem. Cell Biol. 2018, 150, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, J.E.; Sharma, S. Physiology, Pulmonary; StatPearls: Treasure Island, FL, USA; Available online: https://www.statpearls.com/ArticleLibrary/viewarticle/28046 (accessed on 21 September 2020).
- Effros, R. Anatomy, development, and physiology of the lungs. GI Motil. Online 2006. [Google Scholar] [CrossRef]
- Lutfi, M.F. The physiological basis and clinical significance of lung volume measurements. Multidiscip. Respir. Med. 2017, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, H.; Gorain, B.; Pandey, M.; Khurana, R.K.; Kesharwani, P. Strategizing biodegradable polymeric nanoparticles to cross the biological barriers for cancer targeting. Int. J. Pharm. 2019, 565, 509–522. [Google Scholar] [CrossRef]
- Ahmad, I. The Development of Dimple Shape Dry Powder Carrier for Ethambutol Dihydrochloride and Its Antituberculosis Evaluation. Drug Dev. Ind. Pharm. 2014, 41, 791–800. [Google Scholar] [CrossRef]
- Cipolla, D. Will pulmonary drug delivery for systemic application ever fulfill its rich promise? Expert Opin. Drug Deliv. 2016, 13, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.; Young, P.M.; Traini, D. Strategies to enhance drug absorption via nasal and pulmonary routes. Pharmaceutics 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagoba, S.N. A Review on Pulmonary Drug Delivery System. Int. J. Pharm. Pharm. Res. 2018, 12, 438–453. [Google Scholar]
- Ashish, K.A.; Hiralal, C.S.; Prajkta, U.L.; Dheeraj, B.T.; Dinesh, J.K. Pulmonary Drug Delivery System. Int. J. PharmTech Res. CODEN 2012, 4, 293–305. [Google Scholar]
- Verma, R.K.; Ibrahim, M.; Garcia-Contreras, L. Lung Anatomy and Physiology and Their Implications for Pulmonary Drug Delivery. In Pulmonary Drug Delivery; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 1–18. [Google Scholar]
- Nokhodchi, A.; Martin, G.P. (Eds.) Pulmonary Drug Delivery: Advances and Challenges; John Wiley & Sons, Ltd.: Chichester, UK, 2015. [Google Scholar]
- Zhang, Z.; Kleinstreuer, C.; Donohue, J.F.; Kim, C.S. Comparison of micro- and nano-size particle depositions in a human upper airway model. J. Aerosol Sci. 2005, 36, 211–233. [Google Scholar] [CrossRef]
- Zhang, Z.; Kleinstreuer, C. Airflow structures and nano-particle deposition in a human upper airway model. J. Comput. Phys. 2004, 198, 178–210. [Google Scholar] [CrossRef]
- Horváth, A.; Balásházy, I.; Tomisa, G.; Farkas, Á. Significance of breath-hold time in dry powder aerosol drug therapy of COPD patients. Eur. J. Pharm. Sci. 2017, 104, 145–149. [Google Scholar] [CrossRef]
- Dabbagh, A.; Abu Kasim, N.H.; Yeong, C.H.; Wong, T.W.; Abdul Rahman, N. Critical parameters for particle-based pulmonary delivery of chemotherapeutics. J. Aerosol Med. Pulm. Drug Deliv. 2018, 31, 139–154. [Google Scholar] [CrossRef] [Green Version]
- Peng, T.; Lin, S.; Niu, B.; Wang, X.; Huang, Y.; Zhang, X.; Li, G.; Pan, X.; Wu, C. Influence of physical properties of carrier on the performance of dry powder inhalers. Acta Pharm. Sin. B 2016, 6, 308–318. [Google Scholar] [CrossRef] [Green Version]
- Rojanarat, W.; Changsan, N.; Tawithong, E.; Pinsuwan, S.; Chan, H.-K.; Srichana, T. Isoniazid proliposome powders for inhalation-preparation, characterization and cell culture studies. Int. J. Mol. Sci. 2011, 12, 4414–4434. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.S.; Lau, R. Inhalation performance of pollen-shape carrier in dry powder formulation: Effect of size and surface morphology. Int. J. Pharm. 2011, 413, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Chan, J.G.Y.; Chan, H.K. Pulmonary drug delivery by powder aerosols. J. Control. Release 2014, 193, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Shetty, N.; Cipolla, D.; Park, H.; Zhou, Q.T. Physical stability of dry powder inhaler formulations. Expert Opin. Drug Deliv. 2020, 17, 77–96. [Google Scholar] [CrossRef] [Green Version]
- Lienhardt, C.; Vernon, A.; Raviglione, M.C. New Drugs and New Regimens for the Treatment of Tuberculosis: Review of the Drug Development Pipeline and Implications for National Programmes. Curr. Opin. Pulm. Med. 2010, 16, 186–193. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment; WHO: Geneva, Switzerland, 2019; ISBN 9789241550529. [Google Scholar]
- Muralidharan, P.; Malapit, M.; Mallory, E.; Hayes, D.; Mansour, H.M. Inhalable nanoparticulate powders for respiratory delivery. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1189–1199. [Google Scholar] [CrossRef] [Green Version]
- Parumasivam, T.; Chang, R.Y.K.; Abdelghany, S.; Ye, T.T.; Britton, W.J.; Chan, H.K. Dry powder inhalable formulations for anti-tubercular therapy. Adv. Drug Deliv. Rev. 2016, 102, 83–101. [Google Scholar] [CrossRef]
- Mehta, P.; Bothiraja, C.; Kadam, S.; Pawar, A. Potential of dry powder inhalers for tuberculosis therapy: Facts, fidelity and future. Artif. Cells Nanomed. Biotechnol. 2018, 46, S791–S806. [Google Scholar] [CrossRef]
- Patil, T.S.; Deshpande, A.S.; Deshpande, S.; Shende, P. Targeting pulmonary tuberculosis using nanocarrier-based dry powder inhalation: Current status and futuristic need. J. Drug Target. 2019, 27, 12–27. [Google Scholar] [CrossRef]
- Karmakar, M.; Rodrigues, C.H.M.; Horan, K.; Denholm, J.T.; Ascher, D.B. Structure guided prediction of Pyrazinamide resistance mutations in pncA. Sci. Rep. 2020, 10, 1875. [Google Scholar] [CrossRef]
- Rabbani, N.R.; Seville, P.C. The influence of formulation components on the aerosolisation properties of spray-dried powders. J. Control. Release 2005, 110, 130–140. [Google Scholar] [CrossRef]
- Li, H.Y.; Seville, P.C.; Williamson, I.J.; Birchall, J.C. The use of amino acids to enhance the aerosolisation of spray-dried powders for pulmonary gene therapy. J. Gene Med. 2005, 7, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Muddle, J.; Kirton, S.B.; Parisini, I.; Muddle, A.; Murnane, D.; Ali, J.; Brown, M.; Page, C.; Forbes, B. Predicting the Fine Particle Fraction of Dry Powder Inhalers Using Artificial Neural Networks. J. Pharm. Sci. 2017, 106, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, P.; Stein, S.W.; Myrdal, P.B. Factors Influencing Aerodynamic Particle Size Distribution of Suspension Pressurized Metered Dose Inhalers. AAPS PharmSciTech 2014, 16, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laube, B.L.; Janssens, H.M.; De Jongh, F.H.C.; Devadason, S.G.; Dhand, R.; Diot, P.; Everard, M.L.; Horvath, I.; Navalesi, P.; Voshaar, T.; et al. What the pulmonary specialist should know about the new inhalation therapies. Eur. Respir. J. 2011, 37, 1308–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaewjan, K.; Srichana, T. Nano spray-dried pyrazinamide-l-leucine dry powders, physical properties and feasibility used as dry powder aerosols. Pharm. Dev. Technol. 2016, 21, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.D.; Grégoire, N.; Couet, W.; Gueutin, C.; Fattal, E.; Tsapis, N. Pulmonary delivery of pyrazinamide-loaded large porous particles. Eur. J. Pharm. Biopharm. 2015, 94, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Keshani, S.; Daud, W.R.W.; Woo, M.W.; Nourouzi, M.M.; Talib, M.Z.M.; Chuah, A.L.; Russly, A.R. Reducing the deposition of fat and protein covered particles with low energy surfaces. J. Food Eng. 2013, 116, 737–748. [Google Scholar] [CrossRef]
- Price, R.; Young, P.M.; Edge, S.; Staniforth, J.N. The influence of relative humidity on particulate interactions in carrier-based dry powder inhaler formulations. Int. J. Pharm. 2002, 246, 47–59. [Google Scholar] [CrossRef]
- Coelho, M.C.; Harnby, N. Moisture bonding in powders. Powder Technol. 1978, 20, 201–205. [Google Scholar] [CrossRef]
- You, Y.; Zhao, M.; Liu, G.; Tang, X. Physical characteristics and aerosolization performance of insulin dry powders for inhalation prepared by a spray drying method. J. Pharm. Pharmacol. 2007, 59, 927–934. [Google Scholar] [CrossRef]
- Eedara, B.B.; Tucker, I.G.; Das, S.C. Phospholipid-based pyrazinamide spray-dried inhalable powders for treating tuberculosis. Int. J. Pharm. 2016, 506, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Rojanarat, W.; Nakpheng, T.; Thawithong, E.; Yanyium, N.; Srichana, T. Inhaled pyrazinamide proliposome for targeting alveolar macrophages. Drug Deliv. 2012, 19, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Afinjuomo, F.; Barclay, T.G.; Parikh, A.; Chung, R.; Song, Y.; Nagalingam, G.; Triccas, J.; Wang, L.; Liu, L.; Hayball, J.D.; et al. Synthesis and characterization of pH-sensitive inulin conjugate of isoniazid for monocyte-targeted delivery. Pharmaceutics 2019, 11, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.H. High-performance superplasticizer based on chitosan. In Biopolymers and Biotech Admixtures for Eco-Efficient Construction Materials; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 131–150. ISBN 9780081002148. [Google Scholar]
- Kundawala, A.J.; Patel, V.A.; Patel, H.V.; Choudhary, D. Isoniazid loaded chitosan microspheres for pulmonary delivery: Preparation and characterization. Pharm. Sin. 2011, 2, 88–97. [Google Scholar]
- Akrawi, S.H.; Gorain, B.; Nair, A.B.; Choudhury, H.; Pandey, M.; Shah, J.N.; Venugopala, K.N. Development and optimization of naringenin-loaded chitosan-coated nanoemulsion for topical therapy in wound healing. Pharmaceutics 2020, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Pourshahab, P.S.; Gilani, K.; Moazeni, E.; Eslahi, H.; Fazeli, M.R.; Jamalifar, H. Preparation and characterization of spray dried inhalable powders containing chitosan nanoparticles for pulmonary delivery of isoniazid. J. Microencapsul. 2011, 28, 605–613. [Google Scholar] [CrossRef]
- Oliveira, P.M.; Matos, B.N.; Pereira, P.A.T.; Gratieri, T.; Faccioli, L.H.; Cunha-Filho, M.S.S.; Gelfuso, G.M. Microparticles prepared with 50–190 kDa chitosan as promising non-toxic carriers for pulmonary delivery of isoniazid. Carbohydr. Polym. 2017, 174, 421–431. [Google Scholar] [CrossRef]
- Gelfuso, G.M.; Gratieri, T.; Simáo, P.S.; De Freitas, L.A.P.; Lopez, R.F.V. Chitosan microparticles for sustaining the topical delivery of minoxidil sulphate. J. Microencapsul. 2011, 28, 650–658. [Google Scholar] [CrossRef]
- Banik, N.; Hussain, A.; Ramteke, A.; Sharma, H.K.; Maji, T.K. Preparation and evaluation of the effect of particle size on the properties of chitosan-montmorillonite nanoparticles loaded with isoniazid. RSC Adv. 2012, 2, 10519–10528. [Google Scholar] [CrossRef]
- Devi, N.; Maji, T.K. Preparation and evaluation of gelatin/sodium carboxymethyl cellulose polyelectrolyte complex microparticles for controlled delivery of isoniazid. AAPS PharmSciTech 2009, 10, 1412–1419. [Google Scholar] [CrossRef]
- Dhawan, S.; Singla, A.K.; Sinha, V.R. Evaluation of mucoadhesive properties of chitosan microspheres prepared by different methods. AAPS PharmSciTech 2004, 5, 122–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, A.; Mehta, S.; Yadav, S.; Singh, S.K.; Grobler, A.; Goyal, A.K.; Mehta, A. Pulmonary delivery of antitubercular drugs using spray-dried lipid–polymer hybrid nanoparticles. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Palomino, J.C.; Martin, A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert Horsburgh, C.; Barry, C.E.; Lange, C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery systems. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chang, Y.X.; Hu, X.; Liu, C.Y.; Quan, L.H.; Liao, Y.H. Solid lipid nanoparticles for sustained pulmonary delivery of Yuxingcao essential oil: Preparation, characterization and in vivo evaluation. Int. J. Pharm. 2017, 516, 364–371. [Google Scholar] [CrossRef]
- Nemati, E.; Mokhtarzadeh, A.; Panahi-Azar, V.; Mohammadi, A.; Hamishehkar, H.; Mesgari-Abbasi, M.; Ezzati Nazhad Dolatabadi, J.; de la Guardia, M. Ethambutol-Loaded Solid Lipid Nanoparticles as Dry Powder Inhalable Formulation for Tuberculosis Therapy. AAPS PharmSciTech 2019, 20, 120. [Google Scholar] [CrossRef]
- Rahimpour, Y.; Hamishehkar, H. Lactose engineering for better performance in dry powder inhalers. Adv. Pharm. Bull. 2012, 2, 183–187. [Google Scholar]
- Maghsoodi, M. Physicomechanical properties of naproxen-loaded microparticles prepared from eudragit L100. AAPS PharmSciTech 2009, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Rabea, E.I.; Badawy, M.E.T.; Stevens, C.V.; Smagghe, G.; Steurbaut, W. Chitosan as antimicrobial agent: Applications and mode of action. Biomacromolecules 2003, 4, 1457–1465. [Google Scholar] [CrossRef]
- Ahmad, M.I.; Nakpheng, T.; Srichana, T. The safety of ethambutol dihydrochloride dry powder formulations containing chitosan for the possibility of treating lung tuberculosis. Inhal. Toxicol. 2014, 26, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Chew, N.Y.K.; Chan, H.K.; Raper, J.A. Limitation of determination of surface fractal dimension using N2 adsorption isotherms and modified Frenkel-Halsey-Hill theory. Langmuir 2003, 19, 2632–2638. [Google Scholar] [CrossRef]
- Maas, S.G.; Schaldach, G.; Littringer, E.M.; Mescher, A.; Griesser, U.J.; Braun, D.E.; Walzel, P.E.; Urbanetz, N.A. The impact of spray drying outlet temperature on the particle morphology of mannitol. Powder Technol. 2011, 213, 27–35. [Google Scholar] [CrossRef]
- Geldart, D.; Abdullah, E.C.; Hassanpour, A.; Nwoke, L.C.; Wouters, I. Characterization of powder flowability using measurement of angle of repose. China Particuol. 2006, 4, 104–107. [Google Scholar] [CrossRef]
- Marsac, P.J.; Konno, H.; Taylor, L.S. A comparison of the physical stability of amorphous felodipine and nifedipine systems. Pharm. Res. 2006, 23, 2306–2316. [Google Scholar] [CrossRef]
- Ahmad, M.I.; Ungphaiboon, S.; Srichana, T. The development of dimple-shaped chitosan carrier for ethambutol dihydrochloride dry powder inhaler. Drug Dev. Ind. Pharm. 2015, 41, 791–800. [Google Scholar] [CrossRef]
- Alfarisi, O.; Alghamdi, W.A.; Al-Shaer, M.H.; Dooley, K.E.; Peloquin, C.A. Rifampin vs. rifapentine: What is the preferred rifamycin for tuberculosis? Expert Rev. Clin. Pharmacol. 2017, 10, 1027–1036. [Google Scholar] [CrossRef]
- Khadka, P.; Hill, P.C.; Zhang, B.; Katare, R.; Dummer, J.; Das, S.C. A study on polymorphic forms of rifampicin for inhaled high dose delivery in tuberculosis treatment. Int. J. Pharm. 2020, 587, 119602. [Google Scholar] [CrossRef]
- Gervelas, C.; Serandour, A.L.; Geiger, S.; Grillon, G.; Fritsch, P.; Taulelle, C.; Le Gall, B.; Benech, H.; Deverre, J.R.; Fattal, E.; et al. Direct lung delivery of a dry powder formulation of DTPA with improved aerosolization properties: Effect on lung and systemic decorporation of plutonium. J. Control. Release 2007, 118, 78–86. [Google Scholar] [CrossRef]
- El-Gendy, N.; Berkland, C. Combination chemotherapeutic dry powder aerosols via controlled nanoparticle agglomeration. Pharm. Res. 2009, 26, 1752–1763. [Google Scholar] [CrossRef] [Green Version]
- Mehanna, M.M.; Mohyeldin, S.M.; Elgindy, N.A. Rifampicin-carbohydrate spray-dried nanocomposite: A futuristic multiparticulate platform for pulmonary delivery. Int. J. Nanomed. 2019, 14, 9089–9112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, A.K.; Rajaram, M.V.S.; Schlesinger, L.S. Exploitation of the Macrophage Mannose Receptor (CD206) in Infectious Disease Diagnostics and Therapeutics. J. Cytol. Mol. Biol. 2014, 1, 1000003. [Google Scholar] [PubMed]
- Maretti, E.; Costantino, L.; Rustichelli, C.; Leo, E.; Croce, M.A.; Buttini, F.; Truzzi, E.; Iannuccelli, V. Surface engineering of Solid Lipid Nanoparticle assemblies by methyl α-D-mannopyranoside for the active targeting to macrophages in anti-tuberculosis inhalation therapy. Int. J. Pharm. 2017, 528, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Helal, H.M.; Mortada, S.M.; Sallam, M.A. Paliperidone-Loaded Nanolipomer System for Sustained Delivery and Enhanced Intestinal Permeation: Superiority to Polymeric and Solid Lipid Nanoparticles. AAPS PharmSciTech 2017, 18, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Mulla, J.A.S.; Mabrouk, M.; Choonara, Y.E.; Kumar, P.; Chejara, D.R.; du Toit, L.C.; Pillay, V. Development of respirable rifampicin-loaded nano-lipomer composites by microemulsion-spray drying for pulmonary delivery. J. Drug Deliv. Sci. Technol. 2017, 41, 13–19. [Google Scholar] [CrossRef]
- Meenach, S.A.; Anderson, K.W.; Zach Hilt, J.; McGarry, R.C.; Mansour, H.M. Characterization and aerosol dispersion performance of advanced spray-dried chemotherapeutic PEGylated phospholipid particles for dry powder inhalation delivery in lung cancer. Eur. J. Pharm. Sci. 2013, 49, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.; Koduri, L.V.S.K.; Dhawale, V.; Bhatt, T.D.; Kumar, R.; Grover, V.; Tikoo, K.; Suresh, S. Potential of aerosolized rifampicin lipospheres for modulation of pulmonary pharmacokinetics and bio-distribution. Int. J. Pharm. 2015, 495, 627–632. [Google Scholar] [CrossRef]
- Changsan, N.; Chan, H.K.; Separovic, F.; Srichana, T. Physicochemical characterization and stability of rifampicin liposome dry powder formulations for inhalation. J. Pharm. Sci. 2009, 98, 628–639. [Google Scholar] [CrossRef]
- Changsan, N.; Nilkaeo, A.; Pungrassami, P.; Srichana, T. Monitoring safety of liposomes containing rifampicin on respiratory cell lines and in vitro efficacy against Mycobacterium bovis in alveolar macrophages. J. Drug Target. 2009, 17, 751–762. [Google Scholar] [CrossRef]
- Takeuchi, I.; Tetsuka, Y.; Nii, T.; Shinogase, M.; Makino, K. Inhalable nanocomposite particles using amino acids with improved drug content and humidity resistance. Colloids Surf. A Physicochem. Eng. Asp. 2017, 529, 387–393. [Google Scholar] [CrossRef]
- Takeuchi, I.; Taniguchi, Y.; Tamura, Y.; Ochiai, K.; Makino, K. Effects of L-leucine on PLGA microparticles for pulmonary administration prepared using spray drying: Fine particle fraction and phagocytotic ratio of alveolar macrophages. Colloids Surf. A Physicochem. Eng. Asp. 2018, 537, 411–417. [Google Scholar] [CrossRef]
- Carneiro, S.P.; Carvalho, K.V.; de Oliveira Aguiar Soares, R.D.; Carneiro, C.M.; de Andrade, M.H.G.; Duarte, R.S.; dos Santos, O.D.H. Functionalized rifampicin-loaded nanostructured lipid carriers enhance macrophages uptake and antimycobacterial activity. Colloids Surf. B Biointerfaces 2019, 175, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Gorain, B.; Choudhury, H.; Pandey, M.; Kesharwani, P. Paclitaxel loaded vitamin E-TPGS nanoparticles for cancer therapy. Mater. Sci. Eng. C 2018, 91, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Gorain, B.; Choudhury, H.; Pandey, M.; Nair, A.B.; Iqbal Mohd Amin, M.C.; Molugulu, N.; Deb, P.K.; Tripathi, P.K.; Khurana, S.; Shukla, R.; et al. Dendrimer-Based Nanocarriers in Lung Cancer Therapy. In Nanotechnology-Based Targeted Drug Delivery Systems for Lung Cancer; Academic Press: Cambridge, MA, USA, 2019; pp. 161–192. [Google Scholar]
- Agrawal, A.K.; Gupta, C.M. Tuftsin-bearing liposomes in treatment of macrophage-based infections. Adv. Drug Deliv. Rev. 2000, 41, 135–146. [Google Scholar] [CrossRef]
- Bar-Shavit, Z.; Stabinsky, Y.; Fridkin, M.; Goldman, R. Tuftsin-macrophage interaction: Specific binding and augmentation of phagocytosis. J. Cell. Physiol. 1979, 100, 55–62. [Google Scholar] [CrossRef]
- Ko, J.A.; Park, H.J.; Hwang, S.J.; Park, J.B.; Lee, J.S. Preparation and characterization of chitosan microparticles intended for controlled drug delivery. Int. J. Pharm. 2002, 249, 165–174. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Dünnhaupt, S. Chitosan-based drug delivery systems. Eur. J. Pharm. Biopharm. 2012, 81, 463–469. [Google Scholar] [CrossRef]
- Rawal, T.; Parmar, R.; Tyagi, R.K.; Butani, S. Rifampicin loaded chitosan nanoparticle dry powder presents: An improved therapeutic approach for alveolar tuberculosis. Colloids Surf. B Biointerfaces 2017, 154, 321–330. [Google Scholar] [CrossRef]
- Berkenfeld, K.; McConville, J.T.; Lamprecht, A. Inhalable formulations of rifampicin by spray drying of supersaturated aqueous solutions. Eur. J. Pharm. Biopharm. 2020, 153, 14–22. [Google Scholar] [CrossRef]
- Desai, K.G.H.; Park, H.J. Preparation and characterization of drug-loaded chitosan-tripolyphosphate microspheres by spray drying. Drug Dev. Res. 2005, 64, 114–128. [Google Scholar] [CrossRef]
- Pai, R.V.; Jain, R.R.; Bannalikar, A.S.; Menon, M.D. Development and Evaluation of Chitosan Microparticles Based Dry Powder Inhalation Formulations of Rifampicin and Rifabutin. J. Aerosol Med. Pulm. Drug Deliv. 2016, 29, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.; Eleutério, C.V.; Gonalves, L.M.D.; Cruz, M.E.M.; Almeida, A.J. Lipid nanoparticles containing oryzalin for the treatment of leishmaniasis. Eur. J. Pharm. Sci. 2012, 45, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Mancini, G.; Lopes, R.M.; Clemente, P.; Raposo, S.; Gonçalves, L.M.D.; Bica, A.; Ribeiro, H.M.; Almeida, A.J. Lecithin and parabens play a crucial role in tripalmitin-based lipid nanoparticle stabilization throughout moist heat sterilization and freeze-drying. Eur. J. Lipid Sci. Technol. 2015, 117, 1947–1959. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Faria, V.; Gonçalves, L.M.D.; Taboada, P.; Remuñán-López, C.; Almeida, A.J. Rifabutin-loaded solid lipid nanoparticles for inhaled antitubercular therapy: Physicochemical and in vitro studies. Int. J. Pharm. 2016, 497, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Lau, C.M.; Thomas, S.N.; Gray Jerome, W.; Maron, D.J.; Dickerson, J.H.; Hubbell, J.A.; Giorgio, T.D. Size- and charge-dependent non-specific uptake of PEGylated nanoparticles by macrophages. Int. J. Nanomed. 2012, 7, 799–813. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, P.; Hickey, A.J. Respirable PLGA microspheres containing rifampicin for the treatment of tuberculosis: Manufacture and characterization. Pharm. Res. 2000, 17, 955–961. [Google Scholar] [CrossRef]
- Zhou, Q.T.; Morton, D.A.V.; Yu, H.H.; Jacob, J.; Wang, J.; Li, J.; Chan, H.K. Colistin powders with high aerosolisation efficiency for respiratory infection: Preparation and in vitro evaluation. J. Pharm. Sci. 2013, 102, 3736–3747. [Google Scholar] [CrossRef]
- Müller, R.H.; Rühl, D.; Lück, M.; Paulke, B.R. Influence of fluorescent labelling of polystyrene particles on phagocytic uptake, surface hydrophobicity, and plasma protein adsorption. Pharm. Res. 1997, 14, 18–24. [Google Scholar] [CrossRef]
- Parumasivam, T.; Leung, S.S.Y.; Quan, D.H.; Triccas, J.A.; Britton, W.J.; Chan, H.K. Rifapentine-loaded PLGA microparticles for tuberculosis inhaled therapy: Preparation and in vitro aerosol characterization. Eur. J. Pharm. Sci. 2016, 88, 1–11. [Google Scholar] [CrossRef]
- Rojanarat, W.; Nakpheng, T.; Thawithong, E.; Yanyium, N.; Srichana, T. Levofloxacin-proliposomes: Opportunities for use in lung tuberculosis. Pharmaceutics 2012, 4, 385–412. [Google Scholar] [CrossRef]
- Kanchan, V.; Panda, A.K. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials 2007, 28, 5344–5357. [Google Scholar] [CrossRef] [PubMed]
- Patil-Gadhe, A.; Pokharkar, V. Single step spray drying method to develop proliposomes for inhalation: A systematic study based on quality by design approach. Pulm. Pharmacol. Ther. 2014, 27, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Coombs, P.J.; Taylor, M.E.; Drickamer, K. Two categories of mammalian galactose-binding receptors distinguished by glycan array profiling. Glycobiology 2006, 16, 1c–7c. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez-Santoscoy, A.V.; Roychoudhury, R.; Pohl, N.L.B.; Wannemuehler, M.J.; Narasimhan, B.; Ramer-Tait, A.E. Tailoring the immune response by targeting C-type lectin receptors on alveolar macrophages using “pathogen-like” amphiphilic polyanhydride nanoparticles. Biomaterials 2012, 33, 4762–4772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- East, L.; Isacke, C.M. The mannose receptor family. Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 364–386. [Google Scholar] [CrossRef]
- Rodrigues, S.; Alves, A.D.; Cavaco, J.S.; Pontes, J.F.; Guerreiro, F.; da Costa, A.M.R.; Buttini, F.; Grenha, A. Dual antibiotherapy of tuberculosis mediated by inhalable locust bean gum microparticles. Int. J. Pharm. 2017, 529, 433–441. [Google Scholar] [CrossRef]
- Srichana, T.; Ratanajamit, C.; Juthong, S.; Suwandecha, T.; Laohapojanart, N.; Pungrassami, P.; Padmavathi, A.R. Evaluation of proinflammatory cytokines and adverse events in healthy volunteers upon inhalation of antituberculosis drugs. Biol. Pharm. Bull. 2016, 39, 1815–1822. [Google Scholar] [CrossRef] [Green Version]
- Tse, J.Y.; Kadota, K.; Hirata, Y.; Taniguchi, M.; Uchiyama, H.; Tozuka, Y. Characterization of matrix embedded formulations for combination spray-dried particles comprising pyrazinamide and rifampicin. J. Drug Deliv. Sci. Technol. 2018, 48, 137–144. [Google Scholar] [CrossRef]
- Sou, T.; Kaminskas, L.M.; Nguyen, T.H.; Carlberg, R.; McIntosh, M.P.; Morton, D.A.V. The effect of amino acid excipients on morphology and solid-state properties of multi-component spray-dried formulations for pulmonary delivery of biomacromolecules. Eur. J. Pharm. Biopharm. 2013, 83, 234–243. [Google Scholar] [CrossRef]
- Allahham, A.; Stewart, P.J.; Das, S.C. Improving the de-agglomeration and dissolution of a poorly water soluble drug by decreasing the agglomerate strength of the cohesive powder. Int. J. Pharm. 2013, 457, 101–109. [Google Scholar] [CrossRef]
- Zhou, Q.T.; Loh, Z.H.; Yu, J.; Sun, S.P.; Gengenbach, T.; Denman, J.A.; Li, J.; Chan, H.K. How Much Surface Coating of Hydrophobic Azithromycin Is Sufficient to Prevent Moisture-Induced Decrease in Aerosolisation of Hygroscopic Amorphous Colistin Powder? AAPS J. 2016, 18, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Momin, M.A.M.; Tucker, I.G.; Doyle, C.S.; Denman, J.A.; Sinha, S.; Das, S.C. Co-spray drying of hygroscopic kanamycin with the hydrophobic drug rifampicin to improve the aerosolization of kanamycin powder for treating respiratory infections. Int. J. Pharm. 2018, 541, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Necas, J.; Bartosikova, L.; Brauner, P.; Kolar, J. Hyaluronic acid (hyaluronan): A review. Vet. Med. 2008, 53, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.I.; Gomes, E.C.L.; Soares, C.D.V.; Cunha, A.F.; Oliveira, M.A. Thermal Analysis Applied to Verapamil Hydrochloride Characterization in Pharmaceutical Formulations. Molecules 2010, 15, 2439–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluin, O.S.; Busscher, H.J.; Neut, D.; van der Mei, H.C. Poly(trimethylene carbonate) as a carrier for rifampicin and vancomycin to target therapy-recalcitrant staphylococcal biofilms. J. Orthop. Res. 2016, 34, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Rossi, I.; Buttini, F.; Sonvico, F.; Affaticati, F.; Martinelli, F.; Annunziato, G.; Machado, D.; Viveiros, M.; Pieroni, M.; Bettini, R. Sodium hyaluronate nanocomposite respirable microparticles to tackle antibiotic resistance with potential application in treatment of mycobacterial pulmonary infections. Pharmaceutics 2019, 11, 203. [Google Scholar] [CrossRef] [Green Version]
- Momin, M.; Rangnekar, B.; Larson, I.; Sinha, S.; Das, S.C. Dry powder formulation combining bedaquiline with pyrazinamide for latent and drug-resistant tuberculosis. Adv. Powder Technol. 2019, 30, 2473–2482. [Google Scholar] [CrossRef]
- Adler, M.; Unger, M.; Lee, G. Surface composition of spray-dried particles of bovine serum albumin/trehalose/surfactant. Pharm. Res. 2000, 17, 863–870. [Google Scholar] [CrossRef]
- Kaialy, W.; Nokhodchi, A. Engineered Mannitol Ternary Additives Improve Dispersion of Lactose–Salbutamol Sulphate Dry Powder Inhalations. AAPS J. 2013, 15, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Geller, D.E.; Weers, J.; Heuerding, S. Development of an inhaled dry-powder formulation of tobramycin using pulmosphereTM technology. J. Aerosol Med. Pulm. Drug Deliv. 2011, 24, 175–182. [Google Scholar] [CrossRef]
- Vanbever, R.; Mintzes, J.D.; Wang, J.; Nice, J.; Chen, D.; Batycky, R.; Langer, R.; Edwards, D.A. Formulation and physical characterization of large porous particles for inhalation. Pharm. Res. 1999, 16, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Glover, W.; Chan, H.K.; Eberl, S.; Daviskas, E.; Verschuer, J. Effect of particle size of dry powder mannitol on the lung deposition in healthy volunteers. Int. J. Pharm. 2008, 349, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Rangnekar, B.; Momin, M.A.M.; Eedara, B.B.; Sinha, S.; Das, S.C. Bedaquiline containing triple combination powder for inhalation to treat drug-resistant tuberculosis. Int. J. Pharm. 2019, 570, 118689. [Google Scholar] [CrossRef] [PubMed]
- Luca, S.; Mihaescu, T. History of BCG Vaccine. Maedica 2013, 8, 58. [Google Scholar]
- Thakur, A.; Ingvarsson, P.T.; Schmidt, S.T.; Rose, F.; Andersen, P.; Christensen, D.; Foged, C. Immunological and physical evaluation of the multistage tuberculosis subunit vaccine candidate H56/CAF01 formulated as a spray-dried powder. Vaccine 2018, 36, 3331–3339. [Google Scholar] [CrossRef]
- World Health Organization. Report on BCG Vaccine Use for Protection against Mycobacterial Infections Including Tuberculosis, Leprosy, and Other Nontuberculous Mycobacteria (NTM) Infections; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Yoshida, M.; Babensee, J.E. Poly(lactic-co-glycolic acid) enhances maturation of human monocyte-derived dendritic cells. J. Biomed. Mater. Res. Part A 2004, 71, 45–54. [Google Scholar] [CrossRef]
- Yoshida, M.; Babensee, J.E. Differential effects of agarose and poly(lactic-co-glycolic acid) on dendritic cell maturation. J. Biomed. Mater. Res. Part A 2006, 79A, 393–408. [Google Scholar] [CrossRef]
- Yoshida, M.; Mata, J.; Babensee, J.E. Effect of poly(lactic-co-glycolic acid) contact on maturation of murine bone marrow-derived dendritic cells. J. Biomed. Mater. Res. Part A 2007, 80, 7–12. [Google Scholar] [CrossRef]
- Nagpal, P.S.; Kesarwani, A.; Sahu, P.; Upadhyay, P. Aerosol immunization by alginate coated mycobacterium (BCG/MIP) particles provide enhanced immune response and protective efficacy than aerosol of plain mycobacterium against M.tb. H37Rv infection in mice. BMC Infect. Dis. 2019, 19, 568. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Abel, B.; Tameris, M.; Mansoor, N.; Gelderbloem, S.; Hughes, J.; Abrahams, D.; Makhethe, L.; Erasmus, M.; De Kock, M.; Van Der Merwe, L.; et al. The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4+and CD8+ T cells in adults. Am. J. Respir. Crit. Care Med. 2010, 181, 1407–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanahan, E.R.; Pinto, R.; Triccas, J.A.; Britton, W.J.; West, N.P. Cutinase-like protein-6 of Mycobacterium tuberculosis is recognised in tuberculosis patients and protects mice against pulmonary infection as a single and fusion protein vaccine. Vaccine 2010, 28, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.G.Y.; Duke, C.C.; Ong, H.X.; Chan, J.C.Y.; Tyne, A.S.; Chan, H.K.; Britton, W.J.; Young, P.M.; Traini, D. A novel inhalable form of rifapentine. J. Pharm. Sci. 2014, 103, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Muneer, S.; Masood, Z.; Butt, S.; Anjum, S.; Zainab, H.; Anwar, N.; Ahmad, N. Proliposomes as Pharmaceutical Drug Delivery System: A Brief Review. J. Nanomed. Nanotechnol. 2017, 8, 448. [Google Scholar]
- Payne, N.I.; Timmins, P.; Ambrose, C.V.; Ward, M.D.; Ridgway, F. Proliposomes: A novel solution to an old problem. J. Pharm. Sci. 1986, 75, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Shaji, J.; Bhatia, V. Proliposomes: A Brief Overview of Novel Delivery System. Int. J. Pharm. Bio Sci. 2013, 4, 150–160. [Google Scholar]
- Mehta, P. Dry Powder Inhalers: A Focus on Advancements in Novel Drug Delivery Systems. J. Drug Deliv. 2016, 2016, 8290963. [Google Scholar] [CrossRef] [Green Version]
- Omer, H.K.; Hussein, N.R.; Ferraz, A.; Najlah, M.; Ahmed, W.; Taylor, K.M.G.; Elhissi, A.M.A. Spray-Dried Proliposome Microparticles for High-Performance Aerosol Delivery Using a Monodose Powder Inhaler. AAPS PharmSciTech 2018, 19, 2434–2448. [Google Scholar] [CrossRef]
- Patil-Gadhe, A.A.; Kyadarkunte, A.Y.; Pereira, M.; Jejurikar, G.; Patole, M.S.; Risbud, A.; Pokharkar, V.B. Rifapentine-proliposomes for inhalation: In vitro and in vivo toxicity. Toxicol. Int. 2014, 21, 275–282. [Google Scholar]
- Singh, N.; Kushwaha, P.; Ahmad, U.; Abdullah, M. Proliposomes: An Approach for the Development of Stable Liposome. ARS Pharm. 2019, 60, 231–240. [Google Scholar]
- Mitchison, D.A.; Fourie, P.B. The near future: Improving the activity of rifamycins and pyrazinamide. Tuberculosis 2010, 90, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S. Proliposome and Prosurfactosome Formulations for Pulmonary Drug Delivery. Ph.D. Thesis, University of Central Lancashire, Preston, UK, 2015. [Google Scholar]
- Huang, X.; Caddell, R.; Yu, B. Ultrasound-enhanced Microfluidic Synthesis of Liposomes. Anticancer Res. 2010, 30, 463–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.B.; Scarpa, M.V.; Silva, G.V.J.; Rodrigues, D.C.; Santilli, C.V.; Oliveira, A.G. Studies on the encapsulation of diclofenac in small unilamellar liposomes of soya phosphatidylcholine. Colloids Surf. B Biointerfaces 2004, 39, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, A.S.; Kabadi, M.; Gerety, B.; Hickey, A.J.; Fourie, P.B.; Nardell, E. Phase I, single-dose, dose-escalating study of inhaled dry powder capreomycin: A new approach to therapy of drug-resistant tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 2613–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PureIMS Initiates Phase 1 Clinical Trial to Evaluate Dry Powder Inhaler Formulation of Amikacin for Early Eradication Treatment of Tuberculosis—Pure IMS. Available online: https://pureims.com/2020/07/01/pureims-initiates-phase-1-clinical-trial-to-evaluate-dry-powder-inhaler-formulation-of-amikacin-for-early-eradication-treatment-of-tuberculosis/ (accessed on 10 October 2020).
- Pharmacokinetic Evaluation and Local Tolerability of Dry Powder Amikacin Via the CyclopsTM—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04249531 (accessed on 10 October 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
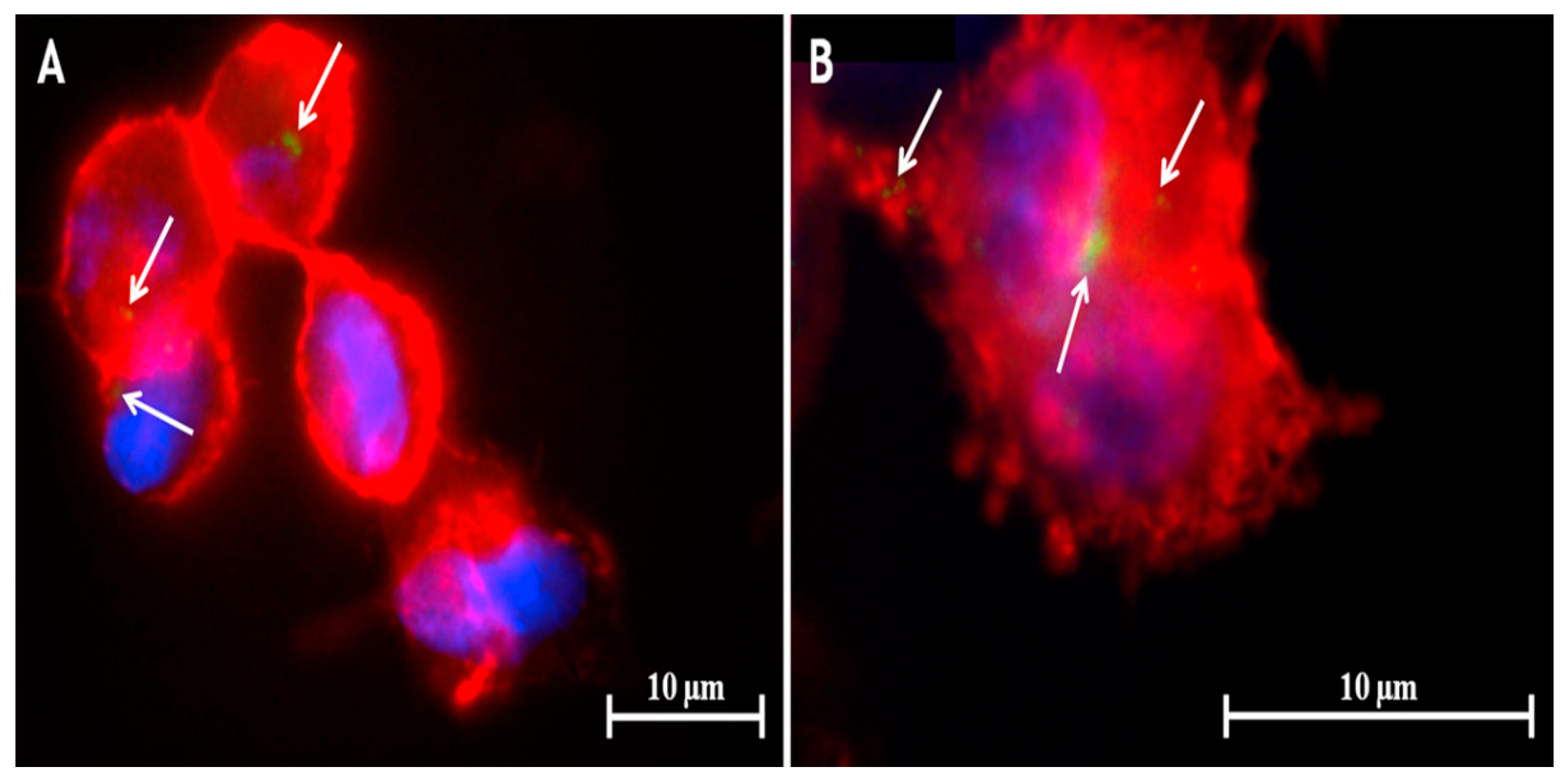{kind=link}
{kind=link}
| Objective | Route of Administration | Type of Formulation | Dosage Form | Method of Preparation | Excipient Used | Cell Line/Animal Model | Outcome | Source |
|---|---|---|---|---|---|---|---|---|
| To investigate the effect of adding l-leucine and using an ethanolic solvent on the physicochemical properties and aerodynamic behaviours of nano spray-dried PZA-l-leucine powders. | Pulmonary | Dry powder inhaler | Nanocarriers | Spray drying method |
| NA |
| [66] |
| To optimize a formulation of PZA as large porous particles for pulmonary delivery by adjusting spray drying parameters | Pulmonary | Dry powder inhaler | Microcarriers | Spray drying method |
| Male Sprague Dawley rats |
| [67] |
| To develop and optimize phospholipid-based PZA spray-dried inhaler powders. | Pulmonary | Dry powder inhaler | Phospholipid-based microparticles | Spray drying method |
| NA |
| [72] |
| To develop PZA-proliposomes in dry powder aerosol form for delivering drugs to AMs. | Pulmonary | Dry powder inhaler | Proliposomes | Spray drying method |
|
|
| [73] |
| To develop proliposomes powder containing INH in a dry powder aerosol form | Pulmonary | Dry powder inhaler | Proliposomes | Spray drying Method |
|
|
| [50] |
| To prepare and characterize INH-loaded chitosan microspheres for pulmonary delivery | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying Method |
| NA |
| [76] |
| To prepare and characterize spray dried inhalable powders containing chitosan nanoparticles for pulmonary delivery of INH. | Pulmonary | Dry powder inhaler | Polymeric nanoparticles | Spray drying Method |
| NA |
| [78] |
| To develop INH-loaded microparticles with 50–190 kDa chitosan as promising nontoxic carriers for pulmonary delivery. | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying Method |
|
|
| [79] |
| To develop pulmonary delivery of antitubercular drugs using spray-dried lipid-polymer hybrid nanoparticles. | Pulmonary | Dry powder inhaler | Lipid-polymer hybrid nanoparticles | Spray drying Method |
|
|
| [84] |
| To assess pulmonary DPI using EMB-loaded SLNs for TB treatment | Pulmonary | Dry powder inhaler | Solid lipid nanoparticles | Homogenization and ultrasonication |
|
|
| [89] |
| To investigate the acceptance of EDH containing chitosan in the form of dry powder formulation for further in vivo studies to target AMs for the treatment of TB | Pulmonary | Dry powder inhaler | Polymeric nanoparticles | Nanospray drying method |
|
|
| [93] |
| A dimple-shaped chitosan carrier was developed to deliver EDH in the form of DPI to the infected lungs | Pulmonary | Dry powder inhaler | Polymeric microcarriers with nanosize drug | Spray drying method |
| NA |
| [98] |
| To investigate the polymorphic forms of RIF for inhaled high dose delivery in TB treatment | Pulmonary | Dry powder inhaler | NA | Spray drying method and crystallization method | NA | NA |
| [100] |
| To formulate RIF loaded carbohydrate spray dried nanocomposite in DPI for TB | Pulmonary | Dry powder inhaler | Microcarriers with nanosize drug | Antisolvent precipitation-ultrasonication method, spray drying method |
|
|
| [103] |
| ||||||||
| To develop RIF loaded mannosylated solid lipid nanoparticle for the active targeting of macrophage in TB therapy | Pulmonary | Dry powder inhaler | Solid lipid nanoparticles | Melt emulsifying technique, freeze drying method |
|
|
| [105] |
| To develop respirable RIF-loaded nano-lipomer composites by microemulsion-spray drying for pulmonary delivery | Pulmonary | Dry powder inhaler | Nanolipomers | Microemulsion-spray drying |
| NA |
| [107] |
| ||||||||
| To formulate RIF loaded phospholipid lipospheres carrier for pulmonary application | Pulmonary | Dry powder inhaler | Phospholipid-based lipospheres | Spray drying method |
|
|
| [109] |
| To improve RIF content in primary nanoparticles and to investigate arginine and leucine for the preparation of nanocomposite particles with low hygroscopicity | Pulmonary | Dry powder inhaler | Polymeric nanoparticles | Emulsion solvent evaporation method |
| NA |
| [112] |
| To study the effect of leucine on the FPF and phagocytotic ratio of AM of RIF loaded PLGA microparticles | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying method, O/W emulsion, lyophilization |
|
|
| [113] |
| To synthesis RIF NLC functionalized with tuftsin-modified peptide to improve TB treatment | Pulmonary | Dry powder inhaler | Nanostructured lipid carriers | Microemulsion technique |
|
|
| [114] |
| To develop RIF-loaded chitosan nanoparticle dry powder to improve therapeutic approach for alveolar TB. | Pulmonary | Dry Powder Inhaler | Polymeric Nanoparticles | Freeze drying method |
|
|
| [121] |
| To develop inhalable formulations of RIF by using supersaturated aqueous solutions. | Pulmonary | Dry powder inhaler | Microparticles | Spray drying Method | NA | NA |
| [122] |
| To develop and evaluate the chitosan microparticles based DPI of RFB. | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying Method |
|
|
| [124] |
| To develop RFB-loaded SLNs for inhaled antitubercular therapy | Pulmonary | Dry powder inhaler | Solid lipid nanoparticles | Lyophilization |
|
|
| [127] |
| To develop RFP-loaded PLGA microparticles for TB inhaled therapy. | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying MethodorO/W single emulsion solvent evaporation |
|
|
| [132] |
| To develop RFP-loaded proliposomes DPI formulation to treat TB | Pulmonary | Dry Powder Inhaler | Proliposomes | Spray drying Method |
|
|
| [135] |
| ||||||||
| To compare inhalable crystalline and amorphous dry powder form of RFP by in vitro test | Pulmonary | Dry powder inhaler | NA | Spray drying method |
|
|
| [167] |
| To formulate dual antibiotherapy of TB by inhalable LBG microparticles. | Pulmonary | Dry powder inhaler | Microcarriers | Spray drying Method |
|
|
| [139] |
| To characterize matrix embedded formulation for combination spray-dried particles comprising PZA and RIF. | Pulmonary | Dry powder inhaler | Polymeric microparticles | Spray drying Method | HBCD |
|
| [141] |
| To investigate the effects of co-spray drying hygroscopic KNM with hydrophobic RIF in improving the aerosolization of KNM powder for treating respiratory infections. | Pulmonary | Dry Powder Inhaler | NA | Spray drying Method | NA |
|
| [145] |
| To develop RIF, INH and VPM loaded sodium hyaluronate nanocomposite respirable microparticles to tackle antibiotic resistance mycobacterial pulmonary infections. | Pulmonary | Dry powder inhaler | Microcarriers with nanosize drug | Spray drying Method | Sodium Hyaluronate | HMDM |
| [149] |
| To develop dry powder formulation combining BDQ with PZA for latent and drug-resistant TB. | Pulmonary | Dry Powder Inhaler | Microcarriers | Spray drying Method |
|
|
| [150] |
| To develop triple combination dry powder formulation combining BDQ, PZA and MX for the treatment of drug-resistant TB. | Pulmonary | Dry powder inhaler | Microcarriers | Spray drying method | l-leucine |
|
| [156] |
| To compare CAF01 co-spray-dried with H56, reconstitution to liquid formulation with the non-spray-dried formulation to induce systemic Th1, Th17 and humoral responses | Pulmonary | Dry powder vaccine | Liposomes | Spray drying method |
|
|
| [158] |
| To encapsulate MIP and BCG into inhalable alginate particles as DPA to evaluate their immunogenic and protective efficacy in animal model of TB | Pulmonary | Dry powder aerosol | Microcarriers | Spray drying method |
|
|
| [163] |
| To investigate inhalable dry powder AERAS-402 vaccine in term of physical stability and aerodynamic properties | Pulmonary | Dry powder inhaler | Microcarriers | Spray drying method |
| NA |
| [165] |
| To formulate inhalable powder vaccine by conjugating Culps1-6 fusion and MPT83 to lipokel for pulmonary delivery | Pulmonary | Dry powder inhaler | NA | Spray drying method | Mannitol | C57BL/6 mice |
| [166] |
| ||||||||
| To formulate LEV-proliposomes DPI using porous mannitol to enhance drug delivery to the lungs | Pulmonary | Dry powder inhaler | Proliposomes | Spray drying method |
|
|
| [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Z.M.; Lai, G.P.; Pandey, M.; Srichana, T.; Pichika, M.R.; Gorain, B.; Bhattamishra, S.K.; Choudhury, H. Novel Approaches for the Treatment of Pulmonary Tuberculosis. Pharmaceutics 2020, 12, 1196. https://doi.org/10.3390/pharmaceutics12121196
Tan ZM, Lai GP, Pandey M, Srichana T, Pichika MR, Gorain B, Bhattamishra SK, Choudhury H. Novel Approaches for the Treatment of Pulmonary Tuberculosis. Pharmaceutics. 2020; 12(12):1196. https://doi.org/10.3390/pharmaceutics12121196
Chicago/Turabian StyleTan, Zhi Ming, Gui Ping Lai, Manisha Pandey, Teerapol Srichana, Mallikarjuna Rao Pichika, Bapi Gorain, Subrat Kumar Bhattamishra, and Hira Choudhury. 2020. "Novel Approaches for the Treatment of Pulmonary Tuberculosis" Pharmaceutics 12, no. 12: 1196. https://doi.org/10.3390/pharmaceutics12121196