
Genetic Characterization of Avian Influenza Viruses Isolated from the Izumi Plain, Japan in 2019/20 Winter Season
 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. AIV Isolation from Crane Roost Site Water on the Izumi Plain during the 2019/20 Winter Season
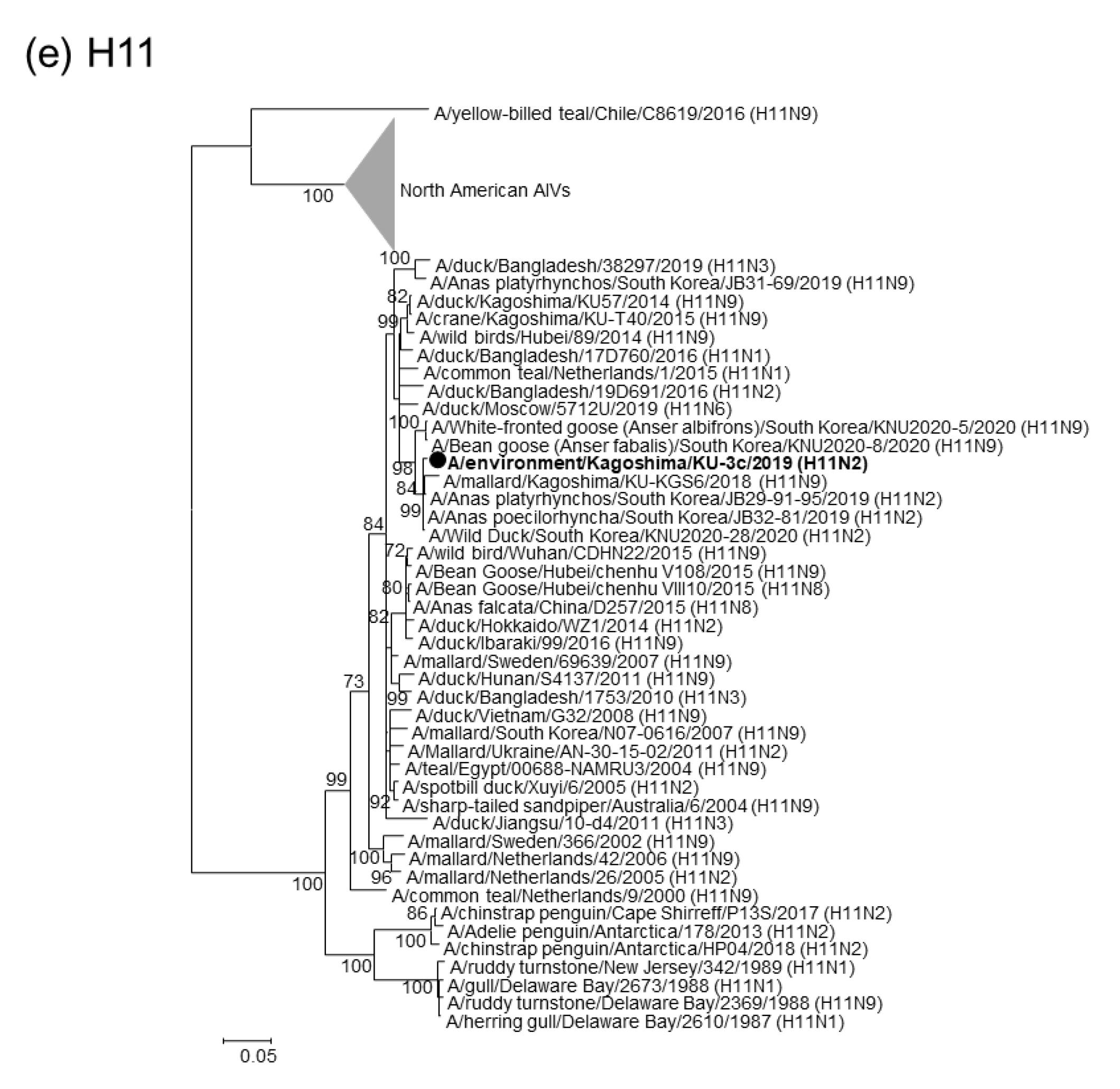
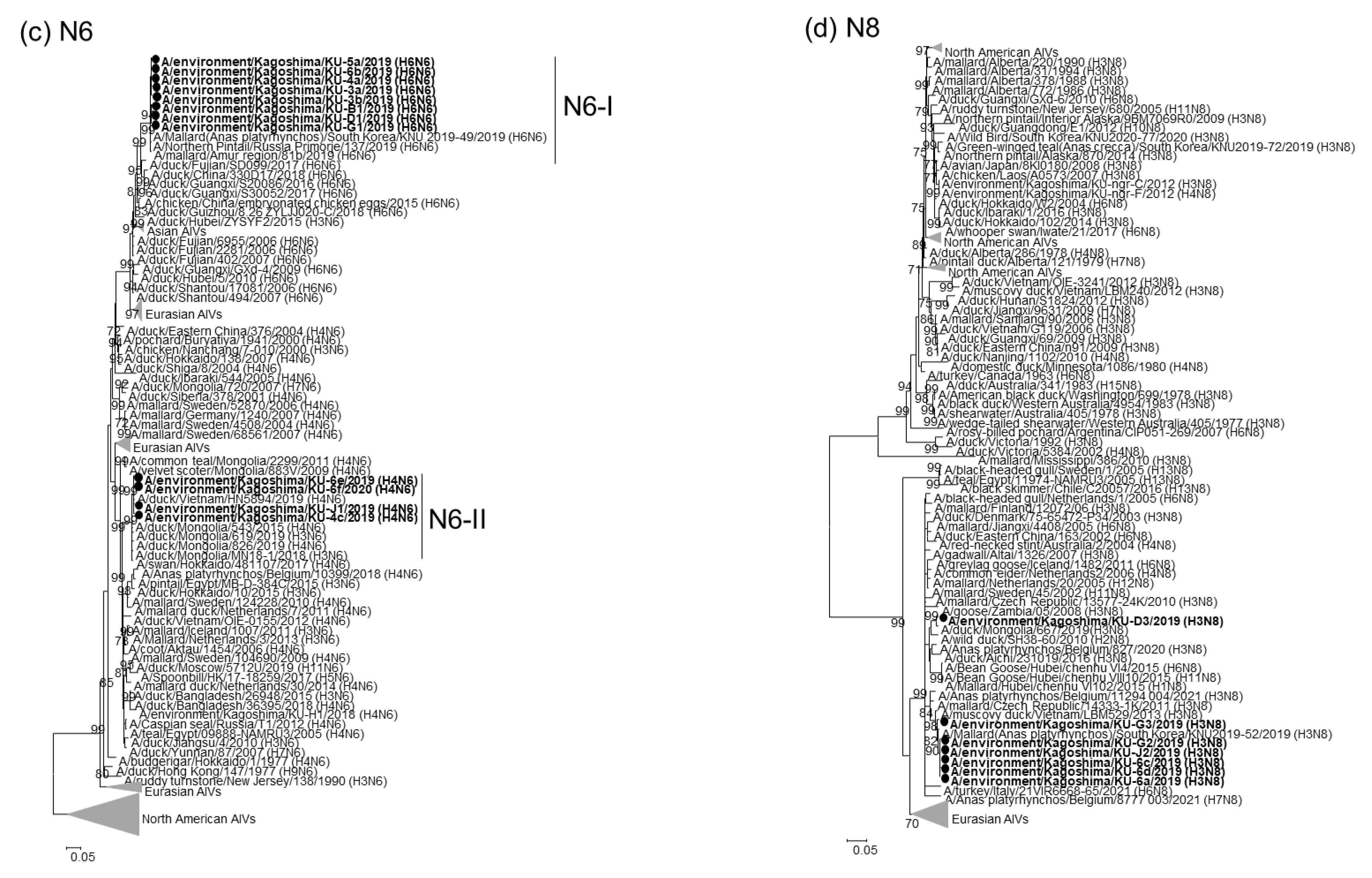
2.2. Phylogenetic Analyses of Our 22 AIV Isolates
2.3. Genotypes of Our 22 AIV Isolates
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. AIV Isolation from Water Samples
4.3. Real Time Reverse Transcription (RT)-PCR and HA and NA Subtyping
4.4. Sequence Analyses of AIV Genes
4.5. Phylogenetic Analyses for AIV Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, S.E.; Cox, N.J.; Klimov, A. Genetic analysis of human H2N2 and early H3N2 influenza viruses, 1957–1972: Evidence for genetic divergence and multiple reassortment events. Virology 2004, 328, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Simonsen, L.; Viboud, C.; Miller, M.A.; Taylor, J.; George, K.S.; Griesemer, S.B.; Ghedin, E.; Sengamalay, N.A.; Spiro, D.J.; et al. Stochastic processes are key determinants of short-term evolution in influenza a virus. PLoS Pathog. 2006, 2, e125. [Google Scholar]
- Kandeil, A.; El-Shesheny, R.; Maatouq, A.; Moatasim, Y.; Cai, Z.; McKenzie, P.; Webby, R.; Kayali, G.; Ali, M.A. Novel reassortant H9N2 viruses in pigeons and evidence for antigenic diversity of H9N2 viruses isolated from quails in Egypt. J. Gen. Virol. 2016, 98, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Jang, Y.; Nguyen, T.D.; Jones, J.; Shepard, S.S.; Yang, H.; Gerloff, N.; Fabrizio, T.; Nguyen, L.V.; Inui, K.; et al. Shifting Clade Distribution, Reassortment, and Emergence of New Subtypes of Highly Pathogenic Avian Influenza A(H5) Viruses Collected from Vietnamese Poultry from 2012 to 2015. J. Virol. 2017, 91, e01708-16. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenstrom, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Okuya, K.; Kawabata, T.; Nagano, K.; Tsukiyama-Kohara, K.; Kusumoto, I.; Takase, K.; Ozawa, M. Isolation and characterization of influenza A viruses from environmental water at an overwintering site of migratory birds in Japan. Arch. Virol. 2015, 160, 3037–3052. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Matsuu, A.; Tokorozaki, K.; Horie, M.; Masatani, T.; Nakagawa, H.; Okuya, K.; Kawabata, T.; Toda, S. Genetic diversity of highly pathogenic H5N8 avian influenza viruses at a single overwintering site of migratory birds in Japan, 2014/15. Eurosurveillance 2015, 20, 21132. [Google Scholar] [CrossRef]
- Okamatsu, M.; Ozawa, M.; Soda, K.; Takakuwa, H.; Haga, A.; Hiono, T.; Matsuu, A.; Uchida, Y.; Iwata, R.; Matsuno, K.; et al. Characterization of Highly Pathogenic Avian Influenza Virus A(H5N6), Japan, November 2016. Emerg. Infect. Dis. 2017, 23, 691–695. [Google Scholar] [CrossRef]
- Nakagawa, H.; Okuya, K.; Kawabata, T.; Matsuu, A.; Takase, K.; Kuwahara, M.; Toda, S.; Ozawa, M. Genetic characterization of low-pathogenic avian influenza viruses isolated on the Izumi plain in Japan: Possible association of dynamic movements of wild birds with AIV evolution. Arch. Virol. 2018, 163, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Fujimoto, Y.; Kojima, I.; Esaki, M.; Ri, K.; Masatani, T.; Matsui, T.; Ozawa, M. Genetic Characterization of H5N8 Highly Pathogenic Avian Influenza Viruses Isolated from Falcated Ducks and Environmental Water in Japan in November 2020. Pathogens 2021, 10, 171. [Google Scholar] [CrossRef]
- Khalil, A.M.; Kojima, I.; Fukunaga, W.; Okajima, M.; Mitarai, S.; Fujimoto, Y.; Matsui, T.; Kuwahara, M.; Masatani, T.; Okuya, K.; et al. Improved method for avian influenza virus isolation from environmental water samples. Transbound. Emerg. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Hatai, H.; Fujimoto, Y.; Kojima, I.; Okajima, M.; Esaki, M.; Kinoshita, K.; Ozawa, M. A Lethal Case of Natural Infection with the H5N8 Highly Pathogenic Avian Influenza Virus of Clade 2.3.4.4 in a Mandarin Duck. Zoonotic Dis. 2022, 2, 32–36. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Ashizawa, H.; Nakanishi, K.; Kaji, N.; Suzuki, K.; Okamatsu, M.; Yamaguchi, S.; Mase, M. Subtyping of avian influenza viruses H1 to H15 on the basis of hemagglutinin genes by PCR assay and molecular determination of pathogenic potential. J. Clin. Microbiol. 2008, 46, 3048–3055. [Google Scholar] [CrossRef]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | Isolate | Collection Date | Isolate ID * |
|---|---|---|---|
| H1N1 | A/environment/Kagoshima/KU-G4/2020 (H1N1) | 27 January 2020 | EPI_ISL_13374949 |
| H3N2 | A/environment/Kagoshima/KU-4b/2019 (H3N2) | 2 December 2019 | EPI_ISL_13374950 |
| H3N8 | A/environment/Kagoshima/KU-6a/2019 (H3N8) | 18 November 2019 | EPI_ISL_13374979 |
| A/environment/Kagoshima/KU-6c/2019 (H3N8) | 25 November 2019 | EPI_ISL_13375104 | |
| A/environment/Kagoshima/KU-6d/2019 (H3N8) | 25 November 2019 | EPI_ISL_13375105 | |
| A/environment/Kagoshima/KU-D3/2019 (H3N8) | 2 December 2019 | EPI_ISL_13375750 | |
| A/environment/Kagoshima/KU-G2/2019 (H3N8) | 2 December 2019 | EPI_ISL_13375751 | |
| A/environment/Kagoshima/KU-G3/2019 (H3N8) | 2 December 2019 | EPI_ISL_13417555 | |
| A/environment/Kagoshima/KU-J2/2019 (H3N8) | 9 December 2019 | EPI_ISL_13418079 | |
| H4N6 | A/environment/Kagoshima/KU-J1/2019 (H4N6) | 25 November 2019 | EPI_ISL_13418433 |
| A/environment/Kagoshima/KU-4c/2019 (H4N6) | 23 December 2019 | EPI_ISL_13418523 | |
| A/environment/Kagoshima/KU-6e/2019 (H4N6) | 23 December 2019 | EPI_ISL_13418524 | |
| A/environment/Kagoshima/KU-6f/2020 (H4N6) | 20 January 2020 | EPI_ISL_13418525 | |
| H6N6 | A/environment/Kagoshima/KU-3a/2019 (H6N6) | 18 November 2019 | EPI_ISL_13437619 |
| A/environment/Kagoshima/KU-5a/2019 (H6N6) | 18 November 2019 | EPI_ISL_13437942 | |
| A/environment/Kagoshima/KU-6b/2019 (H6N6) | 18 November 2019 | EPI_ISL_13437509 | |
| A/environment/Kagoshima/KU-3b/2019 (H6N6) | 25 November 2019 | EPI_ISL_13439033 | |
| A/environment/Kagoshima/KU-4a/2019 (H6N6) | 25 November 2019 | EPI_ISL_13439034 | |
| A/environment/Kagoshima/KU-B1/2019 (H6N6) | 25 November 2019 | EPI_ISL_13439048 | |
| A/environment/Kagoshima/KU-D1/2019 (H6N6) | 25 November 2019 | EPI_ISL_13439049 | |
| A/environment/Kagoshima/KU-G1/2019 (H6N6) | 25 November 2019 | EPI_ISL_13439050 | |
| H11N2 | A/environment/Kagoshima/KU-3c/2019 (H11N2) | 16 December 2019 | EPI_ISL_13439051 |
| Subtype | Isolate | Genetic Clade * of the Following Gene Segment: | Genotype ** | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HA | NA | PB2 | PB1 | PA | NP | M | NS | |||
| H1N1 | A/environment/Kagoshima/KU-G4/2020 (H1N1) | NA | NA | PB2-I | PB1-II | PA-I | NP-II | M-I | NS-I | NA *** |
| H3N2 | A/environment/Kagoshima/KU-4b/2019 (H3N2) | H3-I | N2-I | PB2-I | PB1-II | PA-II | NP-I | M-II | NS-I | NA |
| H3N8 | A/environment/Kagoshima/KU-6a/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-I | PA-II | NP-I | M-II | NS-I | H3N8-I |
| A/environment/Kagoshima/KU-6c/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-II | PA-II | NP-I | M-II | NS-I | H3N8-II | |
| A/environment/Kagoshima/KU-6d/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-II | PA-II | NP-I | M-II | NS-I | H3N8-II | |
| A/environment/Kagoshima/KU-D3/2019 (H3N8) | H3-II | N8-I | PB2-II | PB1-I | PA-III | NP-IV | M-II | NS-I | H3N8-III | |
| A/environment/Kagoshima/KU-G2/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-II | PA-II | NP-I | M-II | NS-I | H3N8-II | |
| A/environment/Kagoshima/KU-G3/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-I | PA-IV | NP-II | M-I | NS-I | H3N8-IV | |
| A/environment/Kagoshima/KU-J2/2019 (H3N8) | H3-I | N8-I | PB2-I | PB1-III | PA-II | NP-IV | M-I | NS-I | H3N8-V | |
| H4N6 | A/environment/Kagoshima/KU-J1/2019 (H4N6) | NA | N6-II | PB2-I | PB1-II | PA-IV | NP-I | M-I | NS-I | H4N6-I |
| A/environment/Kagoshima/KU-4c/2019 (H4N6) | N6-II | PB2-IV | PB1-II | PA-I | NP-IV | M-I | NS-I | H4N6-II | ||
| A/environment/Kagoshima/KU-6e/2019 (H4N6) | N6-II | PB2-I | PB1-II | PA-II | NP-IV | M-I | NS-I | H4N6-III | ||
| A/environment/Kagoshima/KU-6f/2020 (H4N6) | N6-II | PB2-IV | PB1-II | PA-I | NP-IV | M-I | NS-I | H4N6-II | ||
| H6N6 | A/environment/Kagoshima/KU-3a/2019 (H6N6) | NA | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I |
| A/environment/Kagoshima/KU-5a/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| A/environment/Kagoshima/KU-6b/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| A/environment/Kagoshima/KU-3b/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| A/environment/Kagoshima/KU-4a/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| A/environment/Kagoshima/KU-B1/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| A/environment/Kagoshima/KU-D1/2019 (H6N6) | N6-I | PB2-I | PB1-I | PA-II | NP-II | M-I | NS-II | H6N6-II | ||
| A/environment/Kagoshima/KU-G1/2019 (H6N6) | N6-I | PB2-III | PB1-IV | PA-V | NP-III | M-III | NS-III | H6N6-I | ||
| H11N2 | A/environment/Kagoshima/KU-3c/2019 (H11N2) | NA | N2-I | PB2-I | PB1-II | PA-III | NP-I | M-I | NS-I | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuya, K.; Khalil, A.M.; Esaki, M.; Kojima, I.; Nishi, N.; Koyamada, D.; Matsui, T.; Yoshida, Y.; Ozawa, M. Genetic Characterization of Avian Influenza Viruses Isolated from the Izumi Plain, Japan in 2019/20 Winter Season. Pathogens 2022, 11, 1013. https://doi.org/10.3390/pathogens11091013
Okuya K, Khalil AM, Esaki M, Kojima I, Nishi N, Koyamada D, Matsui T, Yoshida Y, Ozawa M. Genetic Characterization of Avian Influenza Viruses Isolated from the Izumi Plain, Japan in 2019/20 Winter Season. Pathogens. 2022; 11(9):1013. https://doi.org/10.3390/pathogens11091013
Chicago/Turabian StyleOkuya, Kosuke, Ahmed Magdy Khalil, Mana Esaki, Isshu Kojima, Natsuko Nishi, Donna Koyamada, Tsutomu Matsui, Yuuhei Yoshida, and Makoto Ozawa. 2022. "Genetic Characterization of Avian Influenza Viruses Isolated from the Izumi Plain, Japan in 2019/20 Winter Season" Pathogens 11, no. 9: 1013. https://doi.org/10.3390/pathogens11091013