1. Introduction
Lettuce is one of the most widely cultivated leafy vegetables in Greece, grown in the field or under protection in multitunnels or in greenhouses [
1]. Lettuce cultivation in Greece covers an area of almost 3.200 hectares, in open fields and under greenhouses, and the production reached 46.643 tons [
2]. A large number of soilborne and foliar pathogens affect lettuce cultivation in Europe [
3]. Moreover, the intensive cultivation of leafy vegetables and the absence of adequate crop rotation, as well as climate change and the globalization of the seed market, constitute significant factors for the proliferation and subsequent spread of new pathogens [
4]. Among these recently recorded pathogens,
Fusarium equiseti could represent a significant plant pathogen for leafy vegetables, including lettuce and wild and cultivated rocket [
3,
5].
F. equiseti is a seedborne pathogen affecting many plant hosts [
6]. The specific soil-inhabiting fungus survives as a saprotroph in soil or crop debris and thus this feature enables the ability of adapting easily to intensive cropping systems [
3,
4].
Lettuce is highly susceptible to
F. equiseti, exhibiting severe yield losses at temperatures ranging from 25 to 35 °C and 1 to 3 h of high relative humidity [
5]. The growers of leafy vegetables make use of chemical fungicides as most times the control of the environmental conditions is not practicable [
5]. In addition,
F. equiseti has the ability to produce a diversity of mycotoxins, thus constituting a potential risk for human and animal health [
7,
8].
Fusarium equiseti is a member of the
Fusarium incarnatum-equiseti species complex (FIESC) which is comprised of species exhibiting significant genetic variability [
9]. This species complex is divided into two clades, named
Incarnatum and
Equiseti [
10,
11]. FIESC includes more than 30 phylogenetically distinct species based on morphological identification and sequencing of more than one gene [
7,
12,
13,
14,
15,
16].
F. equiseti was defined as a distinct phylogenetic species within the subclade FIESC-14 [
17].
For molecular identification, Maryani et al. (2019) used a multi-gene phylogeny that included partial fragments of the beta-tubulin (
Bt), calmodulin (
CAM), translation elongation factor 1-alpha (
TEF1-α), the internal transcribed spacer region of the rDNA (ITS), the large subunit of the rDNA (
LSU), as well as the RNA polymerase II large subunit (
RPB1) and second-largest subunit (
RPB2) genes. ITS,
TEF1-α,
RPB1, and
RPB2 were also used for the description of new FIESC species [
14]. In addition, a species phylogeny inferred from partial nucleotide sequences from four housekeeping genes (
CAM,
RPB2,
TEF1-α, and
Bt) was carried out to identify FIESC species [
7]. Furthermore, a multi-locus approach based on ITS,
TEF1-α,
CAM,
RPB1, and
RPB2 was followed in order to distinguish species within the FIESC [
15], while a three-gene (
TEF1-α,
CAM, and
RPB2) phylogenetic inference was also used for the introduction of Latin binomials to unnamed FIESC phylo-species [
16]. More recently, Matić et al. [
12] identified isolates obtained from leafy vegetables using MLST analyses of four loci (
ΤEF1-α,
CAM,
Bt, and IGS).
Pathogen identification based on morphological characteristics and microscopy is considered to be more or less subjective [
18]. Molecular methods are highly sensitive and can detect and quantify microorganisms present even at low concentrations [
19]. Real-time quantitative PCR (RT-qPCR) provides a reliable quantification of the population densities of several soilborne plant pathogens [
20]. DNA can be extracted from various environmental samples, such as host tissues, soil, water, and air [
20,
21]. For instance, qPCR assays have already been developed for the detection and quantification of
Rhizoctonia solani from soil and plant tissue samples derived from lettuce fields [
22].
Accurate identification of
Fusarium species is complicated and the morphological, biological, and especially phylogenetic approaches used most times lead to controversial results [
23,
24]. Thus, the development of cost-effective RT-qPCR assays for the detection and quantification of specific
Fusarium spp. from soil samples could be of a great importance, especially for species infecting a broad range of plant hosts.
Foliar symptoms on lettuce consisting of circular to angular lesions which later become necrotic and cracked have been associated with anthracnose disease, caused by
Microdochium panattonianum (syn.
Marssonina panattoniana) [
25] in Greece [
26]. Interestingly enough, a recent review study on pathogens infecting leafy vegetables did not discuss anthracnose as an important disease of lettuce [
3]. On the contrary, similar leaf spots caused by
F. equiseti have already been recorded on lettuce in Italy [
27].
Updated knowledge of the exact fungal species causing significant foliar symptoms and yield losses on lettuce cultivations in Greece is currently not available. Hence, the objectives of this research were to (a) associate the foliar symptoms observed on lettuce in the field with specific pathogens, (b) identify the isolates obtained from symptomatic plants using morphological characteristics and sequence analysis of the internal transcribed spacer ITS1-5.8S-ITS2 rDNA region (ITS-rDNA), translation elongation factor 1-alpha (TEF1-a), calmodulin (CAM), beta-tubulin (Bt), and small subunit (SSU) genes, (c) assess the pathogenicity of selected F. equiseti isolates on lettuce, and (d) develop an efficient RT-qPCR assay for the detection and quantification of the pathogen in soil samples collected from fields cultivated intensively with lettuce.
2. Materials and Methods
2.1. Pathogen Isolation
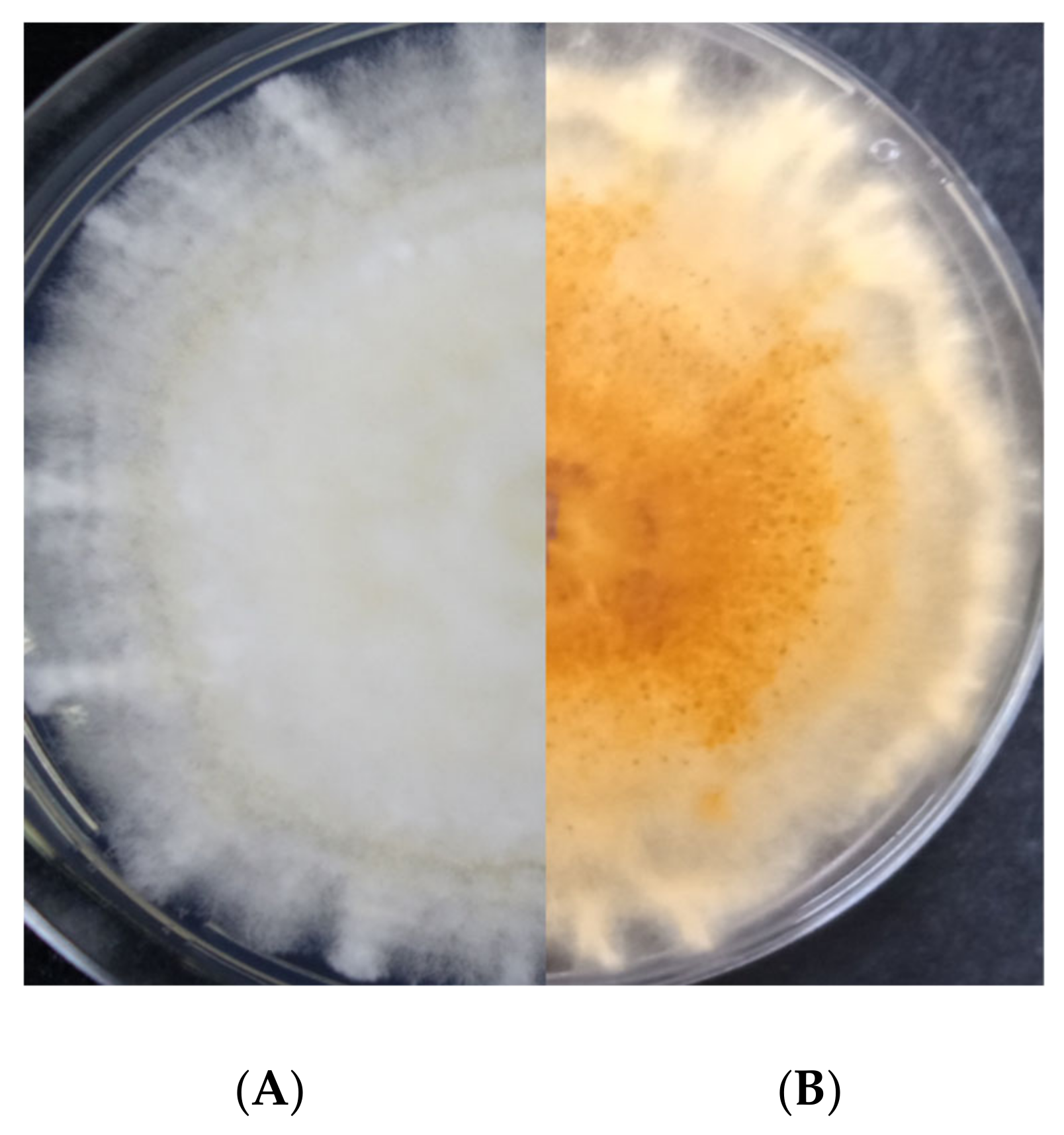
A survey was conducted in the fields of a commercial farm specialized on the cultivation of leafy vegetables (Vezyroglou Farm, Alexandria, Imathia, Central Macedonia, Greece) in order to obtain isolates of the pathogen(s) causing foliar disease symptoms on Romaine lettuce plants. The symptoms recorded on the leaves and stems were small, 1 to 4 mm in diameter, dark yellow to brown, circular to angular leaf spots which later became necrotic, and which were sometimes cracked in the center (
Figure 1). The disease was severe and widespread, and disease symptoms were observed on 20 to 35% of the leaves of almost all lettuce plants cultivated in an area of approximately 10 ha.
Diseased plants were arbitrarily selected at several sites in the fields and were transported to the laboratory in individual polyethylene bags to prevent cross-contamination. Tissues of symptomatic plants, taken from the margins of leaf and stems spots, were surface disinfested with a 10% sodium hypochlorite solution for 1 min and then were washed three times with sterilized distilled water. Tissue pieces were placed on potato dextrose agar (PDA; Oxoid, Thermo Fisher Scientific, Leiden, The Netherlands) amended with 1 mL of lactic acid 10% per 100 mL of PDA (pH 4.5), and incubated for 2 to 3 days, at 24 °C, in the dark. The yielded colonies were transferred onto new PDA dishes and from each petri dish, used for the initial isolation, only one isolate was kept in pure culture. The single-spore isolates obtained in this study were preserved at 4 °C for long-term storage.
2.2. Morphological Characterization
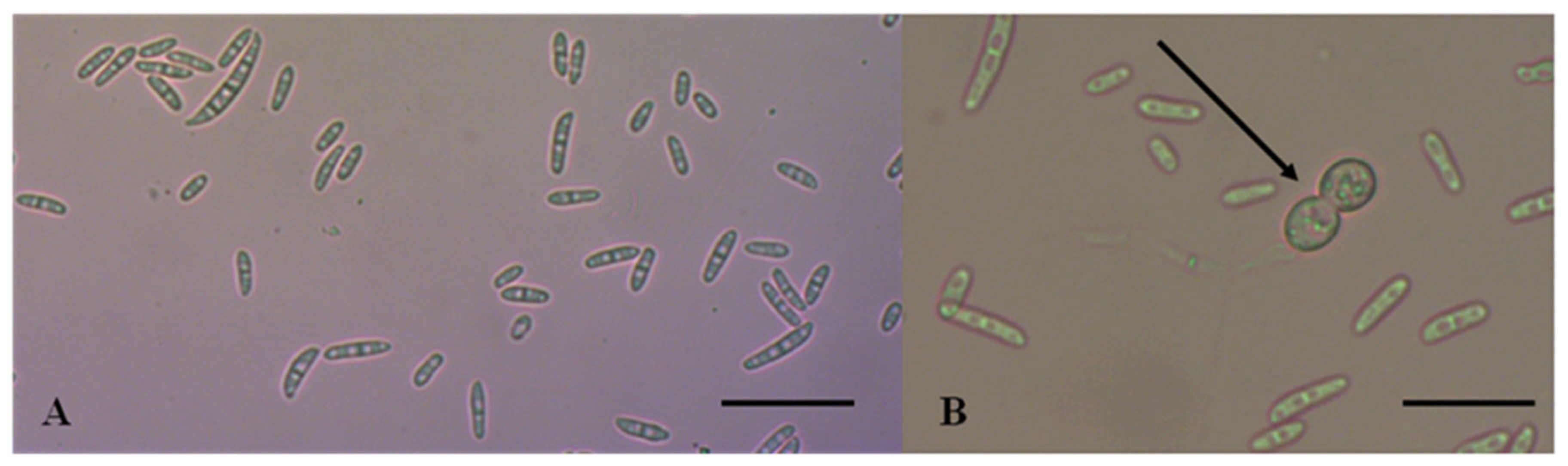
All isolates obtained were morphologically characterized following the protocols described by Leslie and Summerell [
9]. Colony morphology and pigmentation were evaluated on PDA after 7 days of incubation at 24 °C in the dark. The isolates were evaluated for typical characteristics of FIESC species. Mycelial growth was estimated measuring the colony diameter in two perpendicular directions. Micromorphological characteristics of conidial shape and size were examined using water as mounting medium. Fungal structures were observed, captured, and measured (
n = 100) using Zen 10 software of a Carl Zeiss, AXIO Lab A1 microscope.
2.3. Pathogenicity Assays
Lettuce seedlings of Romaine type (cv. Green Towers) were used for pathogenicity assays. Four-week-old lettuce seedlings were planted in plastic pots containing 200 g of sterilized substrate mixture (peat moss and perlite at a rate of 4:1). The candidate isolates were first cultured on PDA for 10 days to achieve sporulation. The conidia were then harvested and diluted to a final concentration of 1 × 106 conidia/mL in sterile distilled water. The inoculum suspension was applied onto the above ground plant organs using an atomizer just before runoff.
Thereafter, the plants were covered with polyethylene bag for 3 days in a growth chamber to maintain high relative humidity. In the growth chamber the mean daily temperature was kept at 23 ± 2 °C. Twenty plants per isolate (three isolates were used in total) were arranged in a randomized block design and the experiment was repeated three times. Twenty plants sprayed with sterilized water were kept as controls. Ten days after inoculation, plants were examined for symptoms developed on their above-ground organs. Hence, in order to fulfill Koch’s postulates, isolations were carried out from non-inoculated plants and from the ones inoculated with the evaluated isolates. Additionally, the isolates derived from artificially inoculated plants were amplified with TEF1-α genes and the amplicons were sequenced.
2.4. DNA Extraction
Petri dishes (diameter 9 cm) containing ca. 20 mL PDA, overlain with sterilized cellophane sheets (gel drying frames, Sigma–Aldrich Chemie GmbH, Taufkirchen, Germany), were inoculated with mycelial plugs (4 mm in diameter) and incubated for 3–5 days in the dark at 24 °C. The mycelium was scraped from each plate, lyophilized, and ground to a fine powder. Genomic DNA was extracted from this material using the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The concentration of the extracted DNA was measured using a P330 nanophotometer (Implen GmbH, Munich, Germany).
2.5. PCR Amplification, Sequencing, and Phylogenetic Analysis
Initially, the ITS1-5.8S-ITS2 region was amplified with primers ITS1/ITS4 [
28]. The translation elongation factor 1-alpha (
TEF-1α) gene was amplified with primers EF-1/EF-2 [
29], while partial amplification of calmodulin (
CAM), and beta-tubulin (
Bt) was performed with CL1/CL2A [
30] and Bt2a/Bt2b [
31] primers, respectively. In addition, partial amplification of small subunit (
SSU) was performed with the specific primers PNS1/NS41 [
32]. Aliquots of the PCR products were loaded on 1.0% agarose gel in Tris-acetate-EDTA buffer with Midori Green Advance gel stain (Nippon, Duren, Germany). PCR products were purified with PureLink PCR Purification Kit (Invitrogen, Carlsbad, CA, USA) and custom sequenced (CEMIA).
All the sequences generated in this study were initially visualized by ChromasLite (Technelysium, South Brisbane, Australia), then visually aligned and two representative sequences were deposited in the GenBank. The sequence data obtained were compared by BLAST search on the National Center for Biotechnology Information (NCBI) database to determine the species, searching for similarities between the sequences obtained in this study and already existing sequences in the database.
In accordance with the primer/probe design which is described in
Section 2.6.1, the best hits obtained from the blast searches were downloaded and used in the construction of a phylogenetic tree. Phylogenetic analysis was conducted using MEGA version 7.0, and maximum likelihood (ML) method was used to generate the phylogenetic tree from the
TEF1-α gene. Bootstrap values were obtained from 1000 replicates and distances were calculated using Kimura-2p in both phylogenetic inferences. The sequence of
Didymella pinodella (GenBank accession no. MK525067.1) was used as an outgroup for rooting the phylogenetic tree.
2.6. Development of a Fusarium equiseti-Specific qPCR Assay
2.6.1. RT-qPCR Primers/Probe Design and Specificity
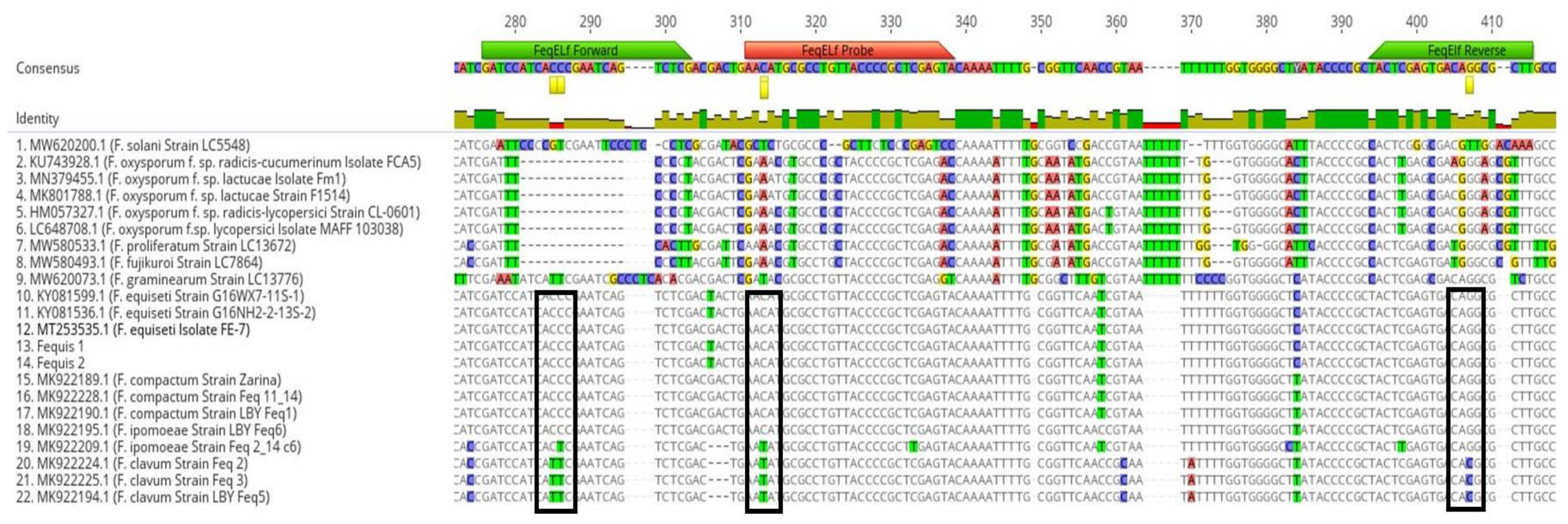
Partial gene sequences of the translation elongation factor 1-alpha (TEF1-α) gene were evaluated for the presence of suitable regions to design primers and a probe specific for F. equiseti. TEF1-α sequences, corresponding to F. equiseti, the Equiseti clade of the FIESC, and to other Fusarium spp., were retrieved from the GenBank database and examined for regions potentially unique to F. equiseti.
The nucleotide alignment of
F. equiseti TEF1-α sequences obtained in this study and retrieved from the NCBI database was used to design the RT-qPCR primers-probe set (
Figure 2). The sequences were aligned using the MAFT alignment tool available in Geneious V9.1.8. Based on the alignment, a set of two primers (FeqELf-F and FeqELf-R) and a probe (FeqELf-Pro) were designed to cover all the variability detected in the selected area, amplifying a PCR product of 128 bp (
Table 1). The TaqMan probe sequence was labeled with the fluorescent reporter dye 6-FAM (6-fluorescein amidate) and the Iowa Black Fluorescent Quencher (IABkFQ) at the 5′ and 3′ ends, respectively, and an internal ZEN quencher. For primer and probe selection, specific criteria were followed as described by Šišić et al. [
18]. Potential secondary structures, such as hairpin, self-dimer, and hetero-dimer interactions were checked using the oligoanalyzer tool from Integrated DNA Technologies (
https://www.idtdna.com/pages/tools/oligoanalyzer (accessed on 15 February 2022)). The specificity of the primers and probe was scanned against the NCBI database using basic local alignment search tools.
The designed primers were subjected to conventional PCR to confirm their specificity. 10 μL of 5×One Taq Standard Reaction Buffer (New England BioLabs Inc., Hitchin, UK), 1.0 μL of dNTPs at 10 μM, 1.0 μL of each primer at 0.5 μM (IDT, Leuven, Belgium), 0.25 μL of Taq DNA polymerase (New England BioLabs Inc., Hitchin, UK), 5 mL of DNA template, and 31.75 mL of sterile distilled water were added to each reaction tube to make a final volume of 50 μL. PCR was performed with a thermocycler (SensoQuest, Labcycler, Germany) with the following amplification conditions: initial denaturation step at 94 °C for 5 min; 30 cycles of denaturation at 94 °C for 30 s, primer annealing at 55 °C for 30 s, and extension at 72 °C for 1 min and a final extension at 72 °C for 5 min. The PCR step also included DNA samples from seven common soilborne fungal pathogens, as well as no-template samples as negative controls.
The primer/probe set was validated for specificity using a fungal species panel by RT-qPCR amplification following the protocol described below in
Section 2.6.2. The validation panel included DNA extracts from a range of fungal and Oomycete species, including
F. equiseti isolates obtained in this study,
Rhizoctonia solani and
Pythium ultimum commonly occurring in lettuce cultivated fields [
1],
F. oxysporum f.sp.
lactucae from lettuce plants showing wilt symptoms, as well as other
Fusarium spp. (
F. proliferatum,
F. fujikuroi,
F. gramineraum,
F. solani,
F. oxysporum f.sp.
radicis-lycopersici, and
F. oxysporum f.sp.
radicis-cucumerinum) available at the fungal collection of Plant Pathology Lab, AUTh. In addition, DNA extracted with the DNeasy Power Soil kit (Qiagen, Hilden, Germany), from soil samples collected from the surveyed field was also included in the assay.
2.6.2. qPCR Conditions of Fusarium equiseti Assay
DNA extracted from single-spore cultures was amplified with the primer/probe set developed in this study. The reaction mixture for each DNA sample, consisted of 3 μL of DNA template, 10 μL of Luna Universal Probe qPCR Master Mix (New England Biolabs, Ipswich, MA, USA), 500 nM of each of the forward and reverse primers, 250 nM of the probe, and 0.1 μL BSA (New England Biolabs, Ipswich, MA, USA) in a final volume of 20 μL. For each sample triplicate reactions were performed and molecular grade water (Panreac, AppliChem, Barcelona, Spain) was used as negative control. The RT-qPCR reactions were performed using a Strategene Mx3005P qPCR System (Agilent Technologies, Santa Clara, CA, USA) on 96-well plates. Amplification conditions consisted of a single cycle at 94 °C for 1 min, followed by 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and 94 °C for 10 s. Data were collected in the last holding stage of each cycle and the results were analyzed using the Strategene MxPro-Mx3005P Software (Version 4.00; Agilent Technologies, Santa Clara, CA, USA).
2.6.3. Preparation of RT-qPCR Standard Curve
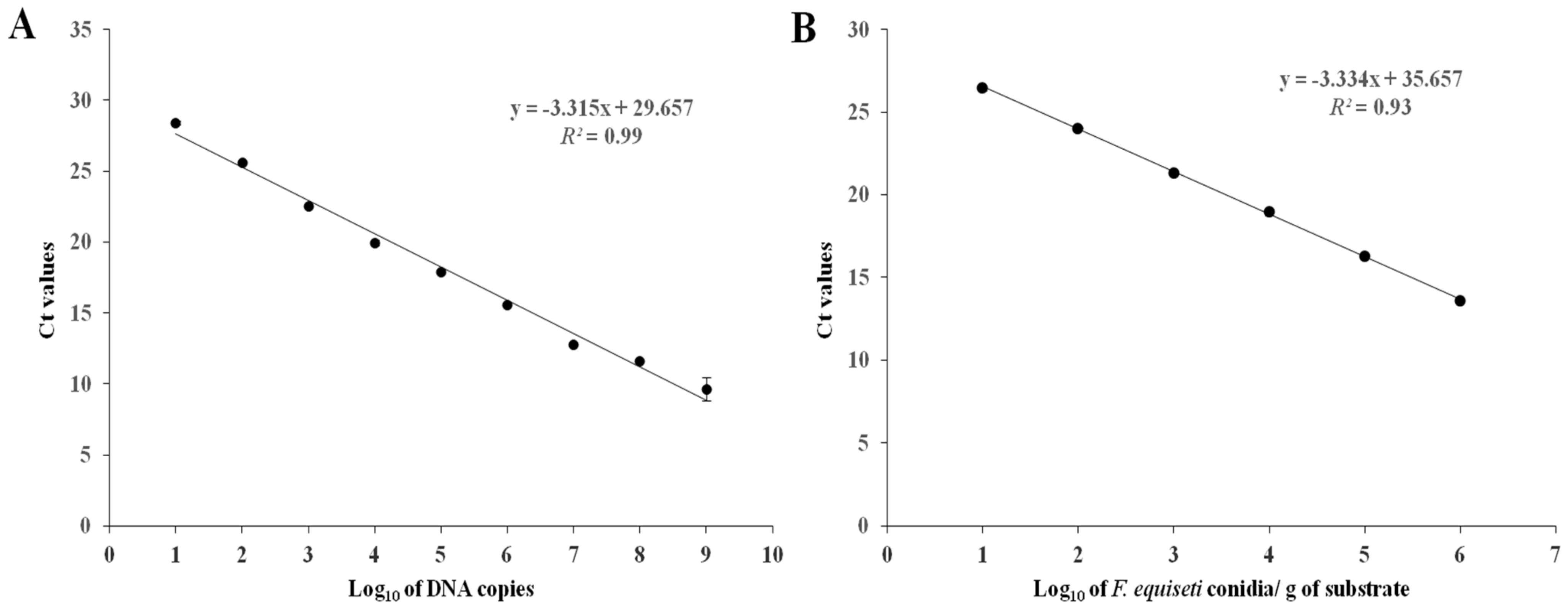
For the preparation of the standard curve, the specific fragment targeted by the qPCR was amplified by conventional PCR using primers FeqELf-F and FeqELf-R. The 128-bp PCR product was purified using PureLink Purification Kit (Invitrogen, Carlsbad, CA, USA), and then inserted in the pUC19 vector contained in the NEB PCR Cloning kit (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions, and cloned into 10-beta Competent Escherichia coli cells contained in the same kit. Transformant colonies were selected by ampicillin resistance. Plasmid DNA purification was carried using the Monarch Plasmid Miniprep Kit (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions and the concentration of the extracted DNA was measured using a P330 nanophotometer (Implen GmbH, Munich, Germany).
The Avogadro constant (6.023 × 1023 molecules/mol) was used to estimate the number of plasmid molecules. Three replicates of ten-fold serial dilutions (109 down to 101 plasmid copies/μL) were prepared and used to generate the standard curve. 1 μL from each dilution was used in RT-qPCR in order to determine the amplification efficiency, the dynamic range of quantification, and the detection limit of the assay. The threshold cycle (Ct) values were automatically calculated with the Strategene MxPro-Mx3005P Software (Version 4.00; Agilent Technologies, Santa Clara, CA, USA) and the standard curve was created by plotting the Ct values versus the logarithm of the concentration of each serial dilution. For each serial dilution, the mean Ct value was estimated, and the number of DNA molecules in each sample was calculated by incorporation of the mean Ct value to the standard curve equation.
2.7. Validation of the Assay in Artificially Inoculated Substrate with F. equiseti Spores
One selected isolate (Fequis 1) identified in this study was used to inoculate a sterile substrate (peat moss, Hawita Professional), autoclaved at 121 °C for 60 min twice, at an interval of 24 h. The pathogen inoculum was prepared as described above in
Section 2.3 of this manuscript. One mL of the
F. equiseti conidial suspensions (10
2, 10
3, 10
4, 10
5, 10
6 conidia mL
−1) were added to the sterile substrate, mixed meticulously, and then placed into 50 mL Falcon tubes. Controls consisting of sterile substrate were inoculated solely with sterile distilled water. The substrate-
F.equiseti conidia mixture was subsequently dried at 60 °C for 24 h. For each concentration, DNA extraction was performed in triplicate using the DNeasy Power Soil kit and amplified using the RT-qPCR assay developed in this study.
4. Discussion
This is the first comprehensive study aiming to identify the pathogen associated with leaf spot disease symptoms on lettuce in Greece. Our results revealed that the pathogen involved in the observed foliar disease is
F. equiseti. Until now, in Greece, the specific disease has been attributed to the pathogen
Microdochium panattonianum (syn.
Marssonina panattoniana) and has been known as ‘‘lettuce anthracnose’’. However, the results of the amplified product of the small ribosomal subunit (
SSU) gene of the isolates obtained in this study showed no homology at all with the existing sequences in the NCBI database. In addition, the morphological characteristics and measurements of the isolates tally with those of
F. equiseti [
9,
12,
15,
16].
Nonetheless, the identification based only on morphology traits is considered insufficient, since genetic methods are required to identify the species precisely [
9,
10,
15,
16,
33]. Hence, in the present study, one nuclear genomic region (ITS-rDNA) and three genes (
TEF1-a,
CAM, and
Bt) were also amplified. A BLAST search of the generated sequences revealed 100% identity with various sequences of
F. equiseti retrieved from the NCBI database.
In previous studies, multi-locus datasets encompassing the internal transcribed spacer region (ITS), the intergenic spacer region (IGS), and/or several genes such as
TEF1-a,
CAM,
Bt,
LSU,
RPB1, and
RPB2 were generated and were very efficient in distinguishing the species belonging to the FIESC [
7,
12,
13,
14,
15,
16]. More specifically, the diversity of FIESC in isolates from lettuce and other leafy vegetables has been presented in the highly informative study of Matić et al. [
12]. These authors characterized the isolates obtained from leafy vegetable hosts using, besides morphological characteristics, MLST analyses of four different loci (
TEF1-a,
CAM,
Bt, and IGS).
Although FIESC has recently been the main objective of several research studies [
7,
12,
13,
14,
15,
16], no information is yet available about its potential detection and subsequent quantification in soil samples. For this reason, we attempted to develop a RT-qPCR assay for the detection and quantification of this emerging plant pathogen, using a newly designed primer pair (FeqELf-F and FeqELf-R) and a probe (FeqELf-Pro). The qPCR showed a high level of specificity and sensitivity for the detection of
F. equiseti in lettuce cultivated fields. The detection limit of the RT-qPCR assay was 10 copies/μL for
F. equiseti, and the linearity of the curve, denoted by the R squared value (
R2) of the developed standard curve, was 0.99, indicating that this standard curve is reliable for quantification of the target organism in soil samples.
Applied detection methods could be crucial for the reliable identification of the pathogens infecting leafy vegetables, enabling the implementation of preventative management methods [
3]. The main method of controlling fungal diseases presented in fields cultivated with leafy vegetables is crop rotation, aiming to keep the inoculum concentration of specific pathogens as low as possible [
5]. However, pathogens with a broad range of plant hosts could constitute problematic cases in terms of control. In cases like this, effective monitoring and reliable quantification could contribute to a more effective disease management. For instance, specific and sensitive quantitative PCR assays were developed for the detection and quantification of
R. solani, causing bottom rot of lettuce, in tissue and soil samples [
22]. In this study, qPCR assay was sensitive enough to detect the lowest soil concentration of
R. solani AG1-IB, capable of inducing symptoms in head lettuce, constituting thus a potential useful tool for the detection and management of lettuce bottom rot disease.
Accurate identification of
Fusarium species using morphological criteria and/or molecular methods is laborious and at most times ambiguous [
9,
23]. In addition, as O’Donnell et al. [
34] have already pointed out, the top BLAST matches upon querying the GenBank database may be imprecise or even confusing due to originally misidentified sequences and/or updated taxonomy. Therefore, the quantitative PCR has turned into a significant laboratory method for the detection and quantification of specific DNA targets, an assumption that has been confirmed recently for
F. avenaceum [
24]. In that study, a primer pair, designed from the
TEF1-α gene sequence, has been effectively used for the quantification of
F. avenaceum, a cosmopolitan pathogen, in soil and seed samples.
Most likely, the recorded emergence of
F. equiseti on lettuce in Greece could be due to its altered pathogenic status, from sporadic weak to principal plant pathogen, and its extended plant host range [
12]. Moreover, the emergence of the specific fungus on new plant hosts could be associated with environmental changes [
12]. Increased disease incidence and severity, caused by
F. equiseti, has already been reported on wild rocket and radish under elevated average temperatures and CO
2 concentrations [
35]. Garibaldi et al. [
36] pointed out the effect of temperature and leaf wetness duration on the incidence and severity of leaf spot caused by
F. equiseti on lettuce and wild rocket under controlled conditions. In their study, lettuce was more susceptible to this pathogen, exhibiting higher disease index and disease severity, at 25–30 °C and after a few hours (1–3 h) of leaf wetness. On the contrary, in case of lower temperatures (15–30 °C), at least 12 h of leaf wetness were needed in order to achieve favorable conditions for disease development and significant losses.
In addition,
F. equiseti could be transmitted by seeds of several leafy vegetable hosts, such as wild rocket [
3]. This epidemiological aspect, along with the climate change scenario, could constitute a potential cause for the outbreak of
F. equiseti on leafy vegetables [
4,
12]. Interestingly enough, this broad-range plant pathogen, once introduced in a field, can survive on infested plant debris or in soil, making rotations in severely infected areas of limited success [
3,
4].
In our opinion, as the identification of FIESC species based only on morphological characteristics is considered problematic [
7], and the molecular identification requires the amplification of more than one gene [
7,
12,
13,
14,
15,
16], a sensitive, specific, and reproducible RT-qPCR could provide reliable detection of the specific pathogen. Henceforth, the RT-qPCR method developed in this study could be used for the quantification of
F. equiseti in soil samples, collected from fields cultivated with leafy vegetables or even with other plant hosts.
Disease diagnosis and plant pathogen detection are key factors to important agronomic decisions, such as selection of plant species for cultivation and selection of the most efficient management method [
37]. The broad plant host range of
F. equiseti, in addition to the environmental conditions that are favorable for the development of the disease, make its control difficult and most times inefficient [
5]. As it is impractical to control environmental parameters (mainly temperature and relative humidity [
36]), growers and agriculturists usually rely on the chemical management of the pathogen, which is still under consideration [
5]. Therefore, the probe-based qPCR assay developed in this manuscript could constitute a potential useful procedure for the detection and quantification of
F. equiseti in soil samples and afterwards the evaluation of the adopted management methods.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}