
Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence
by

Tahrir Alenezi
1,2,3, 
Ying Fu
1,
Bilal Alrubaye
1,
Thamer Alanazi
1,2,
Ayidh Almansour
1,2,
Hong Wang
1,2 and
Xiaolun Sun
1,2,* 1
Center of Excellence for Poultry Science, University of Arkansas, Fayetteville, AR 72701, USA
2
Cell and Molecular Biology, University of Arkansas, Fayetteville, AR 72701, USA
3
College of Medical Applied Sciences, The Northern Border University, Arar 91431, Saudi Arabia
*
Author to whom correspondence should be addressed.
Pathogens 2023, 12(10), 1202; https://doi.org/10.3390/pathogens12101202
Submission received: 24 August 2023
/
Revised: 19 September 2023
/
Accepted: 26 September 2023
/
Published: 28 September 2023
(This article belongs to the Special Issue Clostridium Pathogenesis: Virulence, Host Responses, Microbiome, and Interventions)
Abstract
:Clostridium perfringens is a versatile pathogen, inducing diseases in the skin, intestine (such as chicken necrotic enteritis (NE)), and other organs. The classical sign of NE is the foul smell gas in the ballooned small intestine. We hypothesized that deoxycholic acid (DCA) reduced NE by inhibiting C. perfringens virulence signaling pathways. To evaluate the hypothesis, C. perfringens strains CP1 and wild-type (WT) HN13 and its mutants were cultured with different bile acids, including DCA and isoallolithocholic acid (isoalloLCA). Growth, hydrogen sulfide (H2S) production, and virulence gene expression were measured. Notably, isoalloLCA was more potent in reducing growth, H2S production, and virulence gene expression in CP1 and WT HN13 compared to DCA, while other bile acids were less potent compared to DCA. Interestingly, there was a slightly different impact between DCA and isoalloLCA on the growth, H2S production, and virulence gene expression in the three HN13 mutants, suggesting possibly different signaling pathways modulated by the two bile acids. In conclusion, DCA and isoalloLCA reduced C. perfringens virulence by transcriptionally modulating the pathogen signaling pathways. The findings could be used to design new strategies to prevent and treat C. perfringens-induced diseases.
1. Introduction
Clostridium perfringens is a spore-forming, anaerobic, and Gram-positive bacterium and is responsible for a variety of acute diseases, such as chicken necrotic enteritis (NE), foodborne enteritis, and myonecrosis [1]. It is estimated that NE causes a USD 6 billion loss to the poultry industry worldwide every year [2]. The increased NE incidence mainly resulted from the ongoing restriction of prophylactic antimicrobial usage in poultry production, while no effective alternatives are available [3]. Infection of coccidia of Eimeria (e.g., E. maxima and acervuline) is one of the prevalent predisposed factors in poultry farms [4]. Birds with severe clinical NE are shown with severe diarrhea, foul-smell and distended small intestine, mortality, anorexia, and reduced body weight gain and feed efficiency [5].
C. perfringens strains are classified into seven toxin types (A to G) based on their ability to produce the various combinations of six major toxins, namely alpha (CPA), beta (CPB), epsilon (ETX), iota (ITX), enterotoxin (CPE), and enteritis B-like toxin (NetB) [1,6,7]. Various combinations of the toxins cause necrosis of muscle, subcutaneous, and mucosal tissue in addition to the production of toxic gas of hydrogen sulfide (H2S) [1]. Most C. perfringens isolates produce α-toxin (CPA or PLC) encoded by the plc (or cpa) gene [8]. In the progression of gas gangrene, CPA penetrates the cell membrane with holes and activates arachidonic acid cascade and protein kinase C, resulting in cellular dysfunction [8,9]. In addition, CPA suppresses the immune response by restraining leukocytes entering the infected tissues [10] and reduces the blood supply to infected sites by triggering vasoconstriction, thrombosis, and platelet aggregation [8]. The gene expression of plc, perfringolysin O (pfoA), κ-toxin (colA), alpha-clostripain(ccp), and some extracellular enzymes is regulated by the VirS/VirR system [11,12], small regulatory RNA molecules (sRNAs: vrr, virT, and virU), or the RevR orphan response regulator [13]. virT gene expression is negatively correlated to pfoA and ccp, whereas the virU gene product positively affects the expression of pfoA, virT, ccp, and vrr [14].
Another toxin of C. perfringens enterotoxin (CPE) encoded by the cpe gene is a 35.5 kDa polypeptide [15] and is associated with chicken NE [16]. This toxin is only expressed during C. perfringens sporulation. It is accumulated at the beginning of the sporulation and released after cell lysis at the end of the sporulation [17,18]. CPE production is tightly controlled through the master sporulation regulator Spo0A, the transcriptional factor NanR, and three sigma factors of SigE, SigF, and SigK [19,20]. Spo0A mediates sporulation and biofilm formation through phosphorylation and orphan histidine kinases [21,22,23]. H2S is a volatile gas with a foul odor of rotten eggs that is enzymatically produced by both eukaryotic and prokaryotic cells. H2S is a signaling molecule and modulates animal cellular processes, including cell proliferation, apoptosis, inflammation, and hypoxia [24,25,26]. H2S in bacteria is produced during sulfur amino acid (methionine, cysteine, homocysteine, and taurine) metabolism. Methionine mediates methylation and polyamine biosynthesis for sadenosylmethionine (SAM) and quorum sensing autoinducer 2 (AI-2) [27]. Sulfite (e.g., taurine) is converted into sulfide (e.g., H2S) by anaerobic sulfite reductases (asrABC1 (cpe1438-1440) and asrABC2 (cpe1536-1538)) in C. perfringens [28].
Two main categories of primary bile acids are in the animal gastrointestinal tract: cholic acid (CA) and chenodeoxycholic acid (CDCA). The two primary bile acids are sequentially transformed to their respective bile acid lineages of intermediate bile and secondary bile acids of deoxycholic acid (DCA) and lithocholic acid (LCA), respectively [29,30]. The bile acids are also transformed by intestinal microbiota into 3/7/12-oxo-, iso-, -oxoallo-, isoallo-, or allo-forms such as isoallolithocholic acid (isoalloLCA) [30]. Because of the amphipathic property of bile acids, the physiological action of bile acids on cells is traditionally assumed through the non-selective detergent activity of disturbing the permeabilization of the cellular lipid membrane. Recent research progression has shed insight into specific signaling pathways of bile acids in cells. G-protein-coupled BA receptor (TGR5) is activated by both conjugated and unconjugated bile acids at the decreasing potency: LCA > DCA > CDCA > CA [31,32]. TGR5 activated by bile acids induces a cascade of signaling pathways of cAMP, PKA, or the exchange protein directly activated by cAMP (EPAC) [32,33,34]. Bile acids are the natural ligands of the nuclear receptor of farnesoid X receptor (FXR) with activation potency of CDCA > CA > DCA and UDCA [35], while LCA is an antagonist [36]. On the microorganism side, bile acids induce c-di-AMP degradation in C. difficile, which derepresses genes encoding a solute import system protecting from hyperosmotic stress and bile salts [37]. Bile acids are essential for in vitro C. difficile germination [38] and signal through the cspC germinant receptor with TCA the highest potency [39]. C. perfringens deconjugates conjugated bile acids [40] and transforms CDCA > CA into three beta isoforms [41]. Various bile acids also mediate C. perfringens growth, sporulation, and germination [42,43,44,45]. In a previous study [46], we found that only DCA significantly reduced C. perfringens strain CP1-induced chicken NE with improved growth performance and histopathology compared to ox bile (TCA and GCA), chicken bile (TCDCA and TCA), and LCA. In this study, we aimed to investigate how bile acids influenced C. perfringens virulence signaling pathways.
2. Materials and Methods
2.1. Strains, Media, Growth Condition, and Mariner-Based Transposon Mutagenesis
C. perfringens strain CP1, isolated from our previous chicken studies, was confirmed positive for various toxins, including cpa, cpe, and netB [1,3]. Bile acids CA, CDCA, DCA, and LCA were purchased from VWR (all from Alfa Aesar), while isoalloLCA was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). The C. perfringens strains were grown in Brain Heart Infusion (BHI, Legacy biologicals, Mt Prospect, IL, USA) broth for preparing inoculum under anaerobic conditions for 24 h at 37 °C.
The C. perfringens strain HN13 generated at Dr. Nariya’s lab [47] and mariner-based transposon mutagenesis plasmid pHLL24 were kindly provided by Dr. Melville [48] at Virginia Tech. To generate a transposon mutant library following the reported procedures [48], briefly, pHLL24 was transformed to HN13 by electroporation. The resultant HN13 was selected on a BHI plate supplemented with 20 g/mL chloramphenicol and 30 g/mL erythromycin. The cells on the plate were collected, inoculated in BHI broth with the same antibiotic concentration, and grown to the mid-log phase. The cells were washed twice with PBS and cultured in BHI broth with 1 mM of lactose. After the 2 h culture, the cells were washed 3 times with PBS and incubated at 37 °C after 100-fold dilution in BHI. After 4 h, the 2XYT broth (VWR, Solon, OH, USA) with 30 g/mL erythromycin and 3% galactose was used to dilute the previous culture at 100-fold, and the cells were incubated. After 4 h, the cells were plated on a 2XYT plate with galactose and erythromycin. Because colonies resisting galactose and erythromycin had transposon mutagenesis in the genome, the cells were collected in 20% glycerol and stored at −80 °C as HN13 transposon mutant library. To further select mutants resistant to DCA, the HN13 mutant library was inoculated in BHI broth with 3 mM DCA overnight. The cells were serially diluted and plated on 2XYT plates with galactose and erythromycin. Individual colonies from diluted plates or collective cells (non-diluted plate) were collected and stored at −80 °C. In this study, three HN13 mutants resistant to DCA were randomly selected for use.
2.2. Bile Acid Inhibition of C. perfringens Growth Assay
C. perfringens CP1 and HN13 at around 3.9 log10 colony forming unit (CFU) were inoculated into 1 mL TSB in the presence of CA, CDCA, DCA, LCA, and isoalloLCA at 0, 0.001, 0.01, 0.5, or 1 mM, and cultured for 24 h under anaerobic conditions. The bacterial growth was measured by CFU enumeration. To count CFU, the cells were serially diluted and plated on Tryptic Soy Agar (TSA, Criterion, Santa Maria, CA, USA) with sodium thioglycolate, D-Cycloserine, and sheep blood. Coculturing HN13 mutants with bile was similar to that of wild-type cells, except that the mutant cells were plated on 2XYT plates with galactose and erythromycin. Because of the precipitation of bile in cultured broth, the OD600 results were not consistent with the actual C. perfringens growth, and we only reported the CFU results.
2.3. Bacterial RNA Extraction and Gene Expression Using Real-Time PCR
C. perfringens CP1, HN13, or HN13 mutants were inoculated on BHI broth in the presence of DCA or isoalloLCA at 0, 0.01, 0.5, or 1 mM and were cultured under anaerobic conditions. After 2 h, the cells were collected, and total RNA was extracted using the TRIzol (ThermoFisher, St. Louis, MO, USA) as described before [16,49]. After cDNA was synthesized using M-MLV (NE Biolab, Ipswich, MA, USA) and random hexamer [50], the virulence gene expression (asrA1, plc, spoOA, virT, and cpe) was determined using SYBR Green PCR Master mix (Bio-Rad Hercules, CA, USA) on a Bio-Rad 384-well real-time PCR system as described before [46]. The primer sequences are shown in Table 1. The gene expression of the fold-change was calculated using the ΔΔCt method [50] and gyrA as an internal control.
2.4. H2S Detection Using Lead Acetate Assay
C. perfringens CP1, HN13, or HN13 mutants were cultured on BHI with bile acids at 0, 0.01, 0.5, and 1 mM. Filter papers, saturated with lead acetate (Pb(OAc)₂), were dried, autoclaved, and placed on the cap of the tube. The bacteria were cultured under anaerobic conditions at 37 °C. After 24 h, H2S production was visualized and imaged. H2S was produced when the paper changed from white to brown color. The darker the color, the more H2S production. The culture medium also smelled like rotten eggs.
2.5. Statistical Analysis
In Figure 1, Figure 2 and Figure 3, we used one-way ANOVA to compare means among multiple groups of bile treatments, all of which shared the same control group. Multiple comparisons of Fisher’s LSD test using Prism 7.0 software were then conducted. In Figure 4, Figure 5 and Figure 6, we used an unpaired t-test with Welch’s correction using Prism 7.0 software to compare means between two bile dose groups in either WT HN13 or its individual mutants, each of which had its own control group. Experiments were considered statistically significant if p-values were <0.05.
3. Results
3.1. DCA Reduces C. perfringens Strain CP1 Growth and H2S Production
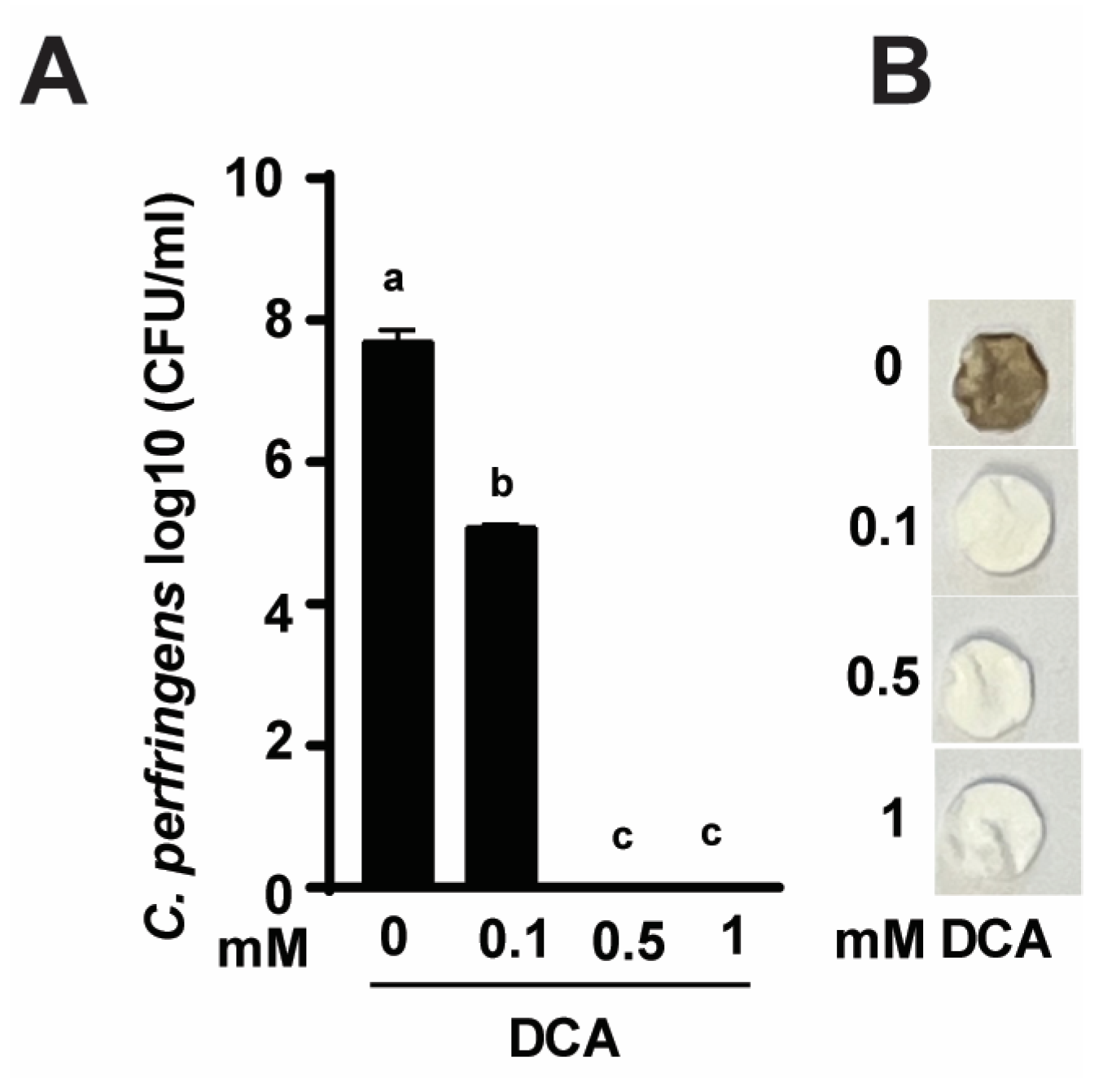
To investigate the mechanism of DCA-reduced C. perfringens CP1-induced NE [46], we conducted an in vitro DCA growth inhibition assay. As shown in Figure 1A, C. perfringens growth reached 7.7 log10 CFU/mL after 24 h culture. Notably, DCA at 0.1, 0.5-, and 1-mM reduced C. perfringens by 33.9 (2.6 log10 CFU/mL), 100 (7.7 log10 CFU/mL), and 100% (7.7 log10 CFU/mL), respectively. At the clinical levels, DCA reduced severe diarrhea and small intestine ballooned with rotten egg-smelling gas in NE birds. We hypothesized that DCA might reduce C. perfringens-produced foul-smell H2S. Using H2S assay, C. perfringens culture had a rotten egg smell and turned disc paper brown (Figure 1B). Consistent with growth inhibition, DCA reduced the foul smell and the dark brown color of the discs in a dose-dependent manner.
3.2. IsoalloLCA Reduces C. perfringens CP1 Growth and H2S Production More Effectively Compared to Other Bile Acids
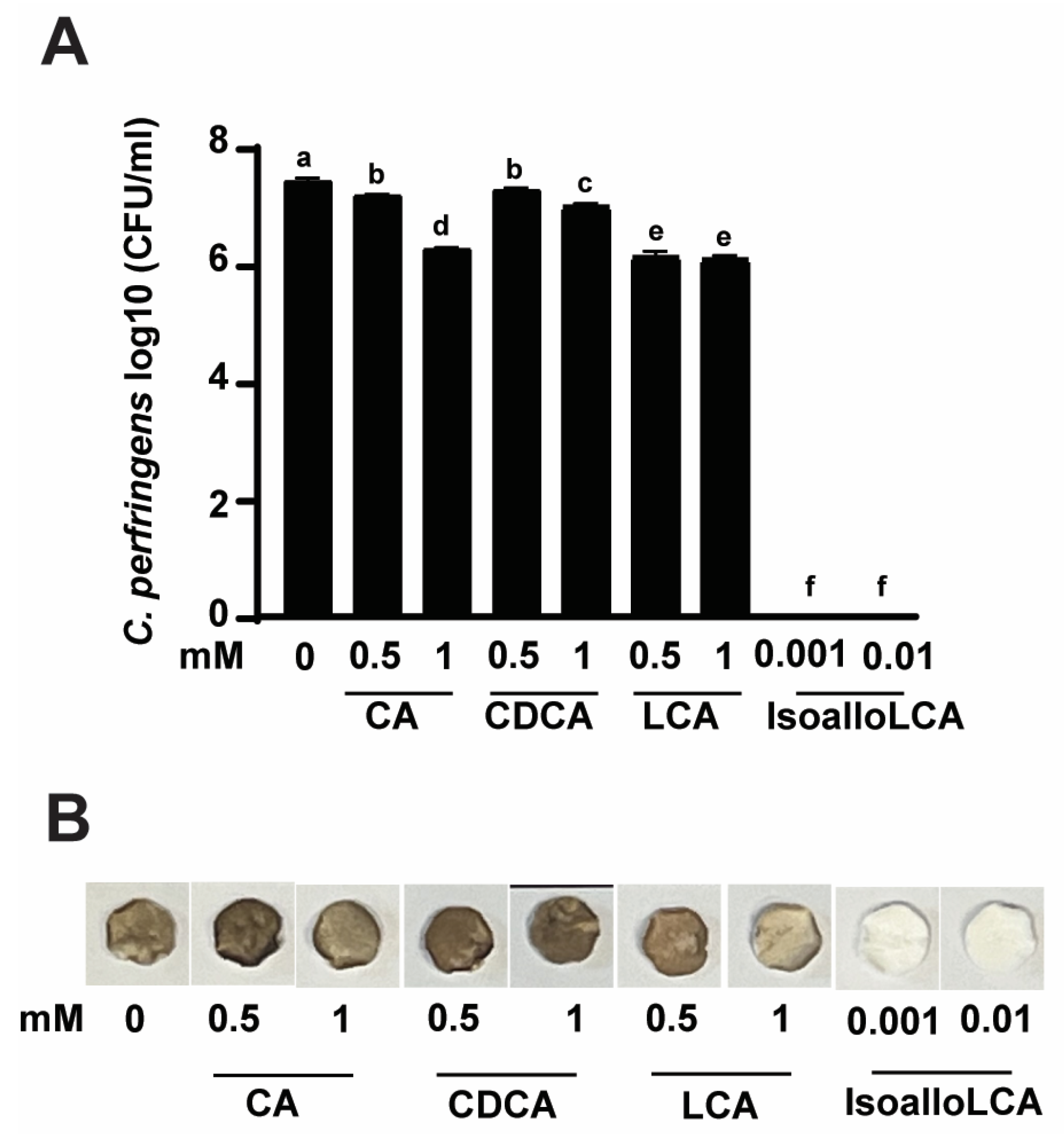
Encouraged by the finding of DCA inhibiting C. perfringens growth and H2S production, we then conducted a growth inhibition assay using the rest of the primary and secondary bile acids with an addition of isoalloLCA, reported as highly potent against C. perfringens growth [30]. As shown in Figure 2A, C. perfringens CP1 growth reached 7.38 log10 CFU after 24 h culture. Interestingly, CA or CDCA at 0.5 vs. 1 mM reduced C. perfringens growth by 3.38 vs. 15.71 (0.25 vs. 1.16 log10 CFU/mL) or 2.16 vs. 5.6% (0.16 vs. 0.41 log10 CFU/mL), respectively. Interestingly, LCA at 0.5 vs. 1 mM comparably reduced the pathogen growth by 17.3% (1.28 log10 CFU/mL). Notably, isoalloLCA at 0.001 and 0.01 mM completely (100%) reduced C. perfringens growth, suggesting its high potency compared to other bile acids. Consistent with the growth inhibition assay, H2S produced from C. perfringens was inhibited by isoalloLCA but barely by CA, CDCA, and LCA (Figure 2B).
3.3. Bile Acids Reduce C. perfringens CP1 Virulence Gene Expression
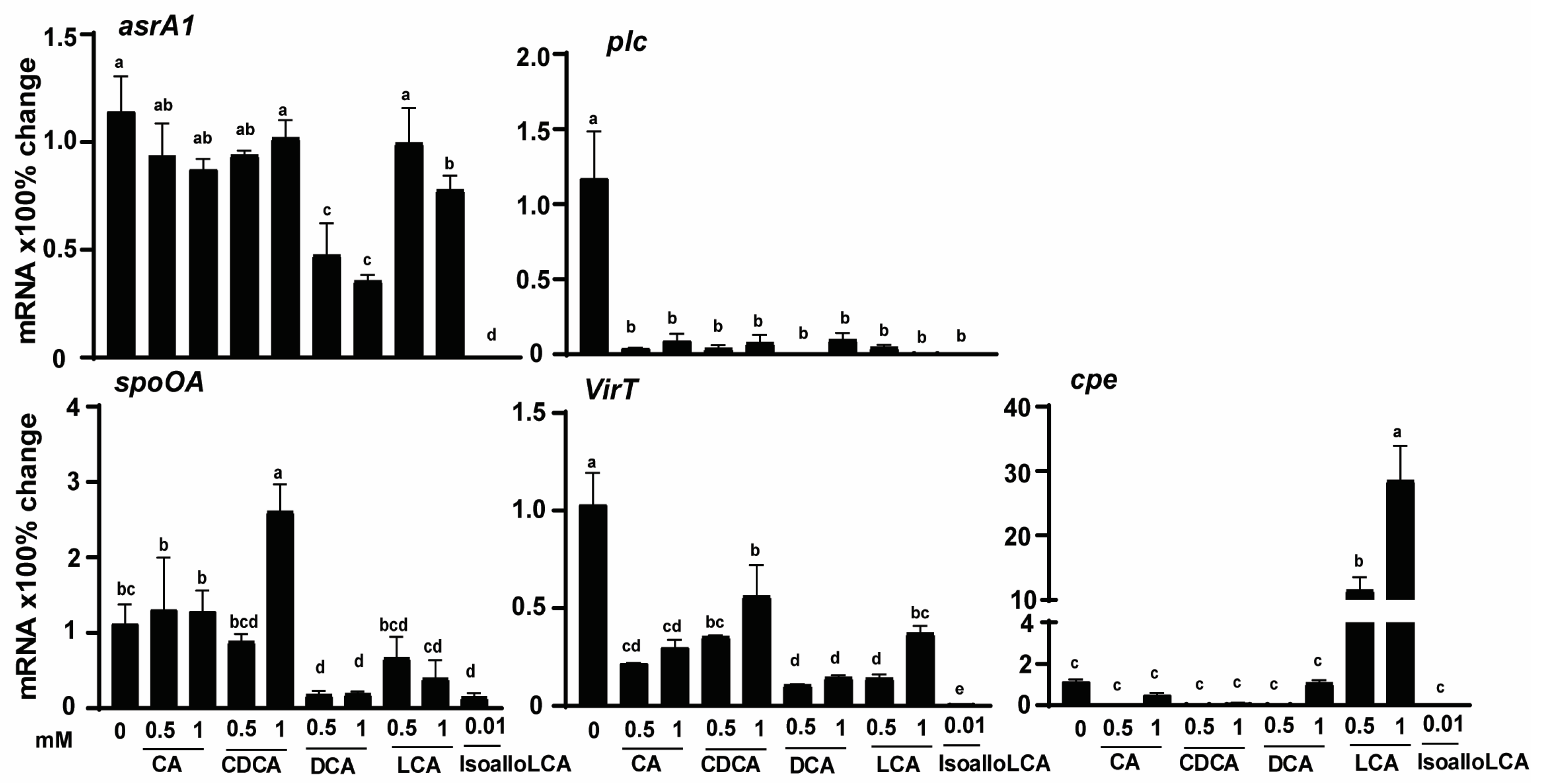
The reduction of H2S production by DCA and isoalloLCA could be mediated at a transcriptional or translational level. Because of the lack of antibodies against most C. perfringens proteins, it was more feasible to measure its transcription vs. translational activities. In addition, we reasoned that the bile acids would modulate other C. perfringens virulent activities. Hence, C. perfringens was cultured with different bile acids, and its RNA was extracted. Gene expressions of virulence genes were measured using real-time PCR. Consistent with the Pb(OAc)2 disc assay, the expression of the H2S-producing gene asrA1 was reduced by 58, 69, and 100% with 0.5 mM DCA, 1 mM DCA, and 0.01 mM isoalloLCA, respectively, while 1 mM LCA reduced asrA1 by 32% (Figure 3). Notably, toxin A gene plc accumulation was reduced universally across all bile at more than 91%. Master regulatory gene spoOA was reduced by 0.5 (84%) and 1 mM (83%) DCA, 1 mM LCA (64%), and 0.01 mM isoalloLCA (86%), while 1 mM CDCA increased the gene by 134%. The regulatory RNA gene virT was reduced by all bile from 45 to 99%, with CDCA and isoalloLCA being the least and most potent ones, respectively. Interestingly, 0.5 and 1 mM LCA increased enterotoxin gene cpe expression by 1157 and 2856%, respectively. These results suggest that bile differentially modulated the H2S production and other virulence activities, at least at the transcriptional level.
3.4. C. perfringens HN13 Mutants Produce H2S in the Presence of DCA
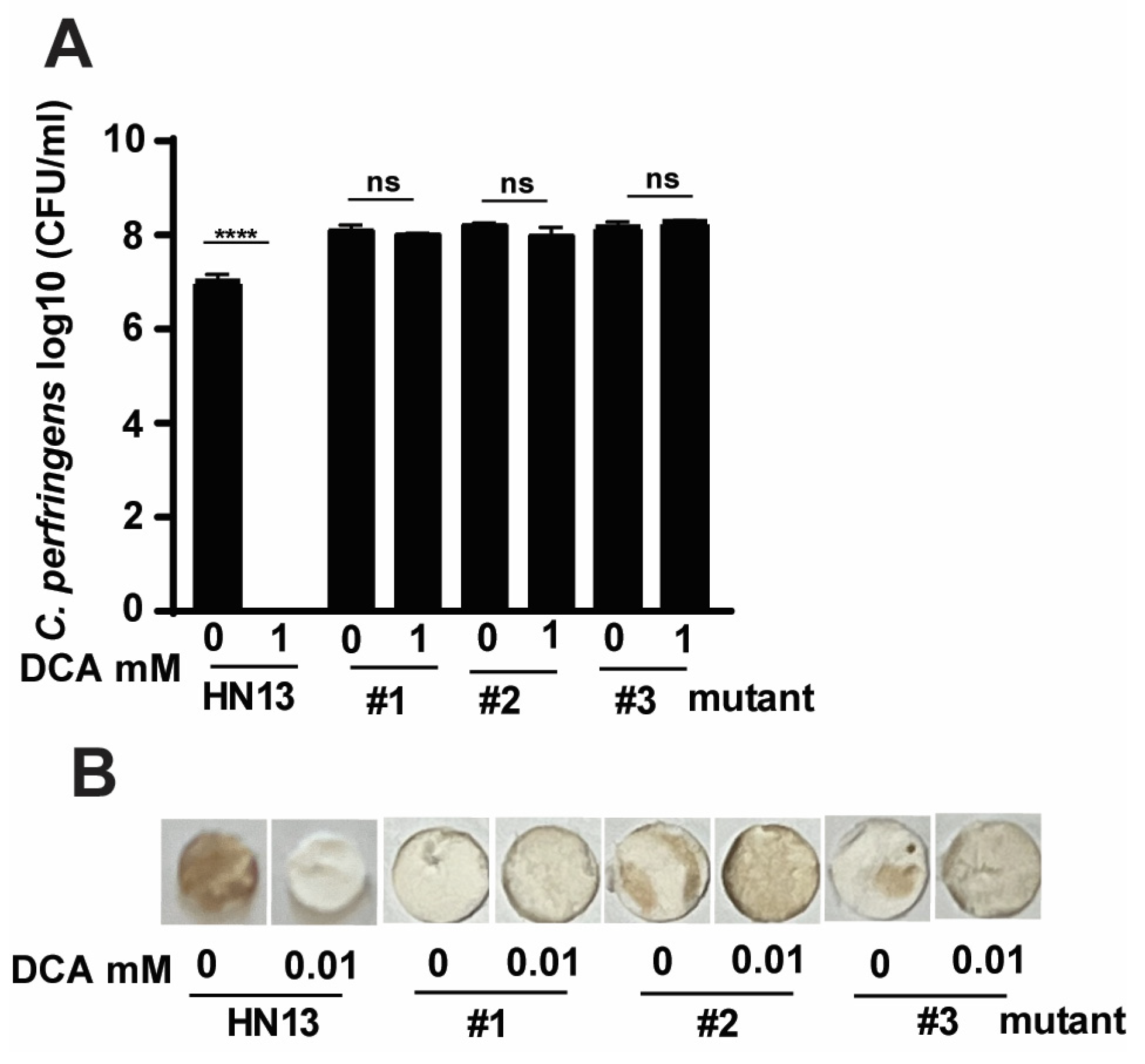
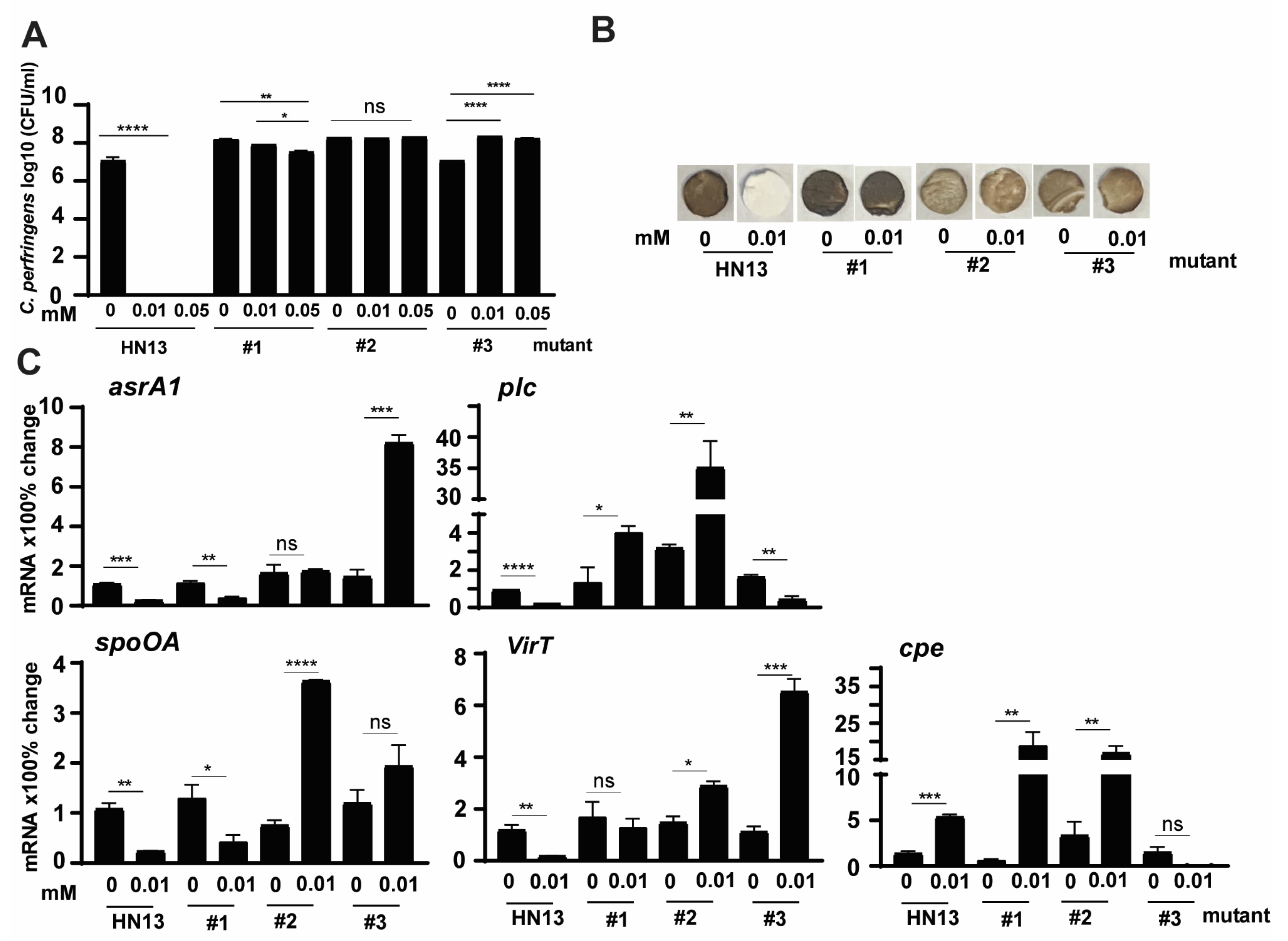
To further confirm that C. perfringens virulence modulated by bile was through specific signaling pathways, we sought to investigate the molecular signaling using C. perfringens strain HN13 and mariner-based transposon mutagenesis mutants [48]. The generated mutants were then selected for bile resistance using 3 M DCA, in which concentration no WT HN13 could grow. Among the thousands of mutant colonies, we randomly picked three colonies for the following assays. Consistent with C. perfringens strain CP1, the growth of wild-type strain HN13 was completely (100%) reduced by 1 mM DCA (Figure 4A). Notably, the growth rates of HN13 mutants #1, #2, and #3 were not changed by 1 mM DCA. We then examined the H2S production in the mutants using Pb(OAc)2 disc assay. As shown in Figure 4B, H2S produced in WT HN13 was greatly reduced by 1 mM DCA, an effect comparable to chicken isolate CP1. Interestingly, H2S production in the mutants was not inhibited but slightly increased by 1 mM DCA, suggesting that DCA modulates C. perfringens signaling pathways related to H2S production.
3.5. C. perfringens HN13 Mutants Express Virulence Genes in the Presence of DCA
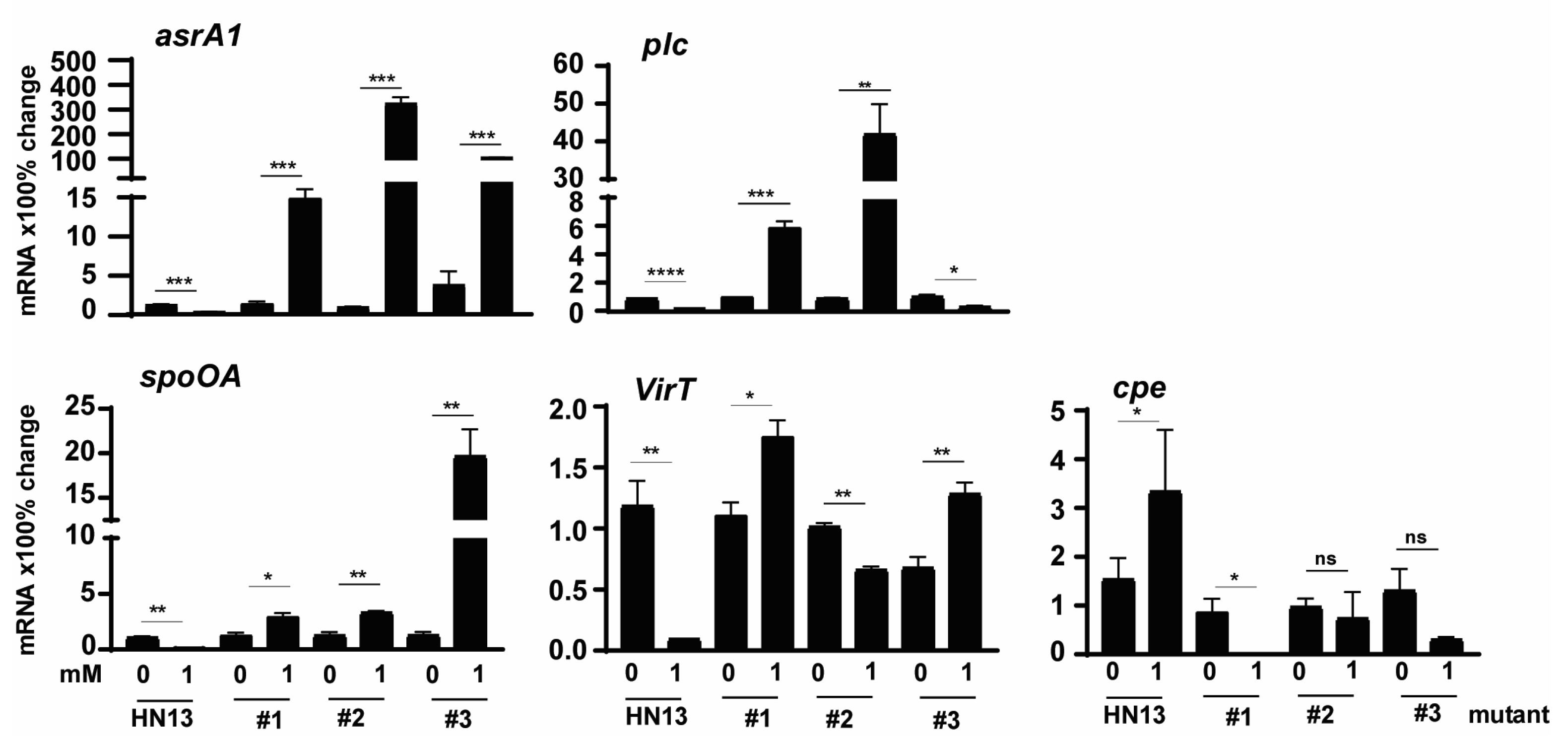
To examine whether the modulation in the mutants was at the transcriptional level, we performed a real-time PCR assay. Notably, C. perfringens mutant isolates increased the expression of H2S production asrA1 gene by 1012, 28, 115, and 805%, respectively, in the presence of 1 mM DCA (Figure 5), while WT HN13 reduced the gene expression by 73% (Figure 5). Notably, plc gene expression in mutants #1 and #2 increased by 469 and 4471% in the presence of DCA, while the gene expression in WT HN13 and mutant #3 reduced by 77 and 68%, respectively. Interestingly, spoOA gene expression in the mutants of # 1, #2, and #3 increased by 133, 164, and 1443%, respectively, in the presence of 1 mM DCA, while the gene reduced by 88% in WT HN13. The gene expression of virT in WT HN13 and mutant #2 reduced by 92 and 35%, respectively, in the presence of 1 mM DCA, while the gene accumulation increased in the mutants of #1 and #3 by 58 and 90%. In addition, 1 mM DCA increased cpe gene expression in WT HN13 by 117%, while the gene expression in mutants #1, #2, and #3 reduced by 100, 24, and 77%, respectively.
3.6. Effects of IsoalloLCA on Growth and Virulence of C. perfringens HN13 Mutants
Based on the results in Figure 1 and Figure 2, isoalloLCA was more potent than DCA in reducing C. perfringens CP1 growth and H2S production. To examine whether isoalloLCA modulated C. perfringens through the same signaling pathways as DCA, we investigated isoalloLCA impacts on WT HN13 and its three mutants. Consistent with the growth reduction by 1 mM DCA in Figure 4A, isoalloLCA at 0.01 and 0.05 mM completely inhibited WT HN13 growth (Figure 6A). Interestingly, the growth of mutant #1 was reduced by isoalloLCA, while isoalloLCA increased the growth of mutant #3. Notably, H2S production in WT HN13 was reduced by 0.01 mM isoalloLCA, while the gas produced by the three mutants was not reduced by the bile (Figure 6B). We then measured the change of C. perfringens virulence gene expression by 0.01 mM isoalloLCA. Similar to virulence gene reduction by 1 mM DCA in Figure 5, 0.01 mM isoalloLCA reduced the gene expression of asrA1, plc, spoOA, and virT in WT HN13 by 78, 80, 78, and 85%, respectively, while the bile increased cpe gene accumulation by 300% (Figure 6C). Interestingly, isoalloLCA reduced asrA1 gene expression in mutant #1 by 67%, while the bile increased the gene expression in mutant #3 by 468%. IsoalloLCA increased plc gene expression by 200 and 1005% in mutants #1 and #2, respectively, while the bile reduced the gene expression in mutant #3 by 74%. IsoalloLCA reduced spoOA gene expression by 68% in mutant #1, while the bile increased the gene expression in mutant #2 by 384%. IsoalloLCA increased virT gene expression by 95 and 482% in mutants #2 and #3. IsoalloLCA increased cpe gene expression by 3033 and 408% in mutants #1 and #2, respectively.
4. Discussion
Dietary DCA effectively reduces C. perfringens-induced clinical [45,46] and subclinical chicken NE [49], but the molecular mechanism of DCA against C. perfringens remains largely elusive. In this study, we reasoned that bile acids were signaling pathway regulators modulating C. perfringens virulence activities. We found that isoalloLCA and DCA were more potent inhibitors toward C. perfringens growth, H2S production, and virulence gene expression compared to CA, CDCA, and LCA. The inhibition of virulence gene expression by the bile indicates regulation, at least at the transcriptional level. Based on the differential impact of DCA and isoalloLCA on the virulence of the three C. perfringens HN13 mutants, the two bile acids might act on slightly different signaling pathways. Together, these results suggest that bile acids regulated C. perfringens virulence signaling pathways with isoalloLCA and DCA as the most potent ones.
Our study results are consistent with the notion that bile acids are signaling regulators. Because bile acid has amphipathic detergent properties, it has traditionally been thought that the cellular toxicity of bile acids was mainly through non-selective detergent-like properties that disturb cellular membranes [51]. That concept was often discrepant with the observations that different bile acids had quite different or even opposite effects on cellular activities despite their comparable detergent properties. Research on animal molecular and cellular activities has revealed that bile acids regulate specific signaling pathways in embryogenesis, development, metabolism, and immunity [52]. G-protein-coupled bile acid receptor (TGR5) is activated by both conjugated and unconjugated bile acids at the decreasing potency of LCA > DCA > CDCA > CA [31,32]. TGR5 activated by bile acids induces a cascade of signaling pathways of cAMP, PKA, or the exchange protein directly activated by cAMP (EPAC) [32,33,34]. Bile acids also directly activate three nuclear receptors: FXR [53], pregnane X receptor (PXR) [54], and vitamin D receptor (VDR) [55]. FXR is activated at the decreasing potency of CDCA > LCA > DCA > CA, while LCA and 3-oxo-LCA activate VDR and FXR [55,56]. Furthermore, bile acids modulate host immunity. 3-oxoLCA inhibits Th17 cell differentiation by binding to retinoid-related orphan receptor-γt (RORγt), and isoalloLCA increases Treg cell differentiation through elevating mitochondrial reactive oxygen species (mitoROS), leading to FOXP3 expression increase [57]. IsoDCA increases Foxp3 expression by reducing FXR-related signaling pathways [58]. These findings highlight the important signaling regulatory role of bile acids on animals. It would be necessary to investigate the role of bile acids on chicken signaling pathways during NE in the future.
Because of the diverse composition of the bacterial community, the effect of bile acids on bacteria is highly variable between bacteria. Bile salts (mainly CA) have been used as the selective ingredient of bacterial growth medium to grow Gram-negative bacteria, such as MacConkey [59]. In this study, only DCA and isoalloLCA effectively inhibited the growth of the Gram-positive bacterium C. perfringens. The inhibition was in the decreasing potency of isoalloLCA > DCA > LCA > CA > CDCA. Because CDCA is the main bile acid of chickens [49], it is not surprising that chickens are susceptible to C. perfringens-induced NE. Besides mediating growth, bile acids also regulate C. perfringens sporulation and germination [42,43,44,45]. Similarly, DCA at 0.01% reduced the growth of another Gram-positive bacterium C. difficile [60], while CA at 2.4 mM promoted C. difficile spore germination but inhibited vegetative growth at 12 mM [61]. Interestingly, CDCA inhibits its spore germination and is a competitive inhibitor of germinant TCA [62]. The cspBAC locus is a major regulator of C. difficile germination, and cspC is the receptor for bile salt (T/GCA, CA) germinants, whereas the related pseudoprotease cspA detected amino acid cogerminants [38,63]. Bile acids (CA) induce c-di-AMP degradation in C. difficile, which derepresses genes encoding a solute import system protecting from hyperosmotic stress and bile salts [37]. Bile acids also induce damage to Gram-negative bacteria. CDCA and DCA induce reactive oxygen species (ROS)-mediated nucleic acid damage in E. coli and activate the SOS response [64]. Ox bile at as high as 20% induced nucleotide substitutions, frameshifts, and chromosomal rearrangements in Salmonella enterica [65]. There is little insight into the molecular mechanism of bile acids in inhibiting C. perfringens growth. In our study, we found that C. perfringens mutants were resistant to growth inhibition by DCA/isoalloLCA, suggesting the involvement of certain molecular signaling pathways. We are working on finding the mutated genes in the three mutants.
Because C. perfringens is a pathogen, we also investigate its virulence changes by the potent bile acids DCA and isoalloLCA. The reduction of toxin gene expression of plc and cpe by DCA was consistent with our studies that DCA reduces C. perfringens sporulation [16] and its induction of chicken NE [45]. Consistently, the cpe regulatory gene spoOA [20] was reduced by DCA. Interestingly, DCA also reduced the regulatory RNA gene virT expression. Because isoalloLCA was more potent in reducing C. perfringens virulence, it would be reasonable to argue that isoalloLCA would be more potent in reducing chicken NE. Besides modulating C. difficile virulence mentioned above, bile acids at 1% induce BrtA-mediated transcription of the multidrug efflux pump genes mdrM and mdrT in Gram-positive Listeria monocytogenes [66]. Bile acids also reduce the virulence in Gram-negative bacteria but not growth inhibition at physiology concentration. DCA or CDCA, but not other bile acids or detergents, enhances Shigella spp. adherence and invasion of HeLa cells [67]. CA at 0.5% and DCA at 0.1% represses S. enterica invasion through postranscriptionally destabilizing the transcription factor HilD [68]. Although bile (59% DCA and 59% CA) does not change Shigella flexneri growth, it induces spE1/ospE2 gene expression and adherence to HeLa cell [69]. DCA modulates the PhoP-PhoQ two-component regulatory system in Salmonella spp. [70]. Ox bile at 3% inhibits the gene expression of the S. enterica pathogenicity island 1 (SPI-1) [71]. Ox bile at 0.2% reduces the gene expression of ctxAB (encoding choleratoxin) and of tcpA in Vibrio cholerae but increases bacterial motility [72]. Bile at 0.05% binds VtrC and induces Vibrio parahaemolyticus virulence type III secretion system 2 (T3SS2) through VtrA/VtrC activating VtrB [73]. DCA at 0.1% increases the gene expression of ciaB, cmeABC, dccR, and tlyA in Campylobacter jejuni [74]. BACTO bile salts at 0.8% reduces the expression of forty-one genes of enterocyte effacement (LEE) pathogenicity island in Escherichia coli O157:H7 but increases the expression of seventeen genes related to iron scavenging and metabolism [75]. Although the bacterial virulence gene expression in response to bile acids is complex due to various bacteria and bile, the accumulated data would generate consensus molecular mechanisms on the interaction of bacteria and bile in the future.
The other important findings in this study are the reduction of asrA1 gene expression and H2S gas generation in C. perfringens by DCA and isoalloLCA. Consistently, DCA reduced the foul smell and ballooned small intestines in NE birds [45]. H2S is a foul-smelling gas [76]. H2S produced by animals and bacteria has been found to have pleiotropic effects on the physiology of organisms since its discovery. The ileums of rabbits, guinea pigs, and rats are relaxed by H2S donors [77]. H2S induces intestinal relaxation through potassium (K) channels, particularly apamin-sensitive small conductance calcium-activated potassium (SK) channels and glybenclamide-sensitive K (ATP) channels [78]. H2S inhibits both L-type calcium channels and BKCa channels in smooth muscle cells of rat colon [79]. There are various other signaling pathways in H2S-mediated motility and relaxation [24]. Hence, C. perfringens-produced H2S may relax and enlarge the intestine, slow digesta movement, and increase bacterial overgrowth, leading to exacerbated NE. It remains unknown whether H2S plays any role in the chicken NE intestinal inflammation, showing ischemia-perfusion-like histopathology [45]. The role of H2S in the intestinal inflammation of other animals is complex and, at times, contradictory. Fecal sulfide levels are increased in patients with ulcerative colitis, and effective treatment of relapsed inflammation is associated with a reduction in sulfide levels [80,81]. The pro-inflammatory effect of a high dose of H2S is shown in an LPS-induced model of endotoxic shock in male Swiss mice, where DL-propargylglycine (H2S synthesis enzyme inhibitor) reduces this effect [82]. Evidence against H2S having a causative role in ulcerative colitis comes from a study that fails to demonstrate elevated fecal sulfide levels in the disease state [83]. Exogenous H2S has a protective role in models of intestinal ischemia [84]. Both endogenous and exogenous H2S protect against nonsteroidal anti-inflammatory drug (NSAID)-induced gastritis [85]. H2S resolves inflammation by reducing leukocyte migration to the site of injury [86] and by enhancing neutrophil apoptosis [87]. Interestingly, C. perfringens α-toxin CPA blocks leukocyte migration and induces neutrophil necrosis [1]. It would be interesting to investigate the histopathology and immunity alterations in the presence of H2S and CPA.
Besides impacting the host, an increasing number of studies have established H2S gas as a major cytoprotectant and redox modulator in microorganisms. H2S influences bacteria activities, such as protecting bacteria from antibiotics and their induction of ROS oxidative stress [88]. The presence of plasmid-borne genetic elements enhances both basal H2S production and antibiotic resistance in multidrug-resistant strains of E. coli [89]. Cystathionine γ-lyase (CSE) generates H2S in two human pathogens, Staphylococcus aureus and Pseudomonas aeruginosa. CSE inhibitors reduce bacterial H2S biogenesis and suppress bacterial tolerance by disrupting biofilm formation and substantially reducing the number of persister bacteria that survive antibiotic treatment [90]. It remains elusive how H2S impacts C. perfringens growth, survival, and virulence.
Together, these results suggest that bile acids are signaling molecules regulating C. perfringens growth, H2S production, and virulence gene expression, with DCA and isoalloLCA as the most potent ones. The bile reduced C. perfringens virulence through transcriptionally modulating the pathogen signaling pathways. The findings in this study could be used to design new strategies to prevent and treat C. perfringens-induced diseases.
Author Contributions
T.A. (Tahrir Alenezi) and X.S. designed the experiments and wrote the manuscript with input from co-authors Y.F., B.A., T.A. (Thamer Alanazi), A.A. and H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants from Arkansas Biosciences Institute, USDA National Institute of Food and Agriculture (NIFA) Hatch project 1012366, NIFA Hatch/Multi State project 1018699, NIFA SAS 2019-69012-29905, and NIFA project 2020-67016-31346 to X.S and B. H. This research was supported by grants of AAES Research Incentive Grant to X.S. This research was also supported by the Poultry Federation Scholarship to T.A., Y.F., and A.A. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
The biosafety protocol No. 17021 was approved by the Institutional Biosafety Committee (IBC) of the University of Arkansas at Fayetteville.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are presented in this paper.
Acknowledgments
We would like to thank Hirofumi Nariya and Stephen B. Melville for kindly providing C. perfringens strain HN13 and mariner-based transposon mutagenesis plasmid pHLL24.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Fu, Y.; Alenezi, T.; Sun, X. Clostridium perfringens-Induced Necrotic Diseases: An Overview. Immuno 2022, 2, 387–407. [Google Scholar] [CrossRef]
- Rood, J.I.; Keyburn, A.L.; Moore, R.J. NetB and necrotic enteritis: The hole movable story. Avian Pathol. 2016, 45, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Kaldhusdal, M.; Benestad, S.L.; Lovland, A. Epidemiologic aspects of necrotic enteritis in broiler chickens - disease occurrence and production performance. Avian Pathol. 2016, 45, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.B. Intercurrent coccidiosis and necrotic enteritis of chickens: Rational, integrated disease management by maintenance of gut integrity. Avian Pathol. 2005, 34, 159–180. [Google Scholar] [CrossRef]
- Latorre, J.D.; Adhikari, B.; Park, S.H.; Teague, K.D.; Graham, L.E.; Mahaffey, B.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Kwon, Y.M.; Ricke, S.C.; et al. Evaluation of the Epithelial Barrier Function and Ileal Microbiome in an Established Necrotic Enteritis Challenge Model in Broiler Chickens. Front. Vet. Sci. 2018, 5, 199. [Google Scholar] [CrossRef]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018, 53, 5–10. [Google Scholar] [CrossRef]
- Fernandes da Costa, S.P.; Mot, D.; Bokori-Brown, M.; Savva, C.G.; Basak, A.K.; Van Immerseel, F.; Titball, R.W. Protection against avian necrotic enteritis after immunisation with NetB genetic or formaldehyde toxoids. Vaccine 2013, 31, 4003–4008. [Google Scholar] [CrossRef]
- Titball, R.W.; Naylor, C.E.; Basak, A.K. The Clostridium perfringens alpha-toxin. Anaerobe 1999, 5, 51–64. [Google Scholar] [CrossRef]
- Marius, N.; Manoilescu, I.; Velnic, A.-A.; Ioan, B. Gas Gangrene: Case Presentation and Literature Data. Jurnalul Chir. 2017, 13. [Google Scholar] [CrossRef]
- Hickey, M.J.; Kwan, R.Y.; Awad, M.M.; Kennedy, C.L.; Young, L.F.; Hall, P.; Cordner, L.M.; Lyras, D.; Emmins, J.J.; Rood, J.I. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008, 4, e1000045. [Google Scholar] [CrossRef]
- Ba-Thein, W.; Lyristis, M.; Ohtani, K.; Nisbet, I.T.; Hayashi, H.; Rood, J.I.; Shimizu, T. The virR/virS locus regulates the transcription of genes encoding extracellular toxin production in Clostridium perfringens. J. Bacteriol. 1996, 178, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Yaguchi, H.; Ohtani, K.; Banu, S.; Hayashi, H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol. 2002, 43, 257–265. [Google Scholar] [CrossRef]
- Hiscox, T.J.; Chakravorty, A.; Choo, J.M.; Ohtani, K.; Shimizu, T.; Cheung, J.K.; Rood, J.I. Regulation of virulence by the RevR response regulator in Clostridium perfringens. Infect. Immun. 2011, 79, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Ohtani, K.; Hayashi, H.; Shimizu, T. Characterization of genes regulated directly by the VirR/VirS system in Clostridium perfringens. J. Bacteriol. 2008, 190, 7719–7727. [Google Scholar] [CrossRef] [PubMed]
- Czeczulin, J.R.; Hanna, P.C.; McClane, B.A. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 1993, 61, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Bansal, M.; Alenezi, T.; Almansour, A.; Wang, H.; Sun, X. Vaccines Using Clostridium perfringens Sporulation Proteins Reduce Necrotic Enteritis in Chickens. Microorganisms 2022, 10, 1110. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Glil, M. In Silico Genome Analysis and Molecular Typing of Clostridium Perfringens. Doctoral Dissertation, Mensch und Buch Verlag, Berlin, Germany, 2019. [Google Scholar]
- Xiao, Y. Clostridium perfringens Sporulation, Germination and Outgrowth in Food: A Functional Genomics Approach; Wageningen University and Research: Wageningen, The Netherlands, 2014. [Google Scholar]
- Ohtani, K.; Hirakawa, H.; Paredes-Sabja, D.; Tashiro, K.; Kuhara, S.; Sarker, M.R.; Shimizu, T. Unique regulatory mechanism of sporulation and enterotoxin production in Clostridium perfringens. J. Bacteriol. 2013, 195, 2931–2936. [Google Scholar] [CrossRef]
- Mi, E.; Li, J.; McClane, B.A. NanR Regulates Sporulation and Enterotoxin Production by Clostridium perfringens Type F Strain F4969. Infect. Immun. 2018, 86, 10–1128. [Google Scholar] [CrossRef]
- Ethapa, T.; Leuzzi, R.; Ng, Y.K.; Baban, S.T.; Adamo, R.; Kuehne, S.A.; Scarselli, M.; Minton, N.P.; Serruto, D.; Unnikrishnan, M. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 2013, 195, 545–555. [Google Scholar] [CrossRef]
- Hamon, M.A.; Lazazzera, B.A. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol. Microbiol. 2001, 42, 1199–1209. [Google Scholar] [CrossRef]
- Hu, W.S.; Woo, D.U.; Kang, Y.J.; Koo, O.K. Biofilm and Spore Formation of Clostridium perfringens and Its Resistance to Disinfectant and Oxidative Stress. Antibiotics 2021, 10, 396. [Google Scholar] [CrossRef]
- Linden, D.R. Hydrogen sulfide signaling in the gastrointestinal tract. Antioxid. Redox Signal 2014, 20, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Yurinskaya, M.M.; Krasnov, G.S.; Kulikova, D.A.; Zatsepina, O.G.; Vinokurov, M.G.; Chuvakova, L.N.; Rezvykh, A.P.; Funikov, S.Y.; Morozov, A.V.; Evgen’ev, M.B. H(2)S counteracts proinflammatory effects of LPS through modulation of multiple pathways in human cells. Inflamm. Res. 2020, 69, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. LuxS quorum sensing: More than just a numbers game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Andre, G.; Haudecoeur, E.; Monot, M.; Ohtani, K.; Shimizu, T.; Dupuy, B.; Martin-Verstraete, I. Global regulation of gene expression in response to cysteine availability in Clostridium perfringens. BMC Microbiol. 2010, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Kumar, D.P.; Mahavadi, S.; Bhattacharya, S.; Zhou, R.; Corvera, C.U.; Bunnett, N.W.; Grider, J.R.; Murthy, K.S. Activation of G protein-coupled bile acid receptor, TGR5, induces smooth muscle relaxation via both Epac- and PKA-mediated inhibition of RhoA/Rho kinase pathway. Am. J. Physiol. -Gastrointest. Liver Physiol. 2013, 304, G527–G535. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.L.; Zhao, A.; Yu, J.; Huang, L.; De Pedro, N.; Pelaez, F.; Wright, S.D.; Cui, J. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J. Biol. Chem. 2004, 279, 8856–8861. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lo, J.L.; Huang, L.; Zhao, A.; Metzger, E.; Adams, A.; Meinke, P.T.; Wright, S.D.; Cui, J. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J. Biol. Chem. 2002, 277, 31441–31447. [Google Scholar] [CrossRef]
- Oberkampf, M.; Hamiot, A.; Altamirano-Silva, P.; Belles-Sancho, P.; Tremblay, Y.D.N.; DiBenedetto, N.; Seifert, R.; Soutourina, O.; Bry, L.; Dupuy, B.; et al. c-di-AMP signaling is required for bile salt resistance, osmotolerance, and long-term host colonization by Clostridioides difficile. Sci. Signal. 2022, 15, eabn8171. [Google Scholar] [CrossRef]
- Francis, M.B.; Allen, C.A.; Shrestha, R.; Sorg, J.A. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog. 2013, 9, e1003356. [Google Scholar] [CrossRef]
- Shen, A. Clostridioides difficile Spore Formation and Germination: New Insights and Opportunities for Intervention. Annu. Rev. Microbiol. 2020, 74, 545–566. [Google Scholar] [CrossRef]
- Gopal-Srivastava, R.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Clostridium perfringens. J. Lipid Res. 1988, 29, 1079–1085. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Hutchison, D.M.; Forrest, T.P.; Bokkenheuser, V.D.; Winter, J.; Holdeman, L.V. Metabolism of primary bile acids by Clostridium perfringens. J. Steroid Biochem. 1983, 18, 97–104. [Google Scholar] [CrossRef]
- Hickey, C.S.; Johnson, M.G. Effects of pH shifts, bile salts, and glucose on sporulation of Clostridium perfringens NCTC 8798. Appl. Environ. Microbiol. 1981, 41, 124–129. [Google Scholar] [CrossRef]
- Heredia, N.L.; Labbe, R.G.; Rodriguez, M.A.; Garcia-Alvarado, J.S. Growth, sporulation and enterotoxin production by Clostridium perfringens type A in the presence of human bile salts. FEMS Microbiol. Lett. 1991, 68, 15–21. [Google Scholar] [CrossRef]
- Yasugi, M.; Okuzaki, D.; Kuwana, R.; Takamatsu, H.; Fujita, M.; Sarker, M.R.; Miyake, M. Transcriptional Profile during Deoxycholate-Induced Sporulation in a Clostridium perfringens Isolate Causing Foodborne Illness. Appl. Environ. Microbiol. 2016, 82, 2929–2942. [Google Scholar] [CrossRef]
- Wang, H.; Latorre, J.D.; Bansal, M.; Abraha, M.; Al-Rubaye, B.; Tellez-Isaias, G.; Hargis, B.; Sun, X. Microbial metabolite deoxycholic acid controls Clostridium perfringens-induced chicken necrotic enteritis through attenuating inflammatory cyclooxygenase signaling. Sci. Rep. 2019, 9, 14541. [Google Scholar] [CrossRef]
- Bansal, M.; Alenezi, T.; Fu, Y.; Almansour, A.; Wang, H.; Gupta, A.; Liyanage, R.; Graham, D.B.; Hargis, B.M.; Sun, X. Specific Secondary Bile Acids Control Chicken Necrotic Enteritis. Pathogens 2021, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nariya, H.; Miyata, S.; Suzuki, M.; Tamai, E.; Okabe, A. Development and application of a method for counterselectable in-frame deletion in Clostridium perfringens. Appl. Environ. Microbiol. 2011, 77, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bouillaut, L.; Sonenshein, A.L.; Melville, S.B. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J. Bacteriol. 2013, 195, 629–636. [Google Scholar] [CrossRef]
- Bansal, M.; Fu, Y.; Alrubaye, B.; Abraha, M.; Almansour, A.; Gupta, A.; Liyanage, R.; Wang, H.; Hargis, B.; Sun, X. A secondary bile acid from microbiota metabolism attenuates ileitis and bile acid reduction in subclinical necrotic enteritis in chickens. J. Anim. Sci. Biotechnol. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Almansour, A.; Bansal, M.; Alenezi, T.; Alrubaye, B.; Wang, H.; Sun, X. Microbiota attenuates chicken transmission-exacerbated campylobacteriosis in Il10(-/-) mice. Sci. Rep. 2020, 10, 20841. [Google Scholar] [CrossRef]
- Moschetta, A.; vanBerge-Henegouwen, G.P.; Portincasa, P.; Renooij, W.L.; Groen, A.K.; van Erpecum, K.J. Hydrophilic bile salts enhance differential distribution of sphingomyelin and phosphatidylcholine between micellar and vesicular phases: Potential implications for their effects in vivo. J. Hepatol. 2001, 34, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Elazhary, M.A.; Saheb, S.A.; Roy, R.S.; Lagace, A. A simple procedure for the preliminary identification of aerobic gram negative intestinal bacteria with special reference to the Enterobacteriaceae. Can. J. Comp. Med. 1973, 37, 43–46. [Google Scholar]
- Usui, Y.; Ayibieke, A.; Kamiichi, Y.; Okugawa, S.; Moriya, K.; Tohda, S.; Saito, R. Impact of deoxycholate on Clostridioides difficile growth, toxin production, and sporulation. Heliyon 2020, 6, e03717. [Google Scholar] [CrossRef]
- Wilson, K.H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 1983, 18, 1017–1019. [Google Scholar] [CrossRef]
- Sorg, J.A.; Sonenshein, A.L. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 2010, 192, 4983–4990. [Google Scholar] [CrossRef]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Kandell, R.L.; Bernstein, C. Bile salt/acid induction of DNA damage in bacterial and mammalian cells: Implications for colon cancer. Nutr. Cancer 1991, 16, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.I.; Ramos-Morales, F.; Casadesus, J. Bile-induced DNA damage in Salmonella enterica. Genetics 2004, 168, 1787–1794. [Google Scholar] [CrossRef]
- Quillin, S.J.; Schwartz, K.T.; Leber, J.H. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol. Microbiol. 2011, 81, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.M.; Reed, K.E.; Payne, S.M. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect. Immun. 1995, 63, 3642–3648. [Google Scholar] [CrossRef]
- Eade, C.R.; Hung, C.C.; Bullard, B.; Gonzalez-Escobedo, G.; Gunn, J.S.; Altier, C. Bile Acids Function Synergistically to Repress Invasion Gene Expression in Salmonella by Destabilizing the Invasion Regulator HilD. Infect. Immun. 2016, 84, 2198–2208. [Google Scholar] [CrossRef]
- Faherty, C.S.; Redman, J.C.; Rasko, D.A.; Barry, E.M.; Nataro, J.P. Shigella flexneri effectors OspE1 and OspE2 mediate induced adherence to the colonic epithelium following bile salts exposure. Mol. Microbiol. 2012, 85, 107–121. [Google Scholar] [CrossRef]
- van Velkinburgh, J.C.; Gunn, J.S. PhoP-PhoQ-regulated loci are required for enhanced bile resistance in Salmonella spp. Infect. Immun. 1999, 67, 1614–1622. [Google Scholar] [CrossRef]
- Prouty, A.M.; Gunn, J.S. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect. Immun. 2000, 68, 6763–6769. [Google Scholar] [CrossRef]
- Gupta, S.; Chowdhury, R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 1997, 65, 1131–1134. [Google Scholar] [CrossRef]
- Li, P.; Rivera-Cancel, G.; Kinch, L.N.; Salomon, D.; Tomchick, D.R.; Grishin, N.V.; Orth, K. Bile salt receptor complex activates a pathogenic type III secretion system. Elife 2016, 5, e15718. [Google Scholar] [CrossRef] [PubMed]
- Malik-Kale, P.; Parker, C.T.; Konkel, M.E. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J. Bacteriol. 2008, 190, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Hamner, S.; McInnerney, K.; Williamson, K.; Franklin, M.J.; Ford, T.E. Bile salts affect expression of Escherichia coli O157:H7 genes for virulence and iron acquisition, and promote growth under iron limiting conditions. PLoS One 2013, 8, e74647. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Choct, M.; Wu, S.B.; Swick, R.A. Necrotic enteritis challenge and high dietary sodium level affect odorant composition or emission from broilers. Poult. Sci. 2018, 97, 39–46. [Google Scholar] [CrossRef]
- Teague, B.; Asiedu, S.; Moore, P.K. The smooth muscle relaxant effect of hydrogen sulphide in vitro: Evidence for a physiological role to control intestinal contractility. Br. J. Pharmacol. 2002, 137, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gallego, D.; Clave, P.; Donovan, J.; Rahmati, R.; Grundy, D.; Jimenez, M.; Beyak, M.J. The gaseous mediator, hydrogen sulphide, inhibits in vitro motor patterns in the human, rat and mouse colon and jejunum. Neurogastroenterol. Motil. 2008, 20, 1306–1316. [Google Scholar] [CrossRef]
- Quan, X.; Luo, H.; Liu, Y.; Xia, H.; Chen, W.; Tang, Q. Hydrogen sulfide regulates the colonic motility by inhibiting both L-type calcium channels and BKCa channels in smooth muscle cells of rat colon. PLoS ONE 2015, 10, e0121331. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Beatty, E.R.; Cummings, J.H. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef]
- Edmond, L.M.; Hopkins, M.J.; Magee, E.A.; Cummings, J.H. The effect of 5-aminosalicylic acid-containing drugs on sulfide production by sulfate-reducing and amino acid-fermenting bacteria. Inflamm. Bowel Dis. 2003, 9, 10–17. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G.; Fiorucci, S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 2007, 132, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; De Rinaldis, P.; Fumarulo, R. Sulfide enhancement of PMN apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef]
- Jones, R.T.; Thai, L.P.; Silver, R.P. Genetic and molecular characterization of an Escherichia coli plasmid coding for hydrogen sulfide production and drug resistance. Antimicrob. Agents Chemother. 1978, 14, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Shatalin, K.; Nuthanakanti, A.; Kaushik, A.; Shishov, D.; Peselis, A.; Shamovsky, I.; Pani, B.; Lechpammer, M.; Vasilyev, N.; Shatalina, E.; et al. Inhibitors of bacterial H(2)S biogenesis targeting antibiotic resistance and tolerance. Science 2021, 372, 1169–1175. [Google Scholar] [CrossRef]
Figure 1.
DCA reduces C. perfringens strain CP1 growth and H2S production. C. perfringens was cultured with DCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate (Pb(OAc)₂) discs. Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–c mean p < 0.05.
Figure 1.
DCA reduces C. perfringens strain CP1 growth and H2S production. C. perfringens was cultured with DCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate (Pb(OAc)₂) discs. Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–c mean p < 0.05.

Figure 2.
IsoalloLCA reduces C. perfringens strain CP1 growth and H2S production. C. perfringens was cultured with CA, CDCA, LCA, or isoalloLCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate discs. Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–f mean p < 0.05.
Figure 2.
IsoalloLCA reduces C. perfringens strain CP1 growth and H2S production. C. perfringens was cultured with CA, CDCA, LCA, or isoalloLCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate discs. Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–f mean p < 0.05.

Figure 3.
C. perfringens CP1 virulence gene expression is reduced by DCA and isoalloLCA. C. perfringens CP1 was cultured with CA, CDCA, DCA, LCA, and isoalloLCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–e mean p < 0.05.
Figure 3.
C. perfringens CP1 virulence gene expression is reduced by DCA and isoalloLCA. C. perfringens CP1 was cultured with CA, CDCA, DCA, LCA, and isoalloLCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. All graphs show mean + SEM. Different letters of a–e mean p < 0.05.

Figure 4.
The effects of DCA on C. perfringens HN13 and its mutant growth and H2S production. C. perfringens strain HN13 and its three mutants were cultured with DCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate discs. # means No. All graphs show mean + SEM. ****, p < 0.0001. ns, not significant.
Figure 4.
The effects of DCA on C. perfringens HN13 and its mutant growth and H2S production. C. perfringens strain HN13 and its three mutants were cultured with DCA for 24 h under anaerobic conditions. (A) C. perfringens enumeration by serial dilution and plating. (B) Detection of H2S production via the lead acetate discs. # means No. All graphs show mean + SEM. ****, p < 0.0001. ns, not significant.

Figure 5.
The effect of DCA on C. perfringens HN13 mutant virulence gene expression. C. perfringens HN13 and its three mutants were cultured with DCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. # means No. All graphs show mean + SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. ns, not significant.
Figure 5.
The effect of DCA on C. perfringens HN13 mutant virulence gene expression. C. perfringens HN13 and its three mutants were cultured with DCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. # means No. All graphs show mean + SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. ns, not significant.

Figure 6.
The effects of isoalloLCA on C. perfringens HN13 mutant growth, H2S production, and the virulence gene expression. (A) C. perfringens strain HN13 and its three mutants were cultured with isoalloLCA for 24 h under anaerobic conditions. C. perfringens enumeration by serial dilution and plating. (B) C. perfringens strain HN13 and its three mutants were cultured with isoalloLCA for 24 h under anaerobic conditions. Detection of H2S production via the lead acetate discs. (C) C. perfringens HN13 and its three mutants were cultured with isoalloLCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. # means No. All graphs show mean + SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. ns, not significant.
Figure 6.
The effects of isoalloLCA on C. perfringens HN13 mutant growth, H2S production, and the virulence gene expression. (A) C. perfringens strain HN13 and its three mutants were cultured with isoalloLCA for 24 h under anaerobic conditions. C. perfringens enumeration by serial dilution and plating. (B) C. perfringens strain HN13 and its three mutants were cultured with isoalloLCA for 24 h under anaerobic conditions. Detection of H2S production via the lead acetate discs. (C) C. perfringens HN13 and its three mutants were cultured with isoalloLCA for 2 h. After RNA extraction and cDNA reverse transcription, the virulence gene accumulation was quantified by real-time PCR (qPCR). Results are representative of 3 independent experiments. # means No. All graphs show mean + SEM. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. ns, not significant.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Primers used for the detection of virulent genes.
| Gene | Primer Name | Sequence |
|---|---|---|
| gyrA | gyrA-F | TGCTTGTTGAC-GGACATGGT |
| gyrA-R | ACAACTGGTTCTTTTTCCTCACC | |
| asrA1 | asrA1-F | ACATCTTCCACATCCTACACACA |
| asrA1-R | TGATGAAATGGCTGGTGGACA | |
| plc | plc- F | TGACACAGGGGAATCACAAA |
| plc- R | CGCTATCAACGGCAGTAACA | |
| spoOA | spoOA- F | ACAGGAATTGCAAAGGATGG |
| spoOA- R | TTTTGTCTTGTCCAACAGCAG | |
| virT | virT- F | TGAAATTGTTCTTTTGGATGAAGA |
| virT- R | GCTTGAAAAGCTCCTGCCTA | |
| cpe | cpe- F | CAACTGCTGGTCCAAATGAA |
| cpe- R | GCATCTTTCGCCAGTTTCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alenezi, T.; Fu, Y.; Alrubaye, B.; Alanazi, T.; Almansour, A.; Wang, H.; Sun, X. Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence. Pathogens 2023, 12, 1202. https://doi.org/10.3390/pathogens12101202
AMA Style
Alenezi T, Fu Y, Alrubaye B, Alanazi T, Almansour A, Wang H, Sun X. Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence. Pathogens. 2023; 12(10):1202. https://doi.org/10.3390/pathogens12101202
Chicago/Turabian StyleAlenezi, Tahrir, Ying Fu, Bilal Alrubaye, Thamer Alanazi, Ayidh Almansour, Hong Wang, and Xiaolun Sun. 2023. "Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence" Pathogens 12, no. 10: 1202. https://doi.org/10.3390/pathogens12101202
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.